Contents
Introduction
Types of Sweet’s syndrome
Medications associated with Sweet’s syndrome
Clinical features
Differential diagnosis and diagnostic work up
Treatment
Conclusions
Introduction
Sweet’s syndrome was originally described by Robert
Douglas Sweet in 1964 as an ‘acute febrile neutrophilic
dermatosis’. Dr Sweet described the cardinal features of a
distinctive and severe illness in eight female patients during the
15-year period from 1949 to 1964 with a similar constellation of
findings: fever, leukocytosis, tender erythematous skin plaques and
nodules (1). When biopsied, these
painful skin lesions revealed dense, neutrophilic dermal infiltrate
into the upper or papillary dermis.
It is now understood that some cases of Sweet’s
syndrome are not limited to the skin and various extracutaneous
manifestations of Sweet’s syndrome have been described (2). Dr Sweet himself preferred the disease
be called as Gomm-Button disease in honor of the first two patients
afflicted with the condition in Dr Sweet’s practice (1). With time, however, the eponymic
‘Sweet’s syndrome’ has taken hold to describe this condition which
may also be referred to by Dr Sweet’s original descriptive ‘acute
febrile neutrophilic dermatosis’.
It is imperative the oncologist not miss the
sentinel nature of this skin lesion, as Sweet’s syndrome can alert
the physician to the diagnosis of cancer or the recurrence of
malignancy. In this report, we will discuss pathophysiology,
diagnosis and challenges in the treatment associated with
malignancy-associated Sweet’s syndrome.
Types of Sweet’s syndrome
There are three main clinical settings in which
Sweet’s syndrome has been described: classical or idiopathic
Sweet’s syndrome, malignancy-associated Sweet’s syndrome and
drug-induced Sweet’s syndrome (2).
Classical or idiopathic Sweet’s
syndrome
The classical Sweet’s syndrome is described by a
constellation of clinical symptoms, physical features and
pathologic findings which include fever, leukocytosis, tender
erythematous skin lesions (papules, nodules and plaques) and a
diffuse infiltrate consisting predominantly of mature neutrophils
typically located in the upper dermis. The symptoms and clinical
manifestations typically respond promptly after initiation of
systemic corticosteroid therapy (1,2). The
syndrome predominately affects women over men at a 4:1 ratio and
most commonly presents between 30–60 years of age (2). In idiopathic Sweet’s syndrome
recurrence occurs in 1/3 of patients (2).
Formal diagnostic criteria for classical Sweet’s
syndrome were introduced by Su and Liu in 1986 (3). They were modified by von den Driesch
in 1994 (4). Major criteria
include rapid onset of characteristic tender skin lesions and
erythematous plaques and nodules with typical histopathologic
features: dense neutrophil infiltration without leukocytoclastic
vasculitis. The minor criteria are fever (>38°C), prior upper
gastrointestinal infection or immunization, the presence of
hematologic or solid neoplasia, inflammatory disorders, or
pregnancy and excellent response to corticosteroids or potassium
iodide. Abnormal laboratory findings include erythrocyte
sedimentation rate >20 mm/h, white blood cell count
>8×109/l, neutrophils >70% and high C-reactive
protein. The diagnosis relies on the presence of at least 3 of
these factors. The presence of both major criteria (1 and 2) and
two of the four minor criteria confirms the diagnosis of classical
Sweet’s syndrome (Table I).
 | Table IDiagnostic criteria of Sweet’s
syndrome (3,4). |
Table I
Diagnostic criteria of Sweet’s
syndrome (3,4).
| Diagnostic criteria
of Sweet’s syndrome |
|---|
| Major |
-
Rapid onset of characteristic skin lesions which
are tender erythematous plaques and nodules
-
Typical histopathologic features: dense neutrophil
infiltration without leukocytoclastic vasculitis
|
| Minor |
-
Pyrexia (>38°C)
-
Association with an underlying hematologic or
visceral malignancy, inflammatory disease, or pregnancy, or
preceded by an upper respiratory or gastrointestinal infection or
vaccination
-
Excellent response to treatment with systemic
corticosteroids or potassium iodide
-
Abnormal laboratory values at presentation (three
of four):
-
Erythrocyte sedimentation rate >20 mm/h
-
High C-reactive protein
-
WBC >8×109/l
-
Neutrophil count >70% of total WBC count
|
Malignancy-associated Sweet’s
syndrome
Malignancy-associated Sweet’s syndrome has equal
incidence in men and women (3). In
1993, a review was published from 15 studies of patients with
Sweet’s syndrome (each containing between 10 and 48 individuals).
This study found that ∼ 21% of patients newly diagnosed with
Sweet’s syndrome were subsequently diagnosed or already diagnosed
with either a hematologic (15%) or solid cancer (6%) (5).
Sweet’s syndrome can be the cutaneous sign of either
an undiagnosed malignancy or the first sign of a cancer recurrence
(6). Approximately 85% of reported
cases of malignancy-associated Sweet’s syndrome had underlying
hemopoietic neoplasia, most commonly acute myeloblastic leukemia.
Other hematological malignancies include myeloproliferative
neoplasms, diffuse large B-cell lymphoma, Hodgkin’s lymphoma,
myelodysplastic syndrome and myelofibrosis. The most common solid
malignancies reported with Sweet’s syndrome are carcinomas of the
genitourinary organs, breast and gastrointestinal tract, most
frequently adenocarcinomas (57%) (5–9).
More recently, the incidence is said to be increasing due to the
awareness of the disease by physicians and also due to the frequent
use of growth factors (6).
Malignancy-associated Sweet’s syndrome was initially
included as a subset of classical Sweet’s syndrome. Therefore,
several researchers and clinicians consider it appropriate to
distinguish malignancy-associated Sweet’s syndrome from the
classical form of this disease (2). The distinct features for
malignancy-associated Sweet’s syndrome are: a) equal frequency in
males and females, b) lack of precedent upper respiratory tract
infection, c) association with newly diagnosed or relapsed
cancer.
The diagnosis of Sweet’s syndrome has a temporal
association with the discovery or relapse of cancer (4). That is, the finding of Sweet’s
syndrome is often the sentinel sign of recurrence in previously
diagnosed malignancy or in the diagnosis of new malignancy
(5,10–14).
Extracutaneous manifestations have been reported to
be present in 50% of patients affected with malignancy-associated
Sweet’s syndrome (7). These
extracutaneous manifestations of Sweet’s syndrome in patients with
malignancy are more likely to be present in hematologic malignancy
compared to solid malignancy (5,6).
These include periorbital mass stimulating periorbital cellulitis
(15), pyoderma gangrenosum
(16), sudden massive swelling of
the tongue in acute myeloid leukemia (17) and erythematous tender plaques and
inflammatory changes in post-mastectomy lymphedemous areas which
may simulate infection (2,18).
Pathogenesis of malignancy-associated
Sweet’s syndrome
The pathogenesis of Sweet’s syndrome remains to be
definitively determined. Circulating autoantibodies, cytokines,
dermal dendrocytes, human leukocyte antigen serotypes, immune
complexes, paraneoplastic phenomena and leukotactic mechanisms, may
contribute to the pathogenesis of Sweet’s syndrome (2,6,19).
Fever and peripheral leukocytosis in the majority of
patients point toward a possible infectious process. This
hypothesis is supported by the observation that the manifestations
of Sweet’s syndrome improve with systemic antibiotics in some
patients with dermatosis-associated culture-confirmed and
serology-confirmed Yersinia enterolitica intestinal
infections (2,20).
Another hypothesis is that Sweet’s syndrome
represents a hypersensitivity reaction to an antigen that is
introduced into the body by either a bacterial, viral or neoplastic
process. This hypothesis is supported by the typical response to
corticosteroids as a means of treatment of Sweet’s syndrome
(6).
The most frequently proposed hypothesis for the
pathogenesis of Sweet’s syndrome in malignancy is the
overproduction and inappropriate regulation of inflammatory
cytokines such as IL-1, IL-3, IL-6, IL-8, granulocyte colony
stimulating factor (G-CSF) and granulocyte macrophage colony
stimulating factor (GM-CSF) (6).
This theory is supported by cases in which Sweet’s syndrome
patients had received G-CSF/GM-CSF, interferon-γ and ATRA with
subsequent development of Sweet’s syndrome (6–8,21).
Some cases have also reported high levels of G-CSF, IL 4, IL 6 and
interferon-γ in solid and hematological malignancies (6,22–28).
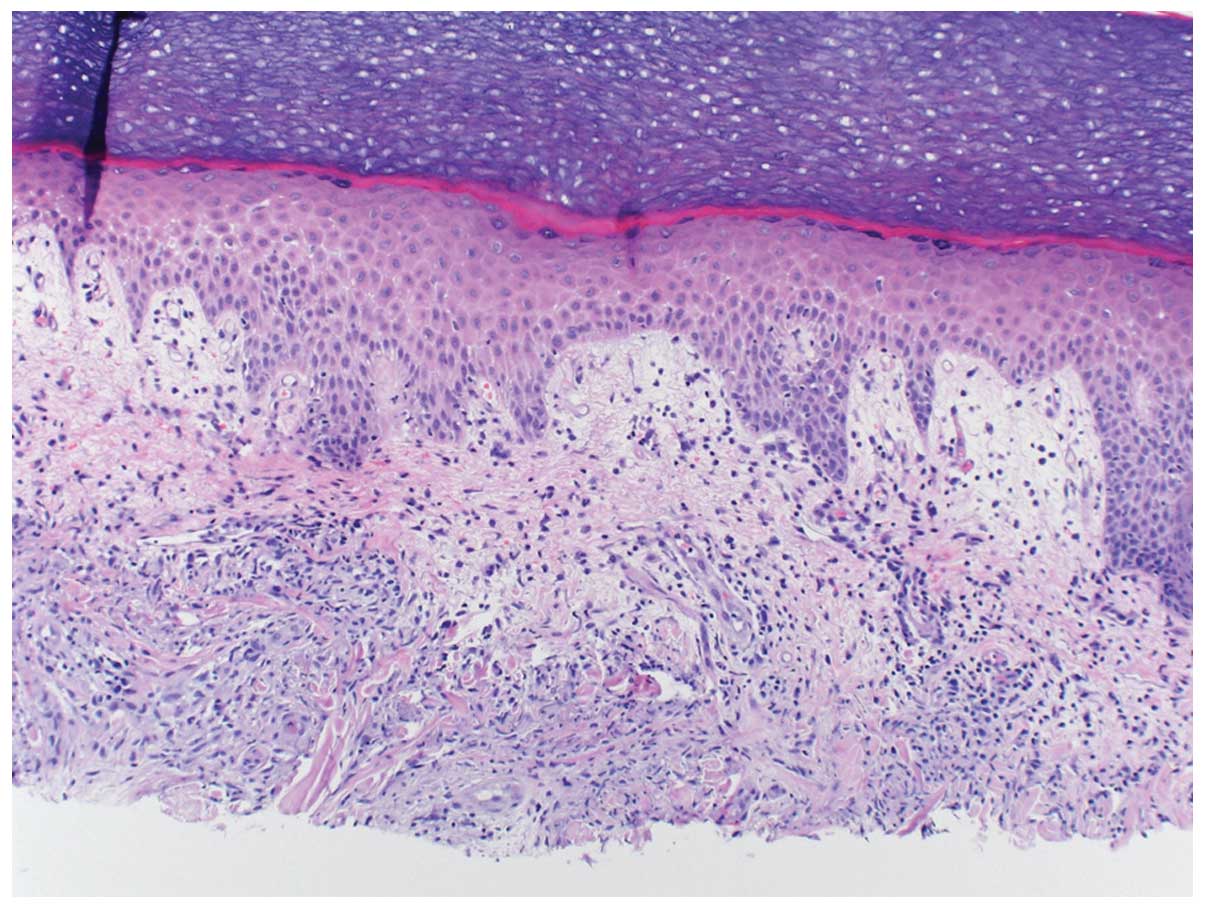
Histopathological findings
The skin lesions of Sweet’s syndrome exhibit a
diffusely distributed inflammatory infiltrate of mature neutrophils
and fragmentation of neutrophil nuclei. This process is referred as
karyorrhexis or leukocytoclasia. The epidermis appears normal and
there is classically no evidence of primary leucocytoclastic
vasculitis such as fibrin deposition or neutrophils within vessel
walls (6). Fig. 1 describes a 60-year-old breast
cancer patient who developed Sweet’s syndrome a week after
receiving the growth factor pegfilgrastim.
Although neutrophilic inflammation is typically
restricted to within the dermis, neutrophils have been observed
within the overlying epidermis (as either neutrophilic spongiotic
vesicles or subcorneal pustules) and within the underlying adipose
tissue (referred to as subcutaneous Sweet’s syndrome) (2). Similar changes have been described in
bones, intestines, liver, aorta, lungs and muscles of patients with
Sweet’s syndrome (6,15,22–27).
Only a small number of patients with Sweet’s
syndrome can also present with skin lesions concurrently
demonstrating leukemia cutis. In leukemia cutis, the dermal
infiltrate consists not only of mature polymorphonuclear cells
(Sweet’s syndrome) but also leukemic blasts (leukemia cutis). Acute
myelocytic leukemia and acute promyelocytic leukemia are the most
frequent hematological malignancies associated with concurrent
leukemia cutis (6,22). A variant of Sweet’s syndrome with
cellular infiltrate, characterized by an abundance of histiocytoid
monocytic cells with intense myeloperoxidase reactivity indicating
a myeloid origin and permitting classification as immature
neutrophilic granulocytes. These infiltrates must be distinguished
from specific leukemic infiltrates (29).
Medications associated with Sweet’s
syndrome
In drug-induced Sweet’s syndrome, there is nearly
always a temporal relationship between medication administration
and symptom development. In 1996, Walker and Cohen described the
diagnostic criteria for drug-induced Sweet’s syndrome (21). All five features should be present
to diagnose drug-induced Sweet’s syndrome (Table II).
| Table IIDiagnostic criteria for
medications-induced Sweet’s syndrome (30,31). |
Table II
Diagnostic criteria for
medications-induced Sweet’s syndrome (30,31).
| Diagnositc criteria
for medications-induced Sweet syndrome |
|---|
-
Acute onset of painful erythematous plaques or
nodules
-
Dermal neutrophilic infiltrate without evidence of
vasculitis on histopathological examination
-
Temperature >38°C
-
Temporal relationship between drug ingestion and
clinical presentation or temporally-related recurrence after
rechallenge
-
Temporally related resolution of lesions after drug
withdrawal or treatment with systemic corticosteroids
|
Anticancer agents associated with Sweet’s
syndrome
The most commonly reported drug causing Sweet’s
syndrome is granulocyte-colony stimulating factor. Other
medications that are frequently associated with Sweet’s syndrome
include bortezomib, hypomethylating agents (azacitidine,
decatibine), imatinib, lenalidomide and all-trans retinoic acid.
Table III describes the most
commonly reported anticancer agents with Sweet’s syndrome (26,31–46).
| Table IIIAnticancer drugs associated with
Sweet’s syndrome. |
Table III
Anticancer drugs associated with
Sweet’s syndrome.
| Anticancer drugs
associated with Sweet’s syndrome |
|---|
-
Granulocyte colony stimulating factor (23,26,32,33)
-
Bortezomib (34,35)
-
Azacitidine (36,37)
-
Decitibine (36,37)
-
Imatinib mesylate (38–40)
-
Lenalidomide (41–44)
-
All-trans retinoic acid (21,45,46)
|
Granulocyte-colony stimulating factor
(G-CSF)
G-CSF is the most commonly associated agent with
drug-induced Sweet’s syndrome. G-CSF is known to induce stem cell
proliferation, differentiation of neutrophils and neutrophil
survival. G-CSF is thus responsible for neutrophil skin
accumulation and seems to show a dose-dependent effect in provoking
Sweet’s syndrome (31). Several
cases have reported G-CSF-associated Sweet’s syndrome (23,31,32).
The majority of these patients were successfully treated with
corticosteroids. These patients usually responded successfully to
40 mg prednisone daily. An interesting case series examined twelve
patients, five with active Sweet’s syndrome and seven without
active Sweet’s syndrome. They found significantly higher levels of
G-CSF with active Sweet’s syndrome than those with inactive Sweet’s
syndrome (26). This suggests that
both exogenous and endogenous G-CSF is associated with this
syndrome.
Bortezomib
Bortezomib (Velcade®) is the first of the new class
of medications called proteasome inhibitors. Bortezomib appears to
act directly on important intracellular processes, such as the
nuclear factor κB (NF-κB) pathway, to cause cell death. Indirectly,
it inhibits growth and survival by acting on the bone environment.
A case has been reported of two patients who developed Sweet’s
syndrome who received bortezomib (34). Both patients responded to
corticosteroids. Another case report suggests that Sweet’s syndrome
induced by bortezomib may not necessarily require cessation of
bortezomib since administration of corticosteroids prevents its
recurrence (35).
Hypomethylating agents
The hypomethylating agents azacitdine and decitibine
are approved for the treatment of patients with myelodysplastic
syndrome (MDS). Azacitidine is thought to induce antineoplastic
activity via inhibition of DNA methyltransferase at low doses
causing hypomethylation of DNA and at high dose incorporated into
both DNA and RNA chains. Decitabine functions in a similar manner
to azacitidine, although decitabine can only be incorporated into
DNA strands while azacitidine can be incorporated into both DNA and
RNA chains.
In 2009, azacitidine associated dense, hemorrhagic,
neutrophilic dermatitis with massive subepidermal and
intraepidermal edema, consistent with a diagnosis of Sweet’s
syndrome was published in a series of myelodysplastic patients
(36). Interestingly, the amount
of neutrophilic involvement of the skin was quantitatively less
than previously reported, possibly because of the neutropenic state
of the patients. All patients initially responded to
corticosteroids. However, one patient on decitibine who did not
respond to high-dose corticosteroids died with recurrent Sweet’s
syndrome. This patient’s course was complicated by a superimposed
acinobacter pneumonia and sepsis. Therefore, it is important that
this syndrome should be noted in the differential and treated early
and aggressively in the course of treatment with these nucleoside
hypomethylating agents. Two other cases have been reported of
Sweet’s syndrome developing in patients on azacitidine (37). In both cases, azacitidine was
stopped and patients were successfully treated with corticosteroids
and antibiotics. After resolution of symptoms, one patient was
treated with decitibine and the other patient was successfully
rechallenged with azacitidine for MDS management.
Imatinib mesylate
Imatinib, a tyrosine kinase inhibitor, is used in
treating chronic myelogenous leukemia (CML), gastrointestinal
stromal tumors and some other diseases. Skin toxicities are a well
recognized side effect of imatinib treatment. The skin toxicities
include Stevens-Johnson syndrome, acute generalized exanthematous
pustulosis, photosensitivity, hypopigmentation and Sweet syndrome
(38).
Liu et al(39) have reported cell infiltration of
the skin in a chronic-phase CML patient who was at the time in
molecular remission and was taking imatinib mesylate. The patient
was treated by her dermatologist with topical steroids and oral
prednisone with gradual resolution of the skin rash. Imatinib was
held starting 4 weeks after the skin rash erupted and discontinued
for a total of 6 weeks. Imatinib was restarted at 300 mg daily due
to positivity for bcr/abl by PCR, although cytogenetics and FISH
studies remained normal with no further recurrence of Sweet’s
syndrome (39).
Another case report demonstrates Sweet’s syndrome
after imatinib in a 53-year-old African American woman with CML. A
skin biopsy specimen showed neutrophilic dermatosis with epidermal
sparing consistent with Sweet’s syndrome. Therapy with prednisone
at 40 mg/d led to complete resolution (40).
Lenalidomide
Lenalidomide (Revlimid) is an amino-substituted
variant of thalidomide that is an immunomodulatory drug. One
patient developed tender vesicles and bullae on the hands 6 days
after starting treatment with lenalidomide for agnogenic myeloid
metaplasia. Biopsy revealed dense superficial inflammatory dermal
neutrophilia. Lenalidomide treatment was discontinued and the
patient was started on prednisone therapy (40 mg/d), which was
gradually tapered over 3 to 4 weeks. The lesions resolved with no
recurrence in 6 months of follow-up (41).
Another interesting presentation of Sweet’s syndrome
was reported with high-dose lenalidomide presenting as multiple
plum-colored, tender ulcerated and crusted nodules predominantly
located on the legs with fewer lesions on the elbows, lower aspect
of back and buttocks. The patient was given the diagnosis of a
neutrophilic dermatosis, lenalidomide was discontinued and
prednisone (60 mg/d) was started with substantial clinical
improvement within 24 h of initiation of therapy (42). This case suggests the possibility
of an immune-complex mediated process, favoring
gravitationally-dependent sites for Sweet’s syndrome with
propensity of higher dose of lenalidomide (25 mg/d).
Another patient is reported with Sweet’s syndrome in
chronic lymphocytic leukemia who was treated with lenalidomide. The
patient responded successfully to prednisone therapy (43). In a recently published study, the
severity of lenalidomide-associated tumor flare reactions in CLL
patients correlated directly with in vitro
lenalidmide-induced upregulation of CD80 on CLL cells and CD69
expression on T cells (T-cell activation) and inversely with
treatment-induced changes in T-cell numbers. A striking increase in
the levels of IL-6 and TNF-α was seen in the patient with the most
severe reaction, suggesting ‘immune-activation’ as the most likely
culprit (43,44).
All-trans retinoic acid (ATRA)
ATRA has been used in the treatment of acute
promyeolocytic leukemia. A review of fourteen case reports of
ATRA-associated Sweet’s syndrome showed the median time for
development of Sweet’s syndrome is 18 days of ATRA therapy (6–34
days). Four patients continued with the ATRA therapy without
interruption, 13 patients were treated with steroids and 12
responded to treatment. One patient improved without any treatment
(45). A possible explanation of
the mechanism of ATRA-associated Sweet’s syndrome involves
alteration of certain functional properties of mature neutrophils,
modifying the migratory capabilities of these cells (45). The occurrence of Sweet’s syndrome
in some patients on granulocyte colony-stimulating factor (G-CSF)
and its increased production by acute promyelocytic leukemia cells
in the presence of ATRA suggest that G-CSF may be involved in the
pathogenesis of these syndromes (21,46).
Clinical features
Fever is the most common complaint in patients with
Sweet’s syndrome. The fever can occasionally precede the skin
lesions by days or even weeks (2).
Notably, 75–90% of patients with classical or idiopathic Sweet’s
syndrome report prior upper respiratory infection preceding the
presentation of Sweet’s syndrome itself. However, only ∼20% of
patients with malignancy-associated Sweet’s syndrome will report
preceding upper respