Introduction
Tumor necrosis factor related apoptosis-inducing
ligand (TRAIL) is a member of the tumor necrosis factor cytokine
superfamily and has a homotrimeric structure. Since TRAIL induces
apoptosis in a variety of transformed and cancer cells, but not in
normal cells, it is a promising target for cancer prevention and
treatment. However, growing evidence suggests that some cancer cell
types such as malignant melanoma, glioma and non-small cell lung
cancer (NSCLC) cells are resistant to TRAIL-induced apoptosis
(1). Moreover, TRAIL-responsive
tumors acquire a resistant phenotype that renders TRAIL therapy
ineffective. Overcoming the TRAIL resistance of cancer cells is
necessary for effective TRAIL therapy and drugs potentiating TRAIL
effectiveness are urgently required (2). TRAIL binds to receptors (DRs) that
contain death domains such as death receptor (DR) 4/TRAIL-receptor
1 (TRAIL-R1) and DR5/TRAIL-R2 (3).
This binding induces oligomerization of the receptors and
conformational changes in the death domains, resulting in the
formation of a death-inducing signaling complex; and subsequent
activation of caspase-8. In turn, activated caspase-8 activates the
effector caspase-3/6/7, which executes the apoptotic process
(4,5). The activation of caspase-8 is also
linked to the intrinsic (mitochondrial) apoptotic pathway.
Activated caspase-8 can cleave and activate the pro-apoptotic
Bcl-2-family molecule Bid, which in turn activates other
Bcl-2-family molecules, Bax and Bak, resulting in their
oligomerization and the formation of megachannels in the outer
mitochondrial membrane. The release of cytochrome c through
the Bax/Bak megachannels into the cytosol induces the assembly of
apoptosome and the activation of caspase-9, resulting in the
activation of caspase-3/6/7 (4).
Recently, various pro-apoptotic receptor agonists such as
recombinant human TRAIL and agonistic antibodies against DR4/DR5
have been subjected to clinical trials in a variety of cancer cell
types, including malignant melanoma and NSCLC cells. Unfortunately,
the results showed that these receptor agonists were disappointedly
only modestly effective (6).
Induction of apoptosis by the intrinsic pathway is considered to be
the major mechanism of conventional chemotherapy and is therefore a
critical target in cancer treatment. However, clinical observations
suggest that amplification of the known apoptotic pathways is not
sufficient for overcoming TRAIL resistance in cancer cells.
Reactive oxygen species (ROS) such as superoxide
(O2−), hydrogen peroxide
(H2O2), and hydroxyl radicals (•OH) are
products of normal metabolism in virtually all aerobic organisms
and are also produced following xenobiotic exposure. Low
physiological levels of ROS function as second messengers in
intracellular signaling and are required for normal cell function,
while excessive ROS cause damage to multiple macromolecules, impair
cell function, and promote cell death (7). Imbalance between ROS production and
the ability of a biological system to detoxify oxidants or to
repair the resulting damage leads to oxidative stress. Several
lines of evidence suggest that intracellular
H2O2 is a mediator of DR ligand-induced
apoptosis in tumor cells. The flavonoid wogonin kills
TNF-α-resistant T-cell leukemia cells and sensitizes them to TNF-α-
or TRAIL-induced apoptosis by increasing intracellular
H2O2 levels (8). LY303511 sensitizes human
neuroblastoma cells to TRAIL, and intracellular
H2O2 generation plays a role in this effect
(9). On the other hand, some
evidence suggests that H2O2 acts as an
anti-apoptotic factor in DR ligand-induced apoptosis. The
continuous presence of low concentrations of
H2O2 inhibits caspase-mediated apoptosis in
Jurkat cells by inactivating pro-caspase-9 (10). It was reported that in human
astrocytoma cells, H2O2 is generated in a
caspase-dependent manner and contributes to resistance to
TRAIL-induced apoptosis (11).
Thus, the role of H2O2 in DR ligand-induced
apoptosis is controversial.
Here we demonstrate that H2O2
induces cell death in TRAIL-resistant melanoma cells via
intracellular O2− generation. Our data
suggest that the intracellular O2− mediates
ER-associated cell death in these cells. Importantly, normal
primary melanocytes were much lesser sensitive than malignant
melanoma cells to oxidative cell death.
Materials and methods
Chemicals and antibodies
Reagents were obtained from the following
manufacturers: soluble recombinant human TRAIL, Enzo Life Sciences
(San Diego, CA); DATS, Wako Pure Chemicals (Osaka, Japan);
thapsigargin, Sigma-Aldrich (St. Louis, MO);
z-VAD-fluoromethylketone (fmk) (VAD), z-DEVD-fmk (DEVD), z-IETD-fmk
(IETD), z-LETD-fmk (LETD) and Mn (III) tetrakis (4-benzonic acid)
porphyrin chloride (MnTBaP), Calbiochem (La Jolla, CA). z-LEVD-fmk
(LEVD) and z-ATAD-fmk (ATAD), BioVision (Mountain View, CA);
dichlorohydrofluorescein diacetate (DCFH-DA), dihydroethidium
(DHE), MitoSOX™ Red (MitoSOX) and
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benz-imidazolocarbocyamine
iodide (JC-1), Life Technologies Japan (Tokyo, Japan). Reagents
were dissolved in dimethylsulfoxide and diluted with Hanks’
balanced salt solution (HBSS; pH 7.4) to a final concentration of
<0.1% before use. Dimethylsulfoxide alone at a concentration of
0.1% (vehicle) showed no effects throughout this study. Polyclonal
antibodies against X-box-binding protein-1 (XBP-1) and
glucose-related protein 78 (GRP78) were purchased from Santa Cruz
Biotechnology (Santa Cruz, CA). All the other chemicals were of
analytical grade.
