Introduction
Pathological angiogenesis and abnormal vessel
structures and functions are common features of solid tumors and
several non-malignant diseases, such as schwannomas and diabetic
retinopathy (DR) (1,2). Vessels in these lesions are highly
disorganized and inefficient, and lack pericytes or normal
attachment between pericytes and endothelial cells (ECs) (3–5).
Such abnormalities of tumor vessels in turn lead to impairment of
endothelial barrier function, decreased blood flow and subsequent
aggravation of hypoxia in tumor tissues, which result in a poor
delivery of anticancer therapeutics and augmentation of
radioresistance in tumors. Similar vessel phenotypes are shown in
DR characterized by breakdown of the blood-retinal barrier (BRB)
that lead to diabetic macular edema and vision loss (6).
By correcting the aberrance in morphological
structure and function of vessels, we could normalize the
microenvironment of retina in favor of disease control and improve
response to other therapies (7,8). The
strategies for vascular normalization are likely multifaceted.
Substantial evidence indicates that anti-angiogenic treatment could
achieve this goal by pharmacological inhibition of vascular
endothelial growth factor (VEGF) signaling and it has become a
widely accepted treatment for several diseases where
neovascularization and permeability plays a pivotal role, including
cancer and retinal disorders (8,9).
Bevacizumab, a humanized, monoclonal anti-VEGF antibody has been
applied clinically for this reason. Nonetheless, other strategies
exist with distinct efficacy.
3-Hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase
inhibitors, or statins, that have been reported to exert impressive
beneficial effects, such as anti-inflammation, anti-oxidation,
protecting against cardiovascular events and DR and anticancer
efficacy via lipid-independent mechanisms (10–12).
Statins have been shown to maintain endothelial tight junctions and
markedly reduce retinal microvascular permeability through
mitigation of oxidative stress and inflammatory status by
downregulating reactive oxygen species (ROS) generation, VEGF and
intercellular adhesion molecule (ICAM-1) expression in diabetic
animals (10,11,13).
Oxidative stress and elevated ROS generation have been incriminated
in many pathological conditions, including diabetes and cancer
(14,15). Hypoxia and other adverse stimuli
inducing high levels of ROS contribute to endothelium dysfunction
and apoptosis in both diabetic retina and tumor microenvironment
(16–18). Additionally, ROS were found to
increase hypoxia inducible factor (HIF)-1α and VEGF expression in
cancer cells, suggesting a role of ROS in regulation of
angiogenesis and tumor growth (19). Statins, act as an anti-oxidant,
scavenging redundant oxygen radicals and hence modulate endothelial
barrier functions. Indeed, biphasic effects of statins on
angiogenesis have been observed in microvascular endothelium.
Low-dose statins promote proliferation and migration of ECs via
stimulation of the PI3K-Akt kinase pathway, resulting in the
activation of endothelial NO synthase (eNOS) and concomitantly
increasing endothelial nitric oxide (NO) production (20,21).
Despite the generally acknowledged impression of NO as a
proangiogenic factor, eNOS-derived NO has been shown to induce
pericyte recruitment and coverage to immature angiogenic vessels
and subsequent stabilization of angiogenic vessels in tumor models
(22), which are representative
hallmarks of vascular normalization. On the other hand, high
concentration of statins exert anti-angiogenic activity by markedly
depleting ROS production and inhibiting synthesis of VEGF and
activation of HIF-1α, key mediators that involve in amelioration of
vascular hyperpermeability (11,20).
Along the pleiotropic actions of statins on
microvascular endothelium mentioned above, in the present study, we
have tested the hypothesis that simvastatin, a second generation of
statins, is a new therapeutic agent that may induce tumor vascular
normalization when given at both low and high doses in syngeneic
C57BL/6 tumor-bearing mice, however, mediated by probably different
underlying mechanisms, as low-dose simvastatin stress on
facilitating mural cell recruitment and vessel stabilization and
high-dose simvastatin mainly acts on attenuating ROS-eliciting
vascular leakage, which was validated by administration of another
strong ROS scavenger, diphenyleneiodonium chloride, in the tumor
models.
Materials and methods
Cells and regents
B16F10 melanoma and Lewis lung carcinoma (LLC) cells
(ATCC, Rockville, MD) were cultured in RPMI-1640 supplemented with
10% FBS (Gibco, Grand Island, NY). Human umbilical vein endothelial
cells (HUVEC, ATCC) were maintained in endothelial cell medium
(ECM, ScienCell, San Diego, CA) adding EC growth supplement and 5%
FBS. For in vitro experiments, simvastatin prodrug (Sigma,
St. Louis, MO) was activated to its active forms as described
(23). FITC-labelled lectin
(Lycopersicon esculentum) was purchased from Vector
Laboratories (Lowelville, OH). Diphenyleneiodonium chloride (DPI)
were purchased from Calbiochem (San Diego, CA).
2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA)
were obtained from Molecular Probes (Invitrogen, Carlsbad, CA).
Measurement of EC permeability
To determine the permeability of HUVEC monolayer
using FITC-albumin penetrate assays, HUVEC were seeded onto
gelatin-coated polystyrene filter inserts (Costar Transwell, no.
3470, 6.5-mm diameter, 0.4-μm pore size) at a density of
3×105 cells/insert in a final volume of 250 μl
ECM with supplements. The cells were grown to high confluence at
37°C. Subsequently, cells were treated with indicated
concentrations (0.1, 2 and 5 μM) of simvaststin for 24 h in
the culture medium under either standard cell culture conditions
(21% O2, normoxia) or hypoxia conditions (3%
O2). After which the inserts were transferred into a new
24-well plate containing serum-free media. FITC-labeled albumin
(Sigma) suspended in serum-free media was added to the EC
monolayers to a final concentration of 100 μM. The transit
extent of FITC-albumin across the monolayer was assessed by
measuring the rise of FITC-albumin in the lower chamber after 60
min and fluorescence density was quantified using a
spectrofluorometer at an excitation wavelength of 485 nm and an
emission wavelength of 535 nm. The data are reported as relative
permeability in which the control group under normoxia condition
was set to one.
Animal tumor models
Murine LLC (2×106) and B16F10 melanoma
cells (2×105) were injected s.c. into the right flank of
syngeneic female C57BL/6 mice (HFK Bioscience Co., Beijing, China)
at 6–8 weeks of age. Tumor volumes (mm3) were measured
with a caliper (length × width2 × π/6). Treatment were
initiated when tumors reached a size of ∼100 mm3 (6 days
for B16 and 8 days for LLC). Animals were randomly selected to
receive daily gastric gavage of simvastatin dissolved in 0.25%
carboxymethylcellulose (CMC)/PBS solution at 0.2 and 10 mg/kg body
weight or same amount of vehicles for 7 days. For all animal
experiments, tumor-bearing mice were anesthetized using 2%
pentobarbital sodium and the experimental protocols were approved
by the Animal Care Committee of Huazhong University of Science and
Technology (SYXK 2010-0057).
Tumor perfusion and vessel
permeability
Perfused tumor vessels were visualized by tail
intravenous injection of 0.05 mg FITC-labeled lectin (1 mg/ml in
0.9% NaCl) in tumor-bearing mice. Fluorophores were allowed to
circulate for 10 min in mice before intracardiac perfusion and
fixation by injection of saline (5 min) and 2% paraformaldehyde (7
min). Tumors were then harvested and immediately frozen in optimum
cutting temperature (OCT) compound (Sakura) in the dark. Thick (40
μm) tumor sections were incubated with rabbit anti-CD31
antibody (1:50; Abcam) at 4°C overnight and subsequently with
DyLight 594-conjugated secondary antibody (1:200; Jackson
ImmunoResearch). Vascular leakage was assessed by i.v. injection of
100 μl of 2% Evans blue dye (Sigma) and allowed to circulate
for 20 min before mice were perfused, the tumors were then excised
and the Evans blue dye was extracted from the tumor by incubation
with 1 ml of formamide at 55°C for 16 h. Concentration of the dye
was quantified by spectrophotometer at 630 nm.
Contrast-enhanced ultrasonography
Tumor blood flow was assessed using a color Doppler
flow imaging system with a broadband 1–5 MHz probe (iU22 SonoCT,
Philips, The Netherlands). Tumor-bearing mice were placed on a 37°C
electric warming plate anesthetized and i.v. injected with 0.1 ml
of microbubble contrast agent (SonoVue, Bracco, Italy) diluted in 5
ml of 0.9% saline and images were recorded starting immediately
before the injection and continuing 30 sec at a frame rate of 0.5
Hz to minimize microbubble destruction. A region of interest that
contained abundant blood flow signals in transverse plane of tumor
was drawn and analyzed by QLAB quantitative technique, vascularity
index (VI) and vascularization flow index (VFI) reflecting the
amount and intensity of blood flow signal were calculated.
Immunohistochemistry and
immunofluorescence
Hypoxia assessment was performed by
immunohistochemical staining for HIF-1α in formalin-fixed,
paraffin-embedded 4-μm thick tumor serial sections using a
rabbit anti-HIF-1α antibody (1:50, Bioss, Beijing, China).
Peroxidase-conjugated goat anti-rabbit IgG antibodies and
avidin-biotin complex-histostain kit (Zhongshan Goldenbridge,
Beijing, China) were used for revelation; sections were finally
counterstained with Mayer’s hematoxylin. For necrosis assay, LLC
and B16 tumor sections were stained with hematoxylin and eosin and
evaluated by pathologist to confirm necrotic area in tumor tissues.
Immunofluorescent staining of pericyte around tumor vessels was
detected in frozen tumor sections that were prepared as mentioned
above. Frozen tissue sections were co-immunostained with rabbit
anti-CD31 (1:50) and Cy3-conjugated mouse anti-α-smooth muscle
actin (α-SMA) (1:400; Sigma) antibodies, followed by a DyLight
488-conjugated goat anti-rabbit secondary antibody (1:200; Jackson
ImmunoResearch). Images were captured using a confocal microscope
(LMS510, Zeiss, Germary). The pericyte coverage index was estimated
and presented as the percentage of blood vessels stained for CD31
with α-SMA positive pericytes. For all immunostaining assays, five
locations from each tumor were randomly sampled and 6–8 tumors per
group were analyzed. All images were analyzed by Image-Pro Plus 6.0
software (Media Cybernetics, MD).
Western blot analysis
To detect eNOS, VEGF and HIF-1α expression level in
tumor tissues, equal amounts of protein extracts (50 μg)
were loaded on 12% SDS-PAGE and transferred onto a nitrocellulose
membrane (Millipore, Billerica, MA). After blocking with 5%
casein/TBST, the membrane was incubated with rabbit anti-eNOS
(1:100; Boster, Wuhan, China), rabbit anti-HIF-1α (1:50), rabbit
anti-VEGF (1:200; Santa Cruz, CA) or mouse anti-β-actin (1:1,000;
Santa Cruz, CA) antibody as an internal control. Protein blots were
visualized using Super Signal Chemiluminescent kit (Pierce,
Rockford, IL). The chemiluminescent signal on X-ray film was
scanned and analyzed by Quantity One software (Bio-Rad, CA).
In situ ROS detection and DPI
treatment
Intratumoral ROS production was measured in
situ on tumor slices as previously described using a
fluorescent ROS probe, H2DCFDA, a substrate without
fluorescence itself, converts to a green fluorescent product when
it is hydrolyzed by intracellular esterases (24). Briefly, tumor tissues of interest
were freshly dissected in 1X PBS and then 60–80 μm thick
frozen tumor sections were prepared for incubation with the freshly
prepared H2DCFDA (10 μM) for 15 min in dark at
room temperature. After three washes with PBS, sections were
immediately monitored by a confocal microscope and the fluorescent
intensity of DCF was semiquantified using Image-Pro Plus 6.0
software (13). To determined
alterations after ROS clearance, tumor-bearing mice received an
intraperitoneal injection of DPI (a NADPH oxidase inhibitor) or
same amount of vehicle for 5 days.
Synergy determination
Subcutaneously implanted LLC and B16 tumors were
established as described above. Tumors were allowed to grow to ∼100
mm3 (LLC, day 8; B16, day 6) and then tumor-bearing mice
were randomly divided into four groups (6–8 mice each group). Group
1 served as control; group 2 received i.p. injection of
chemotherapeutics (cisplatin for LLC tumors at a dose of 1.25 mg/kg
on days 9, 11 and 13; cyclophosphamide (CTX) for B16 tumors at a
dose of 100 mg/ kg on days 7 and 11 and vehicle (0.25% CMC, 0.2 ml)
by gavage; group 3 received cisplatin or CTX (dose regimen as
stated above) 24 h after 0.2 mg/kg simvastatin (dissolved in 0.25%
CMC, by gavage for 7 consecutive days) treatment; group 4 were
given the same chemotherapeutic modality 24 h after 10 mg/kg
simvastatin daily treatment. Tumor volume was determined every
other day till the end of treatment (day 15 for LLC and day 13 for
B16) by measurement with calipers.
Statistical analysis
The data are expressed as mean ± SEM. Differences
among groups were evaluated by ANOVA and the unpaired Student’s
t-test using SPSS 17.0 software. Statistical significance was set
at p<0.05.
Results
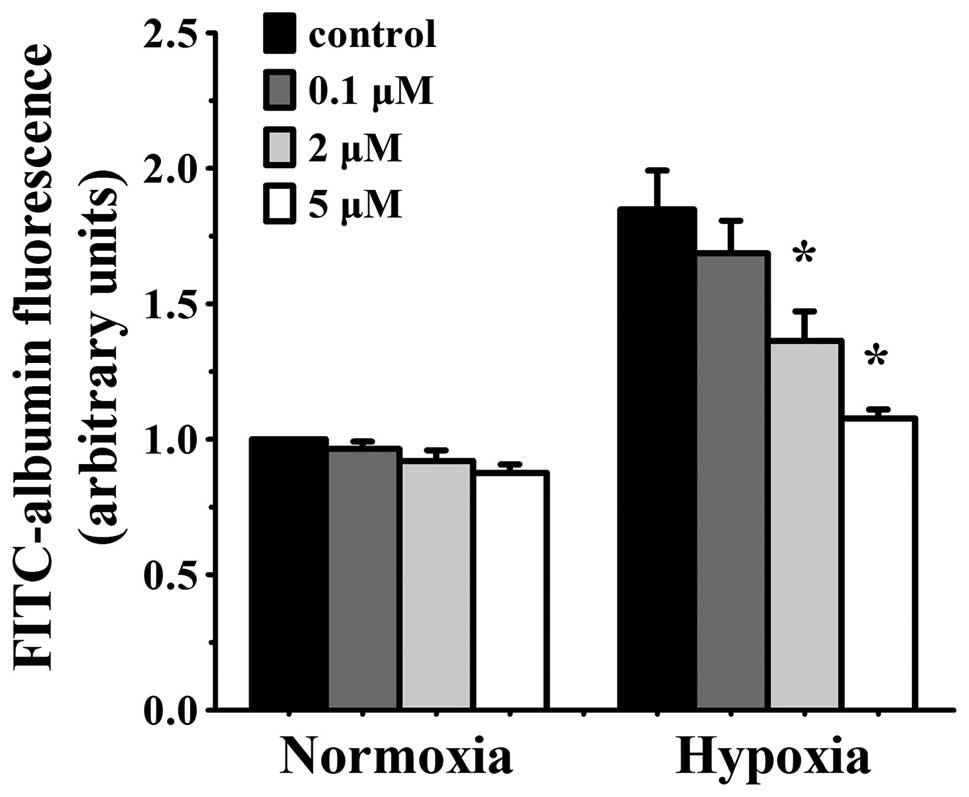
Simvastatin alleviates hypoxia-induced
endothelium permeability in HUVEC in a concentration-dependent
manner
The transfer activity of a labelling high-molecular
protein (FITC-albumin, MW, 66,000) across the EC monolayers was
measured to evaluate the efficacy of simvastatin on endothelial
barrier function in vitro. Permeability increased
significantly in HUVEC monolayers that were exposed to hypoxia for
24 h. Various concentrations of simvastatin had no effect on
normoxia cells, but markedly decreased permeability in hypoxia
cells. The reduction was concentration-dependent. Hypoxia-induced
albumin passage was slightly decreased with low concentration (0.1
μmol/l) of simvastatin but was reduced significantly in 2
μmol/l simvastatin (albumin transmit p<0.05 vs. hypoxia
control cells) and the decline was maximal at 5 μmol/l
simvastatin (p<0.05 vs. hypoxia control cells) (Fig. 1). Higher concentrations were not
available due to their effect on basal EC barrier permeability.
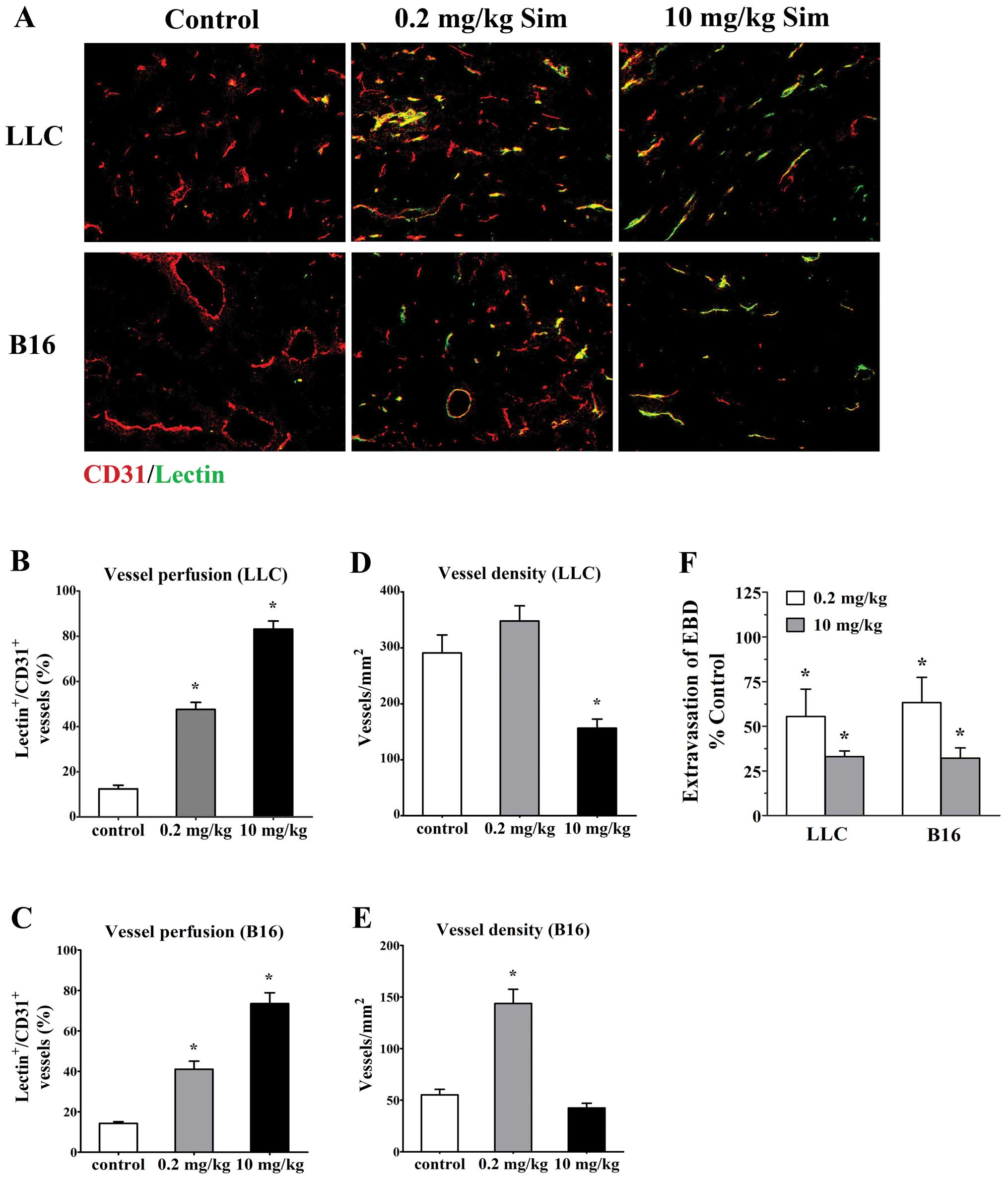
Biphasic doses of simvastatin improve
perfusion and inhibit leakiness of tumor vessels
To investigate whether the tumor vessels treated
with simvastatin (low- and high- doses) were more efficient, we
studied perfusion of tumor vasculature by delivery of FITC-lectin
and measuring the proportion of vessels stained for CD31 (all
vessels, red) that were co-immunostained with FITC-lectin
(functional vessels, green) in the bloodstream using confocal
microscopy in mice bearing LLC tumor and B16 melanoma. Staining for
EC marker CD31 showed that vessels from both untreated tumors had
chaotic structural patterns with increased tortuosity and dilatate
lumina and only very few vessels had lectin staining (12.42±1.66%
for LLC tumors and 14.23±0.91% for B16 tumors, Fig. 2A). In contrast, low-dose (0.2
mg/kg/d) simvastatin significantly increased the amount of lectin
stained vessels (47.62±3.11% for LLC tumors and 41.05±4.05% for B16
tumors, p<0.05 vs. controls, Fig.
2A–C) although overall vessel density was increased
simultaneously to some extent (Fig. 2D
and E). Moreover, after high-dose simvastatin (10 mg/kg/d)
treatment, tumor vessels appeared more sharply and discretely
outlined (Fig. 2A) and the
vascularity was markedly reduced accompanied by a great majority of
vessels perfused as the proportions of lectin-stained vessels in
LLC (83.15±3.57%, p<0.05) and B16 tumors (73.55±5.32%,
p<0.05) were greater than that in control animals (Fig. 2B–E). These data implicate an
effective role of simvastatin at increasing functional blood
vessels in tumor.
Next, the vessel permeability in tumor models was
directly assessed by comparing the extravasation of Evans blue dye
into the interstitium of treated tumors, with control ones and we
found that simvastatin treatment significantly decreased vessel
leakiness in LLC and B16 tumors. After a 7-day treatment, Evans
blue dye extravasation into low- and high-dose treated LLC tumors
were reduced to 55.47±15.36 and 33.03±3.19% (p<0.05), the degree
of Evans blue dye extravasation in size-matched controls;
corresponding data regarding Evans blue dye extravasate reduction
in biphasic-dose treated B16 tumors were 63.33±14.04% (p<0.05)
and 32.17±5.68% (p<0.05), respectively (Fig. 2F).
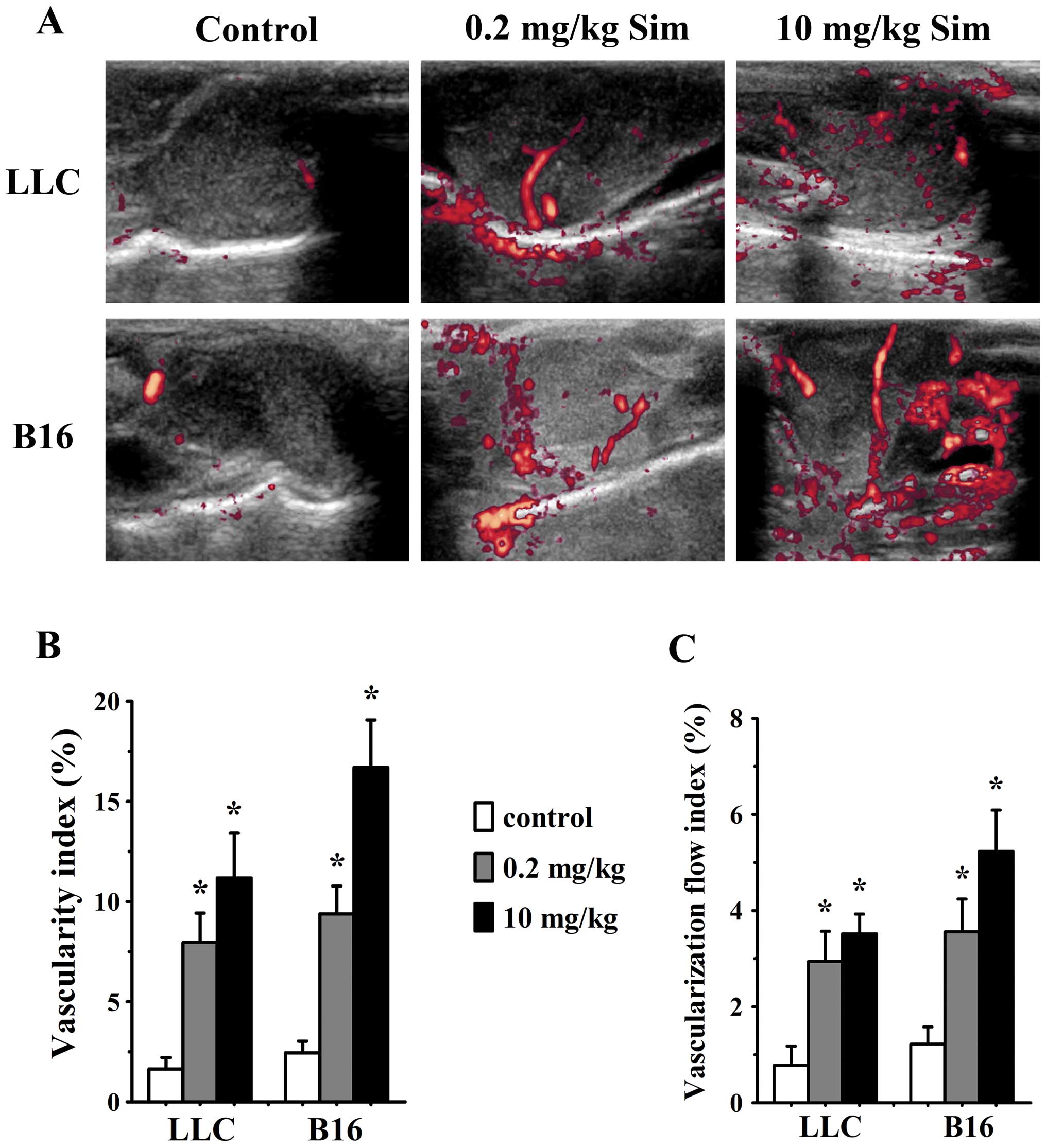
Simvastatin treatment increases tumor
blood flow
In order to further determine whether alterations of
tumor vessel structure and function could affect tumor blood flow
after simvastatin treatment, we used contrast-enhanced Doppler
imaging to evaluate signal amount and intensity of blood flow in
tumor models. Simvastatin treatment increased vascularity index
(VI) and vascularization flow index (VFI) in LLC tumors at 0.2 and
10 mg/kg/d on day 7 (p<0.05 vs. control for both), likewise,
analysis of VI and VFI confirmed a significant increase of blood
flow in 0.2 (p<0.05 vs. controls) and 10 mg/kg/d (p<0.05 vs.
controls) simvastatin-treated B16 tumors (Fig. 3).
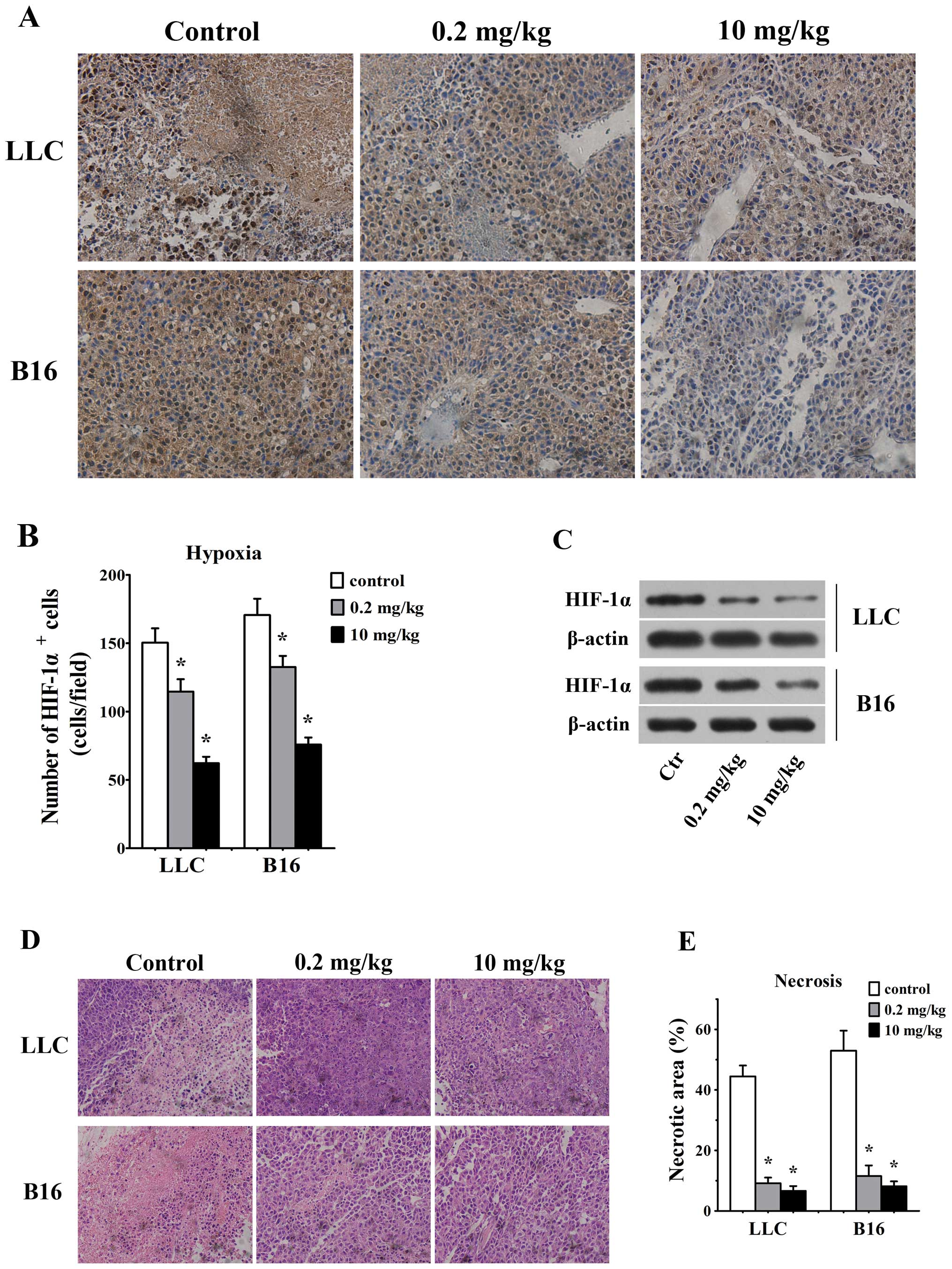
Effect of simvastatin on tissues hypoxia
and necrosis
Considering that correction of vessel structural
abnormalities improves blood flow in simvastatin-treated mice, we
considered that simvastatin, by ameliorating tumor blood flow,
would hence reduce hypoxia and necrosis in tumor tissues. HIF-1α
staining by histochemistry assays in tumor sections was used to
assess the degree of hypoxia in tumor microenvironment. As we
expected, number of HIF-1α-positive cells were reduced in LLC and
B16 tumors in response to 0.2 mg/kg/d simvastatin as compared with
untreated controls (115±9 HIF-1α/field in LLC tumor vs. 150±10
HIF-1α/field in control animals and 133±8 HIF-1α/field in B16
tumors vs. 171±12 HIF-1α/field in control ones, p<0.05, Fig. 4A and B). Also, 10 mg/kg/d
simvastatin groups showed lesser HIF-1α-positive cells in LLC (62±5
HIF-1α/field) and B16 (76±5 HIF-1α/field) tumors than control
animals (p<0.05 for both, Fig. 4A
and B). Our western blotting data further confirmed that HIF-1α
expression was decreased significantly in both LLC and B16 tumors
treated with 0.2 and 10 mg/kg/d simvastatin (Fig. 4C).
As assessed by H&E staining at day 7, 0.2 mg/kg
simvastatin-treated tumors displayed smaller necrotic area (9±2 vs.
44±4% for control LLC tumors and 12±4 vs. 53±7% for control B16
tumors, p<0.05, Fig. 4D and E).
Also, the necrotic tumor area was significantly decreased in 10
mg/kg simvastatin treated LLC (7±2%, p<0.05) and B16 tumors
(8±2%, p<0.05, Fig. 4D and
E).
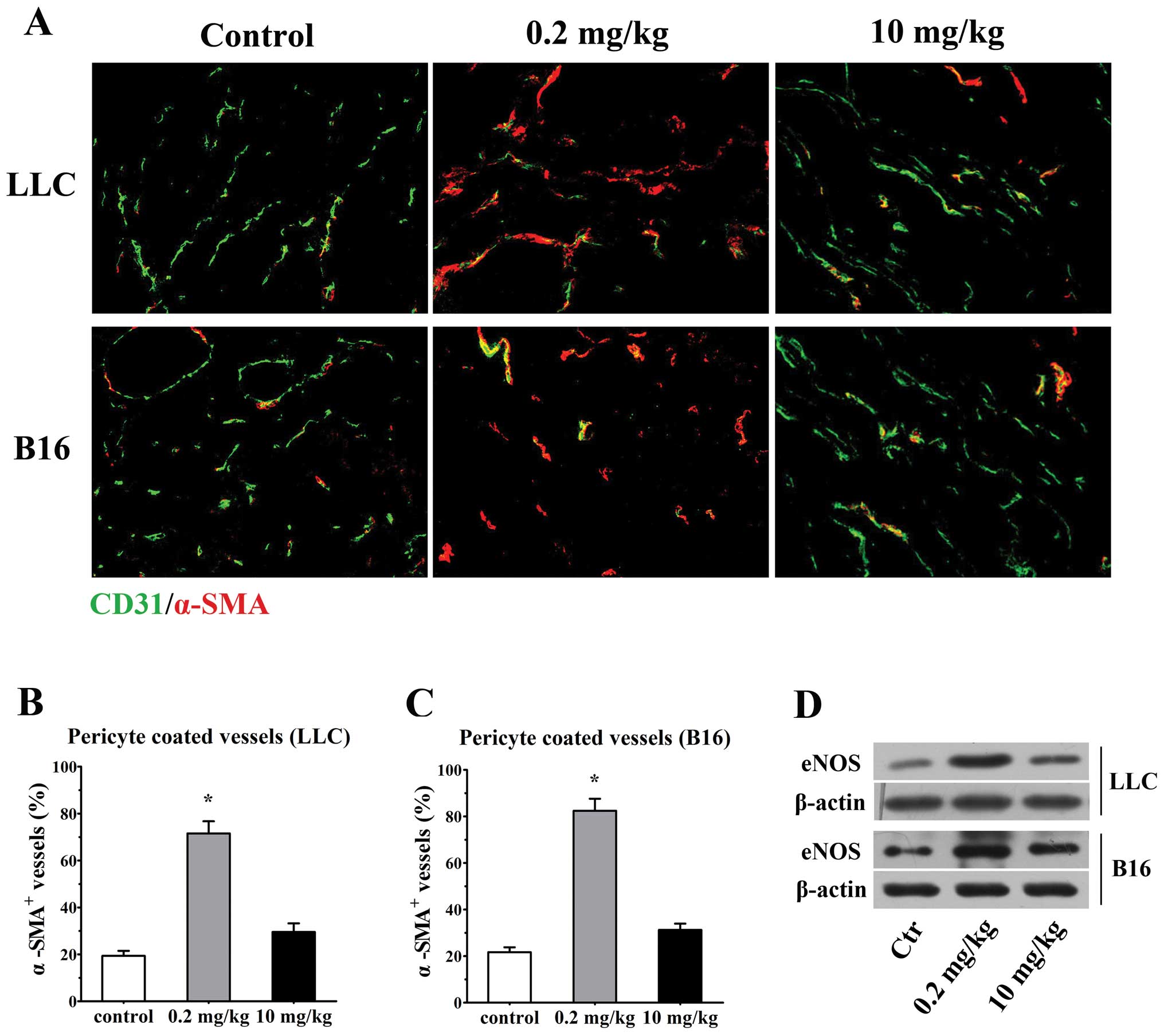
Low-dose simvastatin shows a tendency
toward promotion of mature pericyte recruitment by upregulating
eNOS expression
Pericytes and ECs are two distinct cell types that
constitute a core framework of blood vasculature. Coverage of ECs
by pericytes renders vessels a more mature, tight and stable
pattern and reduces vessel leakiness (25), a favorable feature of tumor vessel
normalization. In this regard, we sought to investigate the effect
of simvastatin treatment on pericyte coverage in tumor blood
vessels. Double stained for CD31 and mature pericyte marker α-SMA
revealed a large proportion of naked vessels without
α-SMA+ cells in untreated LLC and B16 tumors, while only
29.5±3.7 and 31.3±2.7% of the vessels characterized by
discontinuous and incomplete perivascular cell coverage were
observed in 10 mg/kg simvastatin-treated LLC and B16 tumors,
respectively. Encouragingly, after the 0.2 mg/kg simvastatin
administration, tumor vessels that encased compactly with α-SMA+
pericytes were remarkably increased in both tumor types (71.6±5.2%
of α-SMA+ vessels in LLC tumors; 82.5±5.2% in B16
tumors, p<0.001 vs. controls for both, Fig. 5A–C). Since several mechanisms by
which pericytes are recruited have been proposed (22,26),
we further explored a probable mechanism by which simvastatin
promotes pericyte coverage. A potential mechanism for this effect
is that administration of statins with a dosage exerting
proangiogenic effects (low-dose) induces an overexpression of eNOS
(21), the predominant source of
NO and has emerged as a pivotal factor in mediating
endothelium-pericyte interaction and promoting recruitment of
pericytes to tumor vessels (22).
Consistent with this line, we have observed a significant
upregulation of eNOS protein level 7 days after 0.2 mg/kg
simvastatin treatment in LLC and B16 tumors in comparison with low
levels of eNOS expression in both untreated animals and a moderate
elevation of eNOS in high-dose treated animals, detected by western
blotting (Fig. 5D). These data
collectively depicted a tendencious role of low-dose simvastatin in
normalizing tumor vessel by promoting pericyte recruitment.
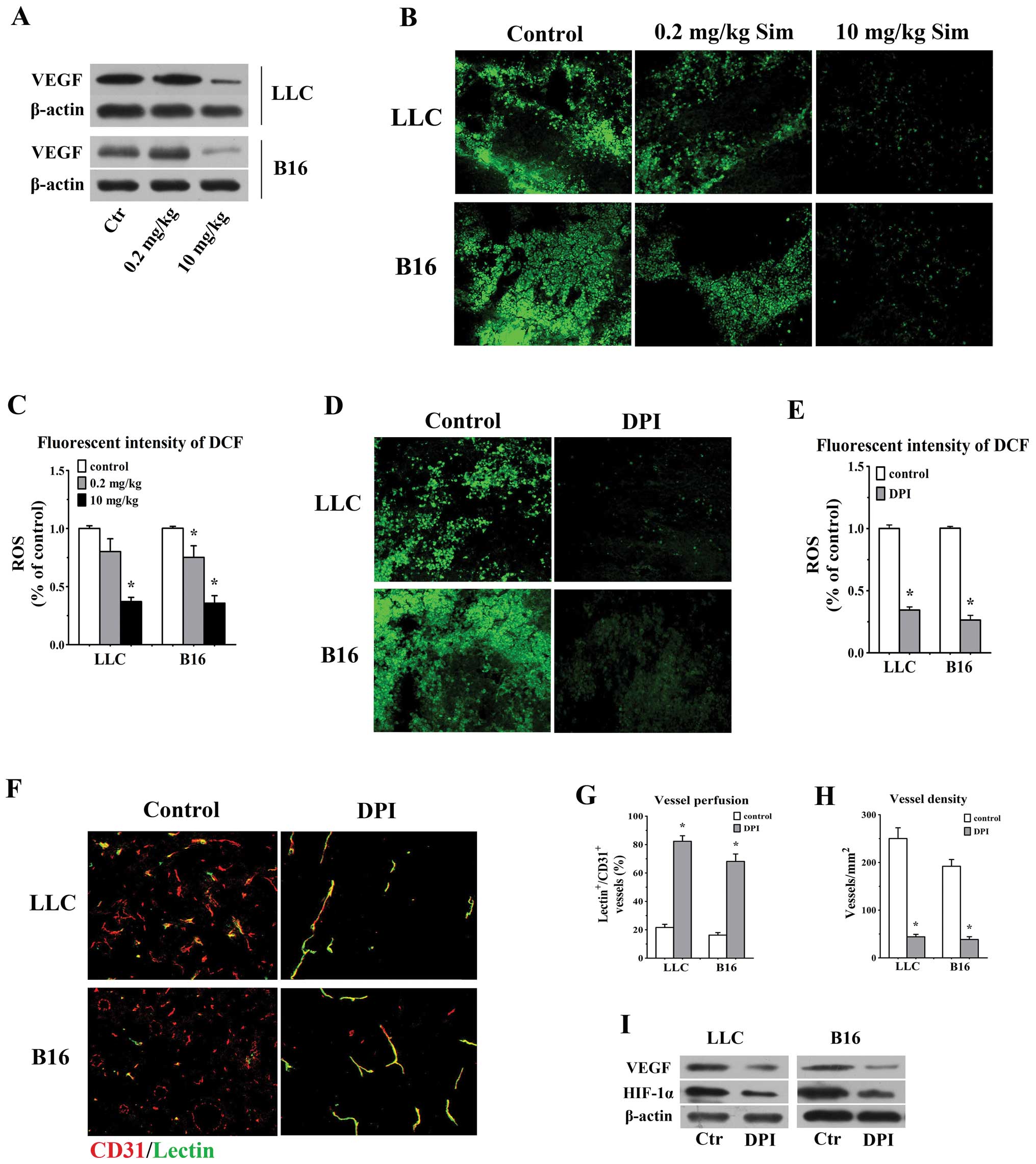
High-dose simvastatin highlights a
favorable effect of attenuation of vascular permeability by
inhibiting VEGF and counteracting ROS generation
Preclinical data demonstrated that high
concentration of statins inhibited ECs proliferation and in
vivo angiogenesis via diminishing VEGF synthesis (20,27).
We reasoned that the anti-angiogenic activity of high-dose
simvastatin would favour its function of tumor vessel
normalization. As is shown in Fig.
2, vessel density and leakiness were significantly reduced in
10 mg/kg groups, concurrently, our western blotting data also
showed a remarkable downregulation of VEGF in 10 mg/kg simvastatin
treated animals bearing LLC and B16 tumors versus control and
low-concentration groups (Fig.
6A). Considering the inhibitory effect of statins on ROS
generation and to further elucidate whether ROS are involved in
statin-mediated anti-angiogenesis in vivo, in situ
ROS production was measured using CM-H2DCFDA staining in tumors
treated with distinct concentrations of simvastatin and a
well-known strong ROS inhibitor (DPI) and then double-staining
assay for CD31 and FITC-lectin was done to assess the alterations
of morphology, density and vessel perfusion in DPI-treated tumors.
The treatment of 10 mg/kg simvastatin significantly reduced the ROS
level by 62.9 and 64.3% in LLC and B16 tumors in comparison with
control ones, respectively (p<0.05 for both, Fig. 6B and C) and ROS generation in
DPI-treated LLC (34.5% of control) and B16 tumors (26.4% of
control) was obviously lower than the values in control animals
(p<0.05, Fig. 6D and E).
Similarly, we observed a greater proportion of lectin-stained
vessels possessed sharp and distinct outline in DPI-treated LLC
(82.3±3.9%) and B16 tumors (68.1±5.3%) accompanied by a remarkably
decreased vessel density (44.2±5.1 vessels/ mm2 in
DPI-treated LLC tumors; 38.6±5.9 vessels/mm2 in
DPI-treated B16 tumors), as compared with the values in controls
(p<0.05 for both comparisons, Fig.
6F–H). Moreover, addition of DPI significantly deceased VEGF
and HIF-1α protein level in both tumors (Fig. 6I). These results confirmed that
high-dose simvastatin mediated anti-angiogenic effect and
downregulation of VEGF, partly through counteracting ROS in tumor
microenvironment, which taken together may explain the ameliorating
efficacy of high-dose simvastatin on tumor vessel
hyperpermeability.
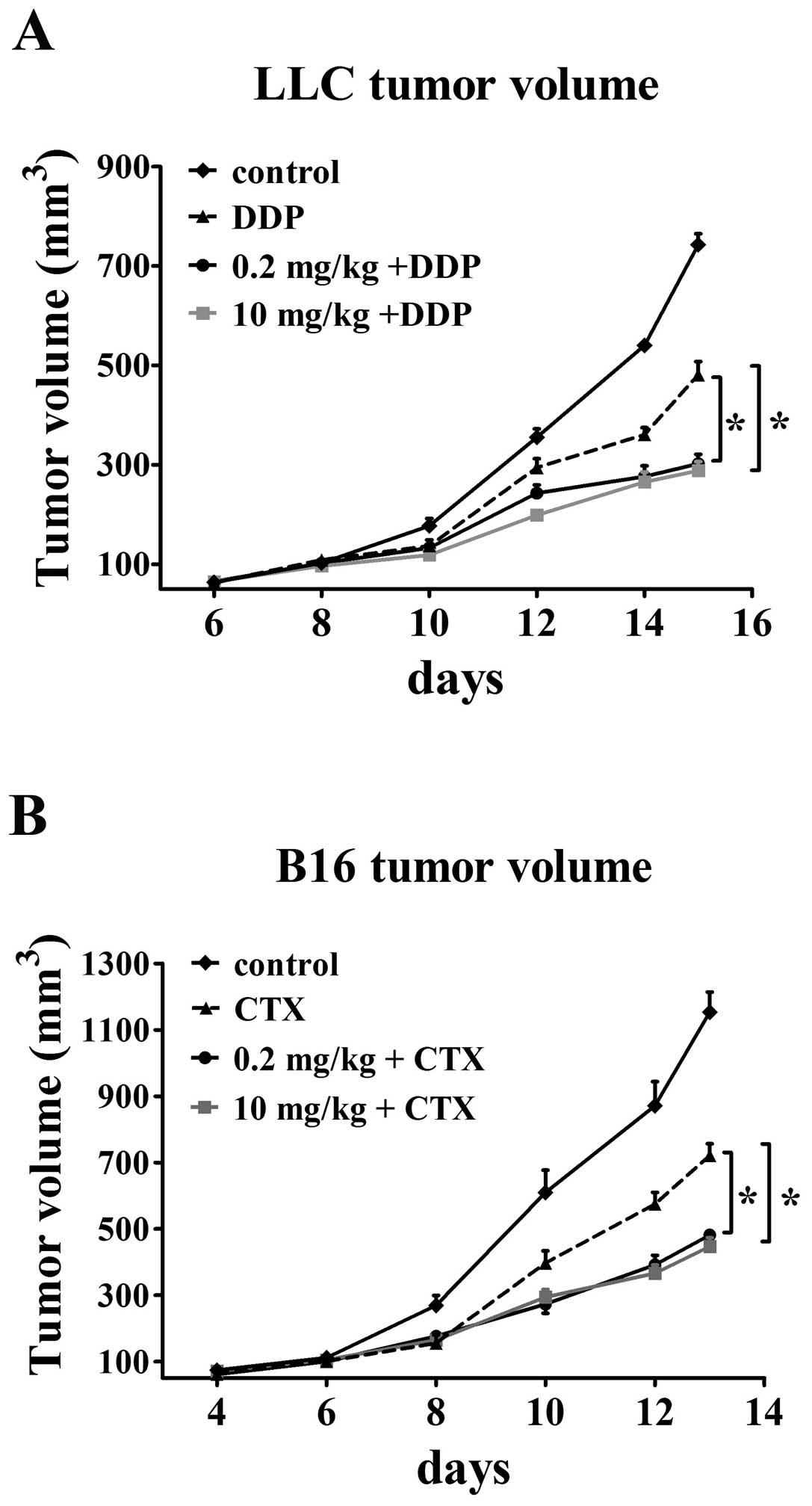
Greater effect of cytotoxic
chemotherapeutics on tumors when given together with
simvastatin
A predicted consequence of increased blood perfusion
to tumor would be enhanced drug delivery, accordingly, we decided
to examine if this change of vessel function following simvastatin
treatment has obvious clinical implications for the combination
therapy of simvastatin and chemotherapeutics. Mice bearing LLC and
B16 tumors were treated with cisplatin and CTX one day after
simvastatin administration. All animals were sacrificed at the end
of treatment and tumor size was then assessed. LLC tumors treated
with combined modality grew slower than those treated with single
chemotherapeutics as the tumor volumes in control and
cisplatin-only treatment groups were 742.94±21.9 mm3 and
480.64±26.9 mm3 at the end of treatment, respectively.
Administration of 0.2 mg/kg simvastatin followed by cisplatin was
effective in reducing the tumor volume (302.03±19.2
mm3). Addition of cisplatin to 10 mg/ kg simvastatin
groups showed maximum inhibition of tumor growth (288.27±18.1
mm3) by 61 and 40% in comparison with control and single
cisplatin-treated groups (p<0.05, Fig. 7A). An increased susceptibility to
cytotoxic agent was also observed in B16 tumors that are sensitive
to CTX. Single CTX treatment had only a modest effect on tumor
growth inhibition (final tumor volume 720.9±35.5 mm3 vs.
1153.4±60.5 mm3 in control groups). CTX treatment
significantly decreased tumor growth by 58% (final tumor volume
481.7±15.6 mm3) when given 24 h after 0.2 mg/kg
simvastatin treatment as compared with control. The synergistic
antitumor effect was improved when CTX was administered together
with 10 mg/kg simvastatin (tumor volume 38.7% of control and 62.6%
of single CTX groups, final volume 446.9±26 mm3,
p<0.05, Fig. 7B). These
findings were consistent with the hypothesis that
simvastatin-mediated increase of tumor vessel perfusion correlates
well with increased efficacy of chemotherapeutic drugs in tumor
tissues.
Discussion
Overall, our data identified a pleiotropic
regulatory role for simvastatin on tumor vascular structure and
function in a dose-dependent manner, which involves a shift from a
hostile pattern of tumor vasculature to a more mature or normalized
phenotype and enhanced response to chemotherapeutics. Underlying
these activities are distinct potential mechanisms varying with
concentrations of simvastatin. On the one hand upregulation of eNOS
and derivate NO, stimulated by low-dose simvastatin, promoted
pericyte recruitment and the subsequent stabilization of tumor
blood vessels, but vessel leakage was remarkably abrogated via
normalization of ROS production and inhibiting VEGF in tumor milieu
treated by high-dose simvastatin. These data provide insight into
the potential use of simvastatin as a vasculoprotective agent.
It is now well established that tumor vessels are
immature, mal-shaped and have a disorderly structure with a leaky
EC lining that leads to elevated interstitial fluid pressure (IFP)
(28). These abnormalities within
tumors not only compromise the delivery of therapeutic agents
(29,30), but also facilitate metastatic
spread (31). Proper incorporation
of mural cells into the vessel wall is an indispensable step in the
process of tumor vessel normalization. Platelet-derived growth
factors (PDGFs), VEGF and angiopoietin 1 (Ang1)/Tie2 are well known
cytokines that are involved in pericyte recruitment (32–34).
It is noteworthy that NO has yet been documented to promote vessel
maturation by increasing mural cell coverage in B16 melanomas
(22). Several reports of both
clinical and experimental tumor models have shown that NO plays an
important role as a proangiogenic factor in mediating branching and
longitudinal extension of blood vessels (22,35),
NO mediates endothelial-mural cell interaction and induces mural
cell recruitment and then a stable functional vessel network is
established in tumors. Additionally, Kashiwagi and collaborators
have confirmed that vessels of B16 tumors grown in
eNOS−/− mice have less mural cell coverage with
relatively larger vessel diameter compared with vessels in
wt B16 tumor-bearing mice, which indicated that eNOS, the
predominant source of NO in ECs, is a key modulator involved in
above-mentioned effect (22).
Here, we have shown that low-dose simvastatin can promote pericyte
recruitment with concomitant overexpression of eNOS in the tumor
models, despite an increase of vessel density was observed. These
actions exactly coincided with the reported regulatory role of eNOS
and NO in tumor vessel maturation. Also, it has been reported that
low-dose simvastatin (1 mg/kg, daily) treatment maintained vascular
integrity in ischemic brain tissues and significantly reduced
blood-brain barrier leakage by increasing Ang1/Tie2 expression,
which further supports the role of low-dose simvastatin in vascular
stabilization (26). Thus, in view
of upregulation of eNOS and protective activities of tumor vessels
after low-dose simvastatin treatment, our data are indirectly
indicative of the hypothesis that low-dose simvastatin promotes
tumor vessel normalization and NO-induced pericyte recruitment is a
probable candidate factor.
As a consequence of maturation of vessel structure,
efficient tumor blood flow is considered to be an important factor
contributing to increased tumor drug delivery. Emerging evidence
supports the promotional role of statins on blood flow both in
preclinical and clinical experiments. As revealed by Doppler
imaging, statins can improve ocular blood flow velocities in
patients with DR and preserve blood flow in ischemic limbs
(36,37). In addition, augmentation of blood
perfusion was reported in subcutaneously inoculated colon cancer
models treated with statins (38).
Similarly, in our experiments, we found that 7 days after both low-
and high-dose simvastatin treatments, tumor blood flow or vessel
perfusion were markedly improved compared with controls. Given that
eNOS-derived NO can serve in its well known capacity as a
vasodilator to reduce vascular resistance and potently increase
microvascular blood volume and flow (39), as well as the pericytes with
abnormal phenotypes could impair tumor blood flow (40), the improvement of tumor blood flow
after low-dose simvastatin treatment accompanied by upregulation of
eNOS is comprehensible. Moreover, eNOS-derived NO functions as a
second messenger in VEGF signaling and is necessary for the
activities of VEGF, such as stimulation of angiogenesis, which is
in part, responsible for the increased tumor blood flow (41). Consequently, these data gave rise
to a speculative conclusion that the concept of vascular
normalization is specialized in the case of low dose simvastatin
treatment and conceived as increased neovessel branches, however,
most of which are concomitantly covered by pericytes and abundant
of blood perfusion.
Impairment of equilibrium of various proangiogenic
and anti-angiogenic factors in tumors mostly contributes to
relentless development of aberrant vessels. Judicious modification
of this imbalance may normalize tumor vasculature (42). One validated modality to correct
these vessel dysfunctions is blockade of VEGF signaling. VEGF, a
well known mediator that promotes proliferation of ECs, contributes
to excessive tumor vessel permeability. Conversely, anti-VEGF
therapy can reduce tumor vessel leakage, resulting in a drop in
intratumoral IFP and in turn improve oxygenation and drug
penetration in tumors (43,44),
providing a rationale for clinical use of VEGF-targeted agents in
terms of vascular normalization in patients with cancer and/or
other pathological angiogenic diseases, exemplified as DR (45,46).
Accumulating reports clearly showed that bevacizumab, administered
by intraocular injection, has a global effect in normalizing the
pathologic intraocular environment and changing the immature,
fenestrated vessels toward a normalized status by neutralizing VEGF
in ocular disease (45,47). Coincidentally, as equally possessed
with antagonistic action against VEGF, high-dose statins have been
widely used in treating DR and complications both in clinical
settings and experimental models, in consideration of its
activities in decreasing retinal neovascularization, retaining
endothelial barrier integrity and eventually reducing the area of
hypoxia (13,48). Thus, it is reasonable to infer that
the beneficial effects of high-dose simvastatin are also applicable
to neovessels of tumor. This hypothesis is in line with our present
work that administration of a high-dose simvastatin to
tumor-bearing mice resulted in significant decrease in the number
of tumor vessels by a mechanism involving abrogation of VEGF.
During the simvastatin-induced profound vascular regression, the
remaining vessels tend to have a more normal structure with
dramatically decreased permeability and it resembled the response
to bevacizumab by destroying immature vessels in case of
pathological ocular and tumor neoangiogenesis. The above might
serve to explain the improvement of blood perfusion or flow in
high-dose simvastatin treated tumor models in our study. Uniquely,
however, as an anti-oxidant, high-dose simvastatin is also
competent in counteracting oxygen radicals. It was reported that
spontaneous overproduction of ROS in tumor cells was required for
inducing angiogenesis and tumor growth through the expression of
VEGF and HIF-1α and this cascade effect was reversible when ROS was
removed by specific inhibitors, such as DPI (19). Consistent with this, we found that
B16 and LLC tumor tissues have much higher generation of ROS and
that high-dose simvastatin or DPI treatment led to a significant
decrease of VEGF and HIF-1α expression when ROS was diminished.
More functional tumor vessels with decreased vessel number were
presented in DPI-treated animals. In addition, elevated ROS was
shown to trigger ECs apoptosis that resulted in the loss of
vascular barrier integrity. Application of anti-oxidants may
abrogate the ROS-induced impairment of endothelial barrier
(16,49). These data are intriguing and
together implicate the critical role of endogenous ROS in
abnormalities of tumor vessel structure and functions. The
anti-angiogenic efficacy of high-dose simvastatin as well as its
underlying mechanism by scavenging redundant ROS might be a novel
potential way to regulate vessel stabilization and normal vascular
barrier function.
In response to improved intratumoral perfusion,
there was a relative lack of hypoxia and necrosis within sections
of simvastatin-treated tumors tissues and these alterations may
indeed lead to a beneficial clinical implication of enhanced
efficacy of systemic chemotherapy, as indicated in our results,
simvastatin has an additive or synergistic effect in cancer therapy
when used in combination with traditional chemotherapeutics.
Statins are commonly-used drugs for treating and/or preventing
cardiovascular diseases due to its cholesterol-lowing effect.
However, increasing evidence suggests that lipid-independent and
pleiotropic activities of statins in anti-angiogenesis, cytostasis
and repression of tumor metastases may facilitate its anticancer
properties in a variety of tumor types (50,51).
In the present study, we have demonstrated that simvastatin with
both low and high dosages can mediate remodeling and stabilization
of tumor vessels, although through differentially ascendant
molecular basis and distinct patterns of manifestation.
Our data strongly support the role of statins, like
simvastatin, as a promising therapeutic option not only for retina
neoangiogenesis but also for use in monotherapy or combination
therapy for the treatment of solid tumors. Given the limitations
that the favorable pattens of vessel phenotype beyond low and
high-dose simvastatin treatments are just some parts of the typical
normalized presentations, as assessed from distinct angles
respectively and it is not yet clear which medication modality
contributes more significantly to tumor vessel normalization,
further investigations should be focused on ascertaining the
appropriate simvastatin dosing regimen that is suitable for
clinical application and optimizing the duration and schedule of
the therapy.
Acknowledgements
We are thankful to Dr Kai Hong
(Department of Medical Ultrasound, Tongji Hospital, Tongji Medical
College of Huazhong University of Science and Technology) for his
excellent technical assistance and would also like to acknowledge
the Department of Experimental Zoology, Tongji Medical College, for
their helpful assistance with animal maintenance. This study was
supported by grants from the National Nature Science Foundation of
China (no. 30973473).
References
|
1
|
Carmeliet P and Jain RK: Principles and
mechanisms of vessel normalization for cancer and other angiogenic
diseases. Nat Rev Drug Discov. 10:417–427. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Crawford TN, Alfaro DV III, Kerrison JB
and Jablon EP: Diabetic retinopathy and angiogenesis. Curr Diabetes
Rev. 5:8–13. 2009. View Article : Google Scholar
|
|
3
|
Fukumura D, Duda DG, Munn LL and Jain RK:
Tumor micro-vasculature and microenvironment: novel insights
through intravital imaging in pre-clinical models.
Microcirculation. 17:206–225. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hamzah J, Jugold M, Kiessling F, Rigby P,
Manzur M, Marti HH, Rabie T, Kaden S, Gröne HJ, Hämmerling GJ,
Arnold B and Ganss R: Vascular normalization in Rgs5-deficient
tumors promotes immune destruction. Nature. 453:410–414. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
De La Cruz JP, Gonzalez-Correa JA,
Guerrero A and de la Cuesta FS: Pharmacological approach to
diabetic retinopathy. Diabetes Metab Res Rev. 20:91–113. 2004.
|
|
6
|
Feng Y, vom Hagen F, Pfister F, Djokic S,
Hoffmann S, Back W, Wagner P, Lin J, Deutsch U and Hammes HP:
Impaired pericyte recruitment and abnormal retinal angiogenesis as
a result of angiopoietin-2 overexpression. Thromb Haemost.
97:99–108. 2007.PubMed/NCBI
|
|
7
|
Durham JT and Herman IM: Microvascular
modifications in diabetic retinopathy. Curr Diab Rep. 11:253–264.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jain RK: Normalizing tumor vasculature
with anti-angiogenic therapy: a new paradigm for combination
therapy. Nat Med. 7:987–989. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mao Y, Kiss S, Boyer JL, Hackett NR, Qiu
J, Carbone A, Mezey JG, Kaminsky SM, D’Amico DJ and Crystal RG:
Persistent suppression of ocular neovascularization with
intravitreal administration of AAVrh.10 coding for bevacizumab. Hum
Gene Ther. 22:1525–1235. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zheng Z, Chen H, Wang H, Ke B, Zheng B, Li
Q, Li P, Su L, Gu Q and Xu X: Improvement of retinal vascular
injury in diabetic rats by statins is associated with the
inhibition of mitochondrial reactive oxygen species pathway
mediated by peroxisome proliferator-activated receptor γ
coactivator 1α. Diabetes. 59:2315–2325. 2010.PubMed/NCBI
|
|
11
|
Li J, Wang JJ, Chen D, Mott R, Yu Q, Ma JX
and Zhang SX: Systemic administration of HMG-CoA reductase
inhibitor protects the blood-retinal barrier and ameliorates
retinal inflammation in type 2 diabetes. Exp Eye Res. 89:71–78.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wong WW, Dimitroulakos J, minden MD and
Penn LZ: HMG-CoA reductase inhibitors and the malignant cell: the
statin family of drugs as triggers of tumor-specific apoptosis.
Leukemia. 16:508–519. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li J, Wang JJ, Yu Q, Chen K, Mahadev K and
Zhang SX: Inhibition of reactive oxygen species by lovastatin
downregulates vascular endothelial growth factor expression and
ameliorates blood-retinal barrier breakdown in db/db mice role of
NADPH oxidase 4. Diabetes. 59:1528–1538. 2010. View Article : Google Scholar
|
|
14
|
Oliveira VN, Bessa A, Jorge ML, Oliveira
RJ, de Mello MT, De Agostini GG, Jorge PT and Espindola FS: The
effect of different training programs on antioxidant status,
oxidative stress and metabolic control in type 2 diabetes. Appl
Physiol Nutr Metab. 37:334–344. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Woo DK, Green PD, Santos JH, D’Souza AD,
Walther Z, Martin WD, Christian BE, Chandel NS and Shadel GS:
Mitochondrial genome instability and ROS enhance intestinal
tumorigenesis in APC(Min/+) mice. Am J Pathol. 180:24–31.
2012.PubMed/NCBI
|
|
16
|
Lin RZ, Wang TP, Hung RJ, Chuang YJ, Chien
CC and Chang HY: Tumor-induced endothelial cell apoptosis: roles of
NAD(P)H oxidase-derived reactive oxygen species. J Cell Physiol.
226:1750–1762. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Deng B, Xie S, Wang J, Xia Z and Nie R:
Inhibition of protein kinase Cβ2 prevents tumor necrosis
factor-α-induced apoptosis and oxidative stress in endothelial
cells: The role of NADPH oxidase subunits. J Vasc Res. 49:144–159.
2012.
|
|
18
|
Kojima H, Otani A, Oishi A, Makiyama Y,
Nakagawa S and Yoshimura N: Granulocyte colony-stimulating factor
attenuates oxidative stress-induced apoptosis in vascular
endothelial cells and exhibits functional and morphologic
protective effect in oxygen-induced retinopathy. Blood.
117:1091–1100. 2011. View Article : Google Scholar
|
|
19
|
Xia C, Meng Q, Liu LZ, Rojanasakul Y, Wang
XR and Jiang BH: Reactive oxygen species regulate angiogenesis and
tumor growth through vascular endothelial growth factor. Cancer
Res. 67:10823–10830. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Weis M, Heeschen C, Glassford AJ and Cooke
JP: Statins have biphasic effects on angiogenesis. Circulation.
105:739–745. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Meda C, Plank C, Mykhaylyk O, Schmidt K
and Mayer B: Effects of statins on nitric oxide/cGMP signaling in
human umbilical vein endothelial cells. Pharmacol Rep. 62:100–112.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kashiwagi S, Izumi Y, Gohongi T, Demou ZN,
Xu L, Huang PL, Buerk DG, Munn LL, Jain RK and Fukumura D: NO
mediates mural cell recruitment and vessel morphogenesis in murine
melanomas and tissue-engineered blood vessels. J Clin Invest.
115:1816–1827. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sadeghi MM, Collinge M, Pardi R and Bender
JR: Simvastatin modulates cytokine-mediated endothelial cell
adhesion molecule induction: involvement of an inhibitory G
protein. J Immunol. 165:2712–2718. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mills EM, Takeda K, Yu ZX, Ferrans V,
Katagiri Y, Jiang H, Lavigne MC, Leto TL and Guroff G: Nerve growth
factor treatment prevents the increase in superoxide produced by
epidermal growth factor in PC12 cells. J Biol Chem.
273:22165–22168. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gerhardt H and Semb H: Pericytes:
gatekeepers in tumour cell metastasis? J Mol Med. 86:135–144. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen J, Cui X, Zacharek A and Chopp M:
Increasing Ang1/Tie2 expression by simvastatin treatment induces
vascular stabilization and neuroblast migration after stroke. J
Cell Mol Med. 13:1348–1357. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vincent L, Chen W, Hong L, Mirshahi F,
Mishal Z, Mirshahi-Khorassani T, Vannier JP, Soria J and Soria C:
Inhibition of endothelial cell migration by cerivastatin, an
HMG-CoA reductase inhibitor: contribution to its anti-angiogenic
effect. FEBS Lett. 495:159–166. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
De Bock K, Cauwenberghs S and Carmeliet P:
Vessel abnormalization: another hallmark of cancer? Molecular
mechanisms and therapeutic implications. Curr Opin Genet Dev.
21:73–79. 2010.PubMed/NCBI
|
|
29
|
Tong RT, Boucher Y, Kozin SV, Winkler F,
Hicklin DJ and Jain RK: Vascular normalization by vascular
endothelial growth factor receptor 2 blockade induces a pressure
gradient across the vasculature and improves drug penetration in
tumors. Cancer Res. 64:3731–3736. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rapisarda A and Melillo G: Role of the
hypoxic tumor micro-environment in the resistance to
anti-angiogenic therapies. Drug Resist Updat. 12:74–80. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pennacchietti S, Michieli P, Galluzzo M,
Mazzone M, Giordano S and Comoglio PM: Hypoxia promotes invasive
growth by transcriptional activation of the met protooncogene.
Cancer Cell. 3:347–361. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Raza A, Franklin MJ and Dudek AZ:
Pericytes and vessel maturation during tumor angiogenesis and
metastasis. Am J Hematol. 85:593–598. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Greenberg JI and Cheresh DA: VEGF as an
inhibitor of tumor vessel maturation: implications for cancer
therapy. Expert Opin Biol Ther. 9:1347–1356. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Winkler F, Kozin SV, Tong RT, Chae SS,
Booth MF, Garkavtsev I, Xu L, Hicklin DJ, Fukumura D, di Tomaso E,
Munn LL and Jain RK: Kinetics of vascular normalization by VEGFR2
blockade governs brain tumor response to radiation: role of
oxygenation, angiopoietin-1 and matrix metalloproteinases. Cancer
Cell. 6:553–563. 2004.
|
|
35
|
Jadeski LC and Lala PK: Nitric oxide
synthase inhibition by NG-nitro-L-arginine methylester inhibits
tumor-induced angiogenesis in mammary tumors. Am J Pathol.
155:1381–1390. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ozkiris A, Erkiliç K, Koç A and Mistik S:
Effect of atorvastatin on ocular blood flow velocities in patients
with diabetic retinopathy. Br J Ophthalmol. 91:69–73. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yu J, deMuinck ED, Zhuang Z, Drinane M,
Kauser K, Rubanyi GM, Qian HS, Murata T, Escalante B and Sessa WC:
Endothelial nitric oxide synthase is critical for ischemic
remodeling, mural cell recruitment and blood flow reserve. Proc
Natl Acad Sci USA. 102:10999–11004. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sata M, Nishimatsu H, Osuga J, Tanaka K,
Ishizaka N, Ishibashi S, Hirata Y and Nagai R: Statins augment
collateral growth in response to ischemia but they do not promote
cancer and atherosclerosis. Hypertension. 43:1214–1220. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang N, Ko SH, Chai W, Li G, Barrett EJ,
Tao L, Cao W and Liu Z: Resveratrol recruits rat muscle
microvasculature via a nitric oxide-dependent mechanism that is
blocked by TNFα. Am J Physiol Endocrinol Metab. 300:E195–E201.
2011.PubMed/NCBI
|
|
40
|
Morikawa S, Baluk P, Kaidoh T, Haskell A,
Jain RK and McDonald DM: Abnormalities in pericytes on blood
vessels and endothelial sprouts in tumors. Am J Pathol.
160:985–1000. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tu YT, Tao J, Liu YQ, Li Y, Huang CZ,
Zhang XB and Lin Y: Expression of endothelial nitric oxide synthase
and vascular endothelial growth factor in human malignant melanoma
and their relation to angiogenesis. Clin Exp Dermatol. 31:413–418.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Jain RK: Normalization of tumor
vasculature: an emerging concept in antiangiogenic therapy.
Science. 307:58–62. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Dickson PV, Hamner JB, Sims TL, Fraga CH,
Ng CY, Rajasekeran S, Hagedorn NL, McCarville MB, Stewart CF and
Davidoff AM: Bevacizumab-induced transient remodeling of the
vasculature in neuroblastoma xenografts results in improved
delivery and efficacy of systemically administered chemotherapy.
Clin Cancer Res. 13:3942–3950. 2007. View Article : Google Scholar
|
|
44
|
Myers AL, Williams RF, Ng CY, Hartwich JE
and Davidoff AM: Bevacizumab-induced tumor vessel remodeling in
rhabdomyosarcoma xenografts increases the effectiveness of adjuvant
ionizing radiation. J Pediatr Surg. 45:1080–1085. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kohno R, Hata Y, Mochizuki Y, Arita R,
Kawahara S, Kita T, Miyazaki M, Hisatomi T, Ikeda Y, Aiello LP and
Ishibashi T: Histopathology of neovascular tissue from eyes with
proliferative diabetic retinopathy after intravitreal bevacizumab
injection. Am J Ophthalmol. 150:223–229. 2010. View Article : Google Scholar
|
|
46
|
Sorensen AG, Emblem KE, Polaskova P,
Jennings D, Kim H, Ancukiewicz M, Wang M, Wen PY, Ivy P, Batchelor
TT and Jain RK: Increased survival of glioblastoma patients who
respond to antiangiogenic therapy with elevated blood perfusion.
Cancer Res. 72:402–407. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sharma RK, Rogojina AT and Chalam KV:
Bevacizumab therapy normalizes the pathological intraocular
environment beyond neutralizing VEGF. Mol Vis. 16:2175–2184.
2010.PubMed/NCBI
|
|
48
|
Gupta A, Gupta V, Thapar S and Bhansali A:
Lipid-lowering drug atorvastatin as an adjunct in the management of
diabetic macular edema. Am J Ophthalmol. 137:675–682.
2004.PubMed/NCBI
|
|
49
|
ten Kate M, van der Wal JB, Sluiter W,
Hofland LJ, Jeekel J, Sonneveld P and van Eijck CH: The role of
superoxide anions in the development of distant tumour recurrence.
Br J Cancer. 95:1497–1503. 2006.PubMed/NCBI
|
|
50
|
Wang C, Tao W, Wang Y, Bikow J, Lu B,
Keating A, Verma S, Parker TG, Han R and Wen XY: Rosuvastatin,
identified from a zebrafish chemical genetic screen for
antiangiogenic compounds, suppresses the growth of prostate cancer.
Eur Urol. 58:418–426. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Mandal CC and Ghosh-Choudhury N, Yoneda T,
Choudhury GG and Ghosh-Choudhury N: Simvastatin prevents skeletal
metastasis of breast cancer by an antagonistic interplay between
p53 and CD44. J Biol Chem. 286:11314–11327. 2011. View Article : Google Scholar : PubMed/NCBI
|