Cell culture
Human A2058 and SK-MEL-2 melanoma cells were
obtained from Health Science Research Resource Bank (Osaka, Japan).
Human A375 melanoma cells were obtained from American Type Culture
Collection (Manassas, VA). These cells were cultured in high
glucose-containing Dulbecco’s modified Eagle’s medium (DMEM;
Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS; JRH
Biosciences, Lenexa, KS) in a 5% CO2-containing
atmosphere. The cells were harvested by incubation in 0.25%
trypsin-EDTA medium (Life Technologies Japan) for 5 min at 37°C.
Normal human epidermal melanocytes were obtained from Cascade
Biologics (Portland, OR) and cultured in DermaLife Basal medium
(Kurabo, Osaka, Japan) supplemented with DermaLife M LifeFactors
(Kurabo) in a 5% CO2-containing atmosphere. The cells
were harvested by incubation in 0.25% trypsin-EDTA medium for 5 min
at 37°C.
Determination of cell death by
fluorescence microscopy
The overall cell death was evaluated by performing
fluorescence microscopy as previously described (12). Briefly, the cells (1×104
cells) were placed on 8-chamber coverslips (Asahi Glass Co., Tokyo,
Japan) and treated with the agents to be tested for 24 h at 37°C in
a 5% CO2-containing atmosphere. The cells were then
stained with 4 μM each of calcein-AM and ethidium bromide
homodimer-1 (EthD-1) to label live and dead cells, respectively,
using a commercially available kit (LIVE/DEAD®
Viability/Cytotoxicity kit; Life Technologies Japan) according to
the manufacturer’s instructions. Images were obtained using a
fluorescence microscope (IX71 inverted microscope, Olympus, Tokyo,
Japan) and analyzed using the LuminaVision software (Mitani
Corporation, Fukui, Japan).
Determination of apoptotic cell
death
Apoptotic cell death was quantitatively assessed as
previously described (12).
Briefly, the cells plated in 24-well plates (2×105
cells/well) were treated with TRAIL and the agents to be tested
alone or together for 20 h in DMEM containing 10% FBS (FBS/DMEM).
Subsequently, the cells were stained with FITC-conjugated Annexin V
and PI using a commercially available kit (Annexin V FITC Apoptosis
Detection Kit I; BD Biosciences, San Jose, CA). The stained cells
were analyzed in a FACSCalibur flow cytometer (BD Biosciences)
using the CellQuest software (BD Biosciences). Four cellular
subpopulations were evaluated: viable cells (Annexin
V−/PI−); early apoptotic cells (Annexin
V+/PI−); late apoptotic cells (Annexin
V+/PI+); and necrotic/damaged cells (Annexin
V−/PI+). Annexin V+ cells were
considered to be apoptotic cells.
Measurements of mitochondrial membrane
potential (ΔΨm) depolarization and caspase-3/7
activation
ΔΨm depolarization and caspase-3/7
activation were simultaneously measured by a previously described
method (12). Briefly, the cells
plated in 24-well plates (2×105 cells/well) were treated
with the agents to be tested in FBS/DMEM for 24 h, stained with the
dual sensor MitoCasp (Cell Technology Inc., Mountain View, CA), and
analyzed for their caspase-3/7 activity and ΔΨm in the
FACSCalibur using the CellQuest software. Changes in ΔΨm
were also measured using the lipophilic cation JC-1 by a previously
described method (13).
Measurement of caspase-12 activation
Activated caspase-12 in living cells was detected
using the caspase-12 inhibitor ATAD conjugated to FITC as
previously described (12). This
compound binds to active caspase-12, but not to inactive
caspase-12. The cells (2×105 cells/ml) were stained with
FITC-ATAD for 30 min at 37°C using a CaspGLOW Fluorescein
Caspase-12 Staining Kit (BioVision) according to the manufacturer’s
protocol. Fluorescence was determined using the FL-1 channel of the
FACSCalibur and analyzed using the CellQuest software.
Measurement of intracellular ROS
The production of intracellular ROS was measured
using the oxidation sensitive DHE and DCFH-DA by flow cytometry by
a previously described method (14). Briefly, cells (4×105/500
μl) resuspended in HBSS were treated with the agents to be
tested and incubated at 37°C for various time periods, then
incubated with 5 μM DHE or DCFH-DA for 15 min at 37°C. The
cells were washed, resuspended in HBSS on ice and centrifuged at
4°C. The green fluorescence (DCFH-DA) and red fluorescence (DHE)
were measured using the FL-1 and FL-2 channels of the FACSCalibur,
respectively, and analyzed using the CellQuest software.
Mitochondrial O2− generation was measured
using the mitochondria-targeting probe MitoSOX Red as previously
described (14). Briefly, the
cells (4×105/500 μl) suspended in HBSS were
treated with the agents to be tested and incubated at 37°C for
various time periods, then incubated with 5 μM MitoSOX Red
for 15 min at 37°C. The cells were washed, resuspended in HBSS on
ice, and centrifuged at 4°C. The red fluorescence was measured
using the FL-2 channels of the FACSCalibur and analyzed by
CellQuest software.
Detection of intercellular ROS by
fluorescent microscopy
The cells (1×104) were plated on
8-chamber cover glasses and treated with the agents to be tested
for 30 min at 37°C in a 5% CO2 containing atmosphere.
After removal of the medium, the cells were stained with 4
μM each of DCFH-DA and DHE to label cells producing
H2O2 and O2−,
respectively. Images were obtained with the fluorescence microscope
and analyzed using LuminaVision software.
Western blot analysis
Western blot analysis was carried out by the
previously described method (12).
The cells in 6-well plates (1×106 cells/ml/well) were
treated with the agents to be tested for 24 h at 37°C, washed and
lysed with SDS-sample buffer. The whole cell lysates were subjected
to SDS-PAGE and transferred to PVDF membranes (Nippon Millipore,
Tokyo, Japan). After blocking the membranes with BlockAce
(Dainippon Sumitomo Pharma, Osaka, Japan) at room temperature for
60 min, GRP78 and XBP-1 proteins on the membranes were detected
using specific antibodies. Antibody-antigen complexes were detected
using the ECL Prime Western Blotting Reagent (GE Healthcare Japan,
Tokyo, Japan). To verify equal loading, the membranes were
re-probed with an antibody against β-actin or GAPDH.
Results
H2O2 induces cell
death in human TRAIL-resistant melanoma cells
First, we examined the effect of exogenously applied
H2O2 on melanoma cell survival. After
treatment with H2O2 at varying
concentrations, A375 cells were stained with calcein-AM and EthD-1
and subjected to fluorescence microscopic analysis. Live cells were
stained green with calcein-AM, while dead cells with compromised
cell membranes were stained red with EthD-1. As shown in Fig. 1A, treatment with 100 μM
H2O2 for 24 h resulted in considerable cell
death, while treatment with 100 ng/ml TRAIL had a marginal effect.
Similarly, A2058 cells and SK-MEL-2 cells were killed by
H2O2, but not by TRAIL (Fig. 1B and C). In addition, more
pronounced cell death was observed in the cells treated with TRAIL
and H2O2 than in the cells treated with
either of the agents alone. The cytotoxic effects of
H2O2 on TRAIL-resistant melanoma cells were
confirmed by apoptosis measurements using Annexin V/PI staining.
H2O2 treatment resulted in a dose- and
time-dependent increase in apoptosis in the A375 cells (Fig. 1D and E). Treatment with
H2O2 up to 30 μM for 24 h resulted in
only a modest (maximum 15%) increase in apoptosis (Annexin
V+ cells) and a moderate (35%) apoptosis was observed
after 72 h. Treatment with 100 μM H2O2
for 24 h caused a moderate apoptosis and 70–90% of the cells
underwent apoptosis at 72 h. While 30 μM
H2O2 primarily caused apoptosis with minimal
induction of necrosis, 100 μM H2O2
appeared to also cause necrosis, since not only Annexin
V+ but also Annexin V−/PI+ cells
were increased. The effect varied considerably in different
experiments depending on the basal level of Annexin
V−/PI+ cells. The level varied ranging from
1.6–9.2% and when the level was relatively high, necrotic cells
were increased up to 25%. These data suggest that under certain
circumstances, these cells spontaneously undergo necrosis and that
high concentrations of H2O2 promotes this
process. Consistent with the analysis by fluorescent microscopy,
TRAIL alone caused minimal apoptosis for 72 h. However, higher
amplitude of apoptosis was observed in the cells treated with TRAIL
plus 30 μM H2O2, but not TRAIL plus
100 μM H2O2 (Fig. 1F). H2O2
induced apoptosis in a dose-dependent manner in all the cell lines
tested while substantial amplification of TRAIL-induced apoptosis
by H2O2 was observed in some but not all cell
lines (data not shown). The amplification was more pronounced with
H2O2 at low concentrations than with
H2O2 at high concentrations. These data show
that H2O2 treatment induces cell death in
human TRAIL-resistant melanoma cells. Subsequently, we investigated
the H2O2-induced cell death in greater detail
using A375 cells as a model cell system.
H2O2 induces
melanoma cell death in a caspase-dependent or -independent manner,
depending on the concentration applied
To understand the mechanisms underlying the
H2O2-induced cell death, we examined whether
or not the cell death was caspase-dependent. Analysis using
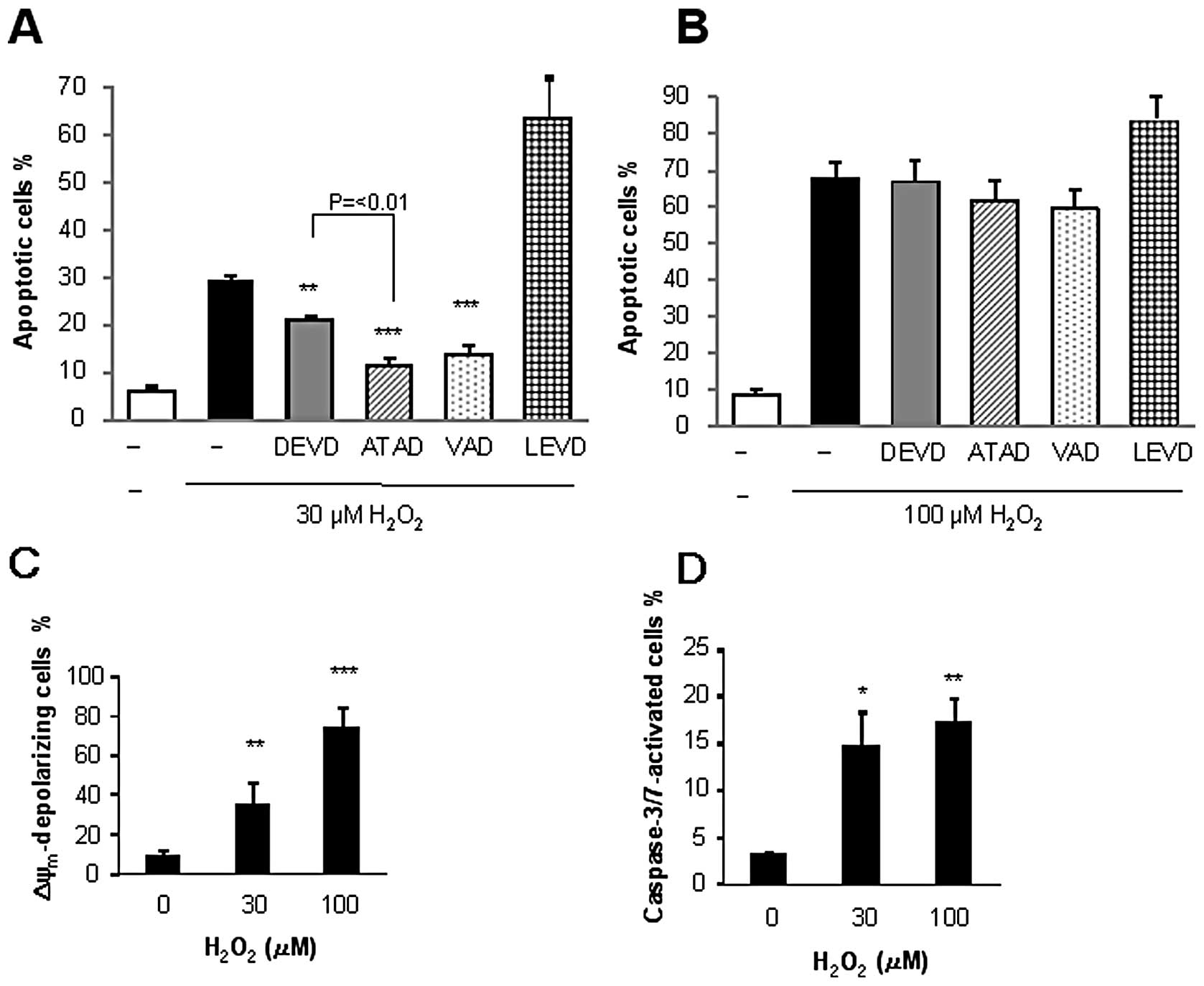
caspase-specific inhibitors revealed that
H2O2-induced apoptosis exhibited discriminate
sensitivities to the given caspase inhibitor depending on the
concentration of the oxidant applied. The general caspase inhibitor
VAD pronouncedly inhibited the apoptosis induced by 30 μM
H2O2 (maximum of 63% inhibition), while the
caspase-3/7 inhibitor DEVD reduced it by 40% (Fig. 2A). Strikingly, the caspase-12
inhibitor ATAD was as effective as VAD and was significantly more
effective than DEVD at inhibiting apoptosis. By contrast, the
caspase-4 inhibitor LEVD enhanced rather than suppressed apoptosis.
Interestingly, all these inhibitors minimally inhibited the
apoptosis induced by 100 μM H2O2
(Fig. 2B). We further examined the
effects of H2O2 on the ΔΨm and
caspase-3/7 activation, as these are hallmarks of the intrinsic
apoptotic pathway. Flow cytometric analysis using the fluorescent
ΔΨm-sensitive dye or the caspase-3/7-specific probe
revealed that H2O2 induced ΔΨm
depolarization and caspase-3/7 activation in a dose-dependent
manner (Fig. 2C and D). These data
show that H2O2 stimulates multiple death
pathways including intrinsic apoptotic and caspase-independent
apoptotic and necrotic death pathways, depending on the
concentration applied.
H2O2 induces
intracellular O2− generation, which mediates
apoptosis in human TRAIL-resistant melanoma cells
To elucidate the potential role of ROS in the
H2O2-induced cell death, we analyzed the
generation of intracellular ROS after H2O2
treatment using the oxidation-sensitive dye DCF or DHE. DCF can
react with multiple oxidants such as H2O2,
peroxynitrite (ONOO−) and •OH, but an increase in DCF
fluorescence can be considered to primarily represent increased
H2O2 level, since H2O2
is the most stable among these DCF-reacting oxidants. On the other
hand, DHE undergoes two-electron-oxidation to form DNA-binding
ethidium bromide; the reaction is mediated by
O2−, but not by H2O2 or
ONOO−. Consequently, DCFH-DA and DHE have been widely
used to assess the generation of intracellular
H2O2 and O2−,
respectively, in various rodent and human non-transformed and
transformed cells (15–18). A375 cells were treated with
H2O2 for 30 min and analyzed for their DCF or
DHE fluorescence by performing fluorescence microscopy. As shown in
Fig. 3A, only a modest increase in
DCF (green) fluorescence, but not DHE (red) fluorescence, was
observed in TRAIL-treated cells. This increase in DCF fluorescence
was transient and declined to below the basal level within 1 h. On
the other hand, unexpectedly, H2O2 treatment
increased not only DCF fluorescence but also DHE fluorescence,
indicating the production of intracellular
O2−. This oxidative response was confirmed by
flow cytometric analysis. The increase in the DHE signal was
initially observed at 1 h (Fig.
3B), and it lasted for at least 4 h; the signal was completely
abolished by superoxide dismutase (SOD) mimetic antioxidant MnTBaP
(Fig. 3C).
Because mitochondria are the major site of ROS
generation under physiological conditions, we examined the role of
this organelle in H2O2-induced
O2− generation. MitoSOX localizes to the
mitochondria and serves as a fluoroprobe for selective detection of
O2− in the organelle (15,19).
As shown in Fig. 3D, a substantial
increase in the MitoSOX signal was initially observed at 1 h after
treatment and the increase lasted for at least 4 h; again, MnTBaP
completely abolished the effect (Fig.
3E). Similar effects were also observed in A2058 cells (data
not shown). To understand the role of intracellular
O2− in H2O2-induced
cell death, we examined the effect of MnTBaP treatment on cell
death. The antioxidant blocked apoptosis in a dose-dependent manner
(Fig. 3F). In addition to
apoptosis, necrotic cell death induced by 100 μM
H2O2 was also reduced. On the contrary,
catalase had no effect on the cell death (data not shown). These
results show that H2O2, but not TRAIL,
induces the generation of intracellular O2−,
including inside the mitochondria in TRAIL-resistant melanoma cells
and that the O2− mediates apoptosis.
H2O2 induces ER
stress responses while scavenging of O2−
inhibits them
Caspase-12 is ubiquitously expressed and is
localized to the ER membrane. It is specifically activated by ER
stress to play a key role in the stress-induced apoptosis (22–25).
Consequently, the data obtained suggested the possible role of ER
stress including caspase-12 activation in oxidative cell death. To
test this view, we examined whether H2O2
modulates caspase-12 activation. Fluorometric analysis using
FITC-ATAD revealed that H2O2 induced
caspase-12 activation in a dose-dependent manner at concentrations
that effectively induced apoptosis (Fig. 4A and B). Furthermore, MnTBaP
treatment blocked H2O2-induced caspase-12
activation. Treatment with MnTBaP (100 μM) almost completely
abolished the effect of 30 μM H2O2 and
reduced the effect of 100 μM H2O2 by
50% (Fig. 4C). These data show
that scavenging of O2− inhibits
H2O2-induced cell death and caspase-12
activation.
To obtain further evidence for the induction of ER
stress, we assessed the levels of 2 unfolded protein response (UPR)
proteins GRP78 and XBP-1, after H2O2
treatment. Western blot analysis showed that treatment with the
positive control thapsigargin considerably upregulated the
expression of GRP78 for 24 h, while H2O2
treatment did not (Fig. 4D). On
the other hand, H2O2 increased the expression
of XBP-1, although the degree varied considerably in different
experiments. Both the active spliced form (XBP-1s) and inactive
unspliced form (XBP-1u) of XBP-1 were increased, and these effects
were totally inhibited by MnTBaP treatment (Fig. 4E). Collectively, these data show
that H2O2 induces ER stress responses and
that scavenging of O2− inhibits them.
H2O2 induces
minimal apoptosis and O2− generation in
primary melanocytes
We examined the cytotoxic effect of
H2O2 on primary normal melanocytes.
Fluorescence microscopic analysis revealed that treatment with 100
ng/ml TRAIL and 100 μM H2O2 alone or
in combination for 24 h resulted in minimal cell death (data not
shown) and apoptosis (Fig. 5A) in
normal melanocytes. In addition, only minimal intracellular and
mitochondrial O2− generation was observed
after 4-h H2O2 treatment (Fig. 5B and C). These data indicate that
melanocytes are resistant to H2O2-induced
cell death and O2− generation.
Discussion
In the present study, we investigated the possible
role of H2O2 in TRAIL-induced apoptosis.
TRAIL induced no or only a marginal increase in intracellular
H2O2 levels in human TRAIL-resistant melanoma
cells. On the other hand, exogenously applied
H2O2 at relatively low concentrations (30–100
μM) substantially killed these cells. In addition, under
certain circumstances, a synergistic induction of apoptosis was
observed when H2O2 and TRAIL were applied in
combination. Collectively, these data indicate that
H2O2 is a modulator rather than primary
mediator of the cytotoxic effect of TRAIL. Interestingly, the
synergism was more clearly observed with low concentrations of
H2O2 than with high concentrations of
H2O2, suggesting that as the concentration
increases, in addition to its intrinsic mechanism,
H2O2 also stimulates apoptotic pathways that
are at least partially shared with TRAIL.
H2O2 induced apoptotic or necrotic cell
death, depending on the concentration of the oxidant applied. The
intrinsic mitochondrial pathway is considered to be the major
mechanism of apoptosis. Consistent with this view, the cell death
induced by low concentrations of H2O2 was
caspase-dependent and was associated with increased ΔΨm
collapse and caspase-3/7 activation. However, inhibition of
caspase-3/7 only partially blocked apoptosis. These data suggest
that while the intrinsic mitochondrial pathway does play a role in
inducing apoptosis, another caspase cascade may also be involved in
this caspase-dependent apoptosis.
ER can initiate cell death through a pathway that
is independent of intrinsic (mitochondria) and extrinsic (death
receptor) pathways. ER-associated cell death is thought to be
mediated by caspase-12 (22–26).
A variety of cellular conditions such as glucose deprivation,
hypoxia, disturbance of calcium homeostasis and excess ROS can
cause ER stress, which is characterized by the accumulation of
unfolded proteins. ER stress activates the adaptive UPR, which
protects cells owing to protein synthesis inhibition, chaperone
protein upregulation and an increase in protein degradation. If UPR
activation is not able to relieve ER stress, the cells undergo
ER-mediated apoptosis (22–26).
Upon ER stress, the chaperone molecule GRP78 dissociates from the
transmembrane proteins, such as inositol requiring enzyme 1α
(IRE1α) and activating transcription factor 6 (ATF6). The free ATF6
translocates to the Golgi apparatus where it is activated. The
active ATF6 in turn enters the nucleus and initiates the expression
of the transcription factor XBP-1. Activated IRE1α splices the
transcribed XBP-1 mRNA to allow translation of the mature XBP-1
protein, which acts as a transcription factor and mediates the
transcriptional upregulation of numerous genes involved in ER
function (20,21,23).
Our data showed that H2O2 induced ER stress,
as shown by caspase-12 activation and upregulated the expression of
the mature XBP-1 protein. Furthermore, inhibition of caspase-12
strongly blocked the H2O2-induced apoptosis.
Collectively, our data suggest that the ER-mediated apoptotic
pathway involving caspase-12 plays a key role in
H2O2-induced apoptosis.
Interestingly, while activation of caspase-12
following the induction of ER stress during apoptosis has been
reported in various mammalian cells including mouse, rat, rabbit
and cow (26), the role of
caspase-12 in ER-mediated apoptosis of human cells is a matter of
debate. This might be because the human caspase-12 gene contains
several mutations that block its expression (27). Nevertheless, an increasing body of
evidence suggests that a caspase-12-like protein exists and is
activated in human cells following the induction of ER stress by
divergent causes, including H2O2, cisplatin,
tetrocarcin A and hyperthermia (12,28–33).
Recently, adaptation to ER stress was suggested to
be a key driver of malignancy and resistance to therapy in cancer
cells, including malignant melanoma cells, with GRP78 playing a key
role in this adaptation (34,35).
GRP78 expression is associated with tumor development and growth
and is correlated with resistance to chemotherapeutic drugs such as
cisplatin and adriamycin (34–36).
In this study, thapsigargin substantially increased GRP78
expression, while H2O2 decreased GRP78
expression in melanoma cells; these cells were killed by
H2O2, but not thapsigargin. On the other
hand, GRP78 expression was minimally increased in
thapsigargin-sensitive Jurkat leukemia cells (Inoue and Suzuki,
unpublished data). GRP78 has been shown to exert its anti-apoptotic
function by inhibiting caspase-4 or caspase-7 activity (36). However, caspase-4 appears to
regulate H2O2-induced apoptosis negatively
rather than positively, as inhibition of the enzymatic activity
significantly enhanced the apoptosis. Given the structural
similarity between caspase-4 and caspase-12, it is possible that
GRP78 may also target caspase-12 to counteract ER-mediated
apoptosis.
ROS levels are controlled by the antioxidant
defense system, including the antioxidant enzymes manganese- or
copper-zinc-containing superoxide dismutase, which catalyze the
dismutation of O2− into
H2O2, and catalase and glutathione
peroxidase, which degrade H2O2. Our data
showed that these enzymes had no effects on
H2O2-induced cell death.
H2O2 is a diffusible molecule that is readily
transported across the cell membrane to the extracellular space.
Consequently, scavenging of extracellular
H2O2 by catalase may eventually result in a
decrease in the intracellular H2O2 level.
Therefore, the ineffectiveness of catalase to suppress
H2O2-induced cell death suggests that
H2O2in situ plays a minor role in the
cell death. The ineffectiveness of MnTBaP to enhance
H2O2-induced cell death supports this view,
since MnTBaP increased intracellular H2O2
levels. Instead, our data showed that O2− is
a key mediator in H2O2-induced cell death.
H2O2 induced persistent intracellular
O2− generation at concentrations that
effectively induced cell death. Consistent with the role of
mitochondria as the most common source of ROS during apoptosis,
H2O2 induced substantial mitochondrial
O2•− generation. Moreover, MnTBaP, a cell
permeable SOD mimetic, reduced the
H2O2-induced mitochondrial
O2− generation and cell death, In addition,
MnTBaP blocked H2O2-induced ER stress
responses such as caspase-12 and XBP-1 activation. Collectively,
these data suggest O2− most likely derived
from the mitochondria mediates ER-mediated apoptosis, thereby
promoting H2O2-induced cell death.
In conclusion, we have demonstrated for the first
time that H2O2 induces cell death in
TRAIL-resistant human melanoma cells via intracellular
O2− generation. Further studies on the
mechanisms by which H2O2 induces this
O2− generation are under way. Since melanoma
cells are much more susceptible to oxidative cell death than normal
primary melanocytes, H2O2 has therapeutic
potential in the treatment of malignant melanoma.
Acknowledgements
We thank Dr M. Murai for her
technical assistance. This study was supported in part by a
Grant-in-Aid from the Ministry of Education, Culture, Sports,
Science and Technology (KAKENHI 23591631; to Y.S.) and
Grants-in-Aid from Nihon University (to Y.S.).
References
|
1
|
Dyer MJ, MacFarlane M and Cohen GM:
Barriers to effective TRAIL-targeted therapy of malignancy. J Clin
Oncol. 25:4505–4506. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Johnstone RW, Frew AJ and Smyth MJ: The
TRAIL apoptotic pathway in cancer onset, progression and therapy.
Nat Rev Cancer. 8:782–798. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
LeBlanc HN and Ashkenazi A: Apo2L/TRAIL
and its death and decoy receptors. Cell Death Differ. 10:66–75.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang S: The promise of cancer therapeutics
targeting the TNF-related apoptosis-inducing ligand and TRAIL
receptor pathway. Oncogene. 27:6207–6215. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sayers TJ: Targeting the extrinsic
apoptosis signaling pathway for cancer therapy. Cancer Immunol
Immunother. 60:1173–1180. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dimberg LY, Anderson CK, Camidge R,
Behbakht K, Thorburn A and Ford HL: On the TRAIL to successful
cancer therapy? Predicting and counteracting resistance against
TRAIL-based therapeutics. Oncogene. May 14–2012.(Epub ahead of
print). View Article : Google Scholar
|
|
7
|
Circu ML and Aw TY: Reactive oxygen
species, cellular redox systems, and apoptosis. Free Radic Biol
Med. 48:749–762. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fas SC, Baumann S, Zhu JY, et al: Wogonin
sensitizes resistant malignant cells to TNFalpha- and TRAIL-induced
apoptosis. Blood. 108:3700–3706. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shenoy K, Wu Y and Pervaiz S: LY303511
enhances TRAIL sensitivity of SHEP-1 neuroblastoma cells via
hydrogen peroxide-mediated mitogen-activated protein kinase
activation and up-regulation of death receptors. Cancer Res.
69:1941–1950. 2009. View Article : Google Scholar
|
|
10
|
Barbouti A, Amorgianiotis C, Kolettas E,
Kanavaros P and Galaris D: Hydrogen peroxide inhibits
caspase-dependent apoptosis by inactivating procaspase-9 in an
iron-dependent manner. Free Radic Biol Med. 43:1377–1387. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Choi K, Ryu SW, Song S, Choi H, Kang SW
and Choi C: Caspase-dependent generation of reactive oxygen species
in human astrocytoma cells contributes to resistance to
TRAIL-mediated apoptosis. Cell Death Differ. 17:833–845. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Suzuki Y, Inoue T, Murai M, et al:
Depolarization potentiates TRAIL-induced apoptosis in human
melanoma cells: role for ATP-sensitive K+ channels and
endoplasmic reticulum stress. Int J Oncol. 41:465–475.
2012.PubMed/NCBI
|
|
13
|
Suzuki Y, Yoshimaru T, Inoue T and Ra C:
Mitochondrial Ca2+ flux is a critical determinant of the
Ca2+ dependence of mast cell degranulation. J Leukoc
Biol. 79:508–518. 2006.
|
|
14
|
Inoue T, Suzuki Y, Yoshimaru T and Ra C:
Reactive oxygen species produced up-or downstreame of calcium
influx regulate proinflammatory mediator release from mast cells:
role of NADPH oxidase and mitochondria. Biochem Biophys Acta.
1783:789–802. 2008. View Article : Google Scholar
|
|
15
|
Robinson KM, Janes MS, Pehar M, et al:
Selective fluorescencet imaging of superoxide in vivo using
ethidium-based probes. Proc Natl Acad Sci USA. 103:15038–15043.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Carter WO, Narayanan PK and Robinson JP:
Intracellular hydrogen peroxide and superoxide anion detection in
endothelial cells. J Leukoc Biol. 55:253–258. 1994.PubMed/NCBI
|
|
17
|
Devadas S, Hinshaw JA, Zaritskaya L and
Williams MS: Fas-stimulated generation of reactive oxygen species
or exogenous oxidative stress sensitize cells to Fas-mediated
apoptosis. Free Radic Biol Med. 35:648–661. 2003. View Article : Google Scholar
|
|
18
|
Suzuki Y, Yoshimaru T, Inoue T and Ra C:
Discrete generations of intracellular hydrogen peroxide and
superoxide in antigen-stimulated mast cells: reciprocal regulation
of store-operated Ca2+ channel activity. Mol Immunol.
46:2200–2209. 2009. View Article : Google Scholar
|
|
19
|
Mukhopadhyay P, Rajesh M, Kashiwaya Y,
Haskó G and Pacher P: Simple quantitative detection of
mitochondrial superoxide production in live cells. Biochem Biophys
Res Commun. 358:203–208. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Breckenridge DG, Germain M, Mathai JP, et
al: Regulation of apoptosis by endoplasmic reticulum pathways.
Oncogene. 22:8608–8618. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Boyce M and Yuan J: Cellular response to
endoplasmic reticulum stress: a matter of life or death. Cell Death
Differ. 13:363–373. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nakagawa T, Zhu H, Morishima N, et al:
Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and
cytotoxicity by amyloid-beta. Nature. 403:98–103. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Groenendyk J and Michalak M: Endoplasmic
reticulum quality control and apoptosis. Acta Biochim Pol.
52:381–395. 2005.PubMed/NCBI
|
|
24
|
Rao RV, Castro-Obregon S, Frankowski H, et
al: Coupling endoplasmic reticulum stress to the cell death
program. An Apaf-1-independent intrinsic pathway. J Biol Chem.
277:21836–21842. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Morishima N, Nakanishi K, Takenouchi H,
Shibata T and Yasuhiko Y: An endoplasmic reticulum stress-specific
caspase cascade in apoptosis. Cytochrome c-independent activation
of caspase-9 by caspase-12. J Biol Chem. 277:34287–34294. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Szegezdi E, Fitzgerald U and Samali A:
Caspase-12 and ER-stress-mediated apoptosis: the story so far. Ann
NY Acad Sci. 1010:186–194. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fischer H, Koenig U, Eckhart L and
Tschachler E: Human caspase 12 has acquired deleterious mutations.
Biochem Biophys Res Commun. 293:722–726. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pallepati P and Averill-Bates DA:
Activation of ER stress and apoptosis by hydrogen peroxide in HeLa
cells: protective role of mild heat preconditioning at 40°C.
Biochim Biophys Acta. 12:1987–1999. 2011.PubMed/NCBI
|
|
29
|
Mandic A, Hansson J, Linder S and Shoshan
MC: Cisplatin induces endoplasmic reticulum stress and
nucleus-independent apoptotic signaling. J Biol Chem.
278:9100–9106. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tinhofer I, Anether G, Senfter M, et al:
Stressful death of T-ALL tumor cells after treatment with the
anti-tumor agent Tetrocarcin-A. FASEB J. 16:1295–1297.
2002.PubMed/NCBI
|
|
31
|
Xie Q, Khaoustov VI, Chung CC, et al:
Effect of tauroursodeoxycholic acid on endoplasmic reticulum
stress-induced caspase-12 activation. Hepatology. 36:592–601. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Trisciuoglio D, Uranchimeg B, Cardellina
JH, et al: Induction of apoptosis in human cancer cells by
candidaspongiolide, a novel sponge polyketide. J Natl Cancer Inst.
100:1233–1246. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shellman Y, Howe WR, Miller LA, et al:
Hyperthermia induces endoplasmic reticulum-mediated apoptosis in
melanoma and non-melanoma skin cancer cells. J Invest Dermatol.
128:949–956. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hersey P and Zhang XD: Adaptation to ER
stress as a driver of malignancy and resistance to therapy in human
melanoma. Pigment Cell Melanoma Res. 21:358–367. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rutkowski DT and Kaufman RJ: That which
does not kill me makes me stronger: adapting to chronic ER stress.
Trends Biochem Sci. 32:469–476. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jiang CC, Mao ZG, Avery-Kiejda KA, Wade M,
Hersey P and Zhang XD: Glucose-regulated protein 78 antagonizes
cisplatin and adriamycin in human melanoma cells. Carcinogenesis.
30:197–204. 2009. View Article : Google Scholar : PubMed/NCBI
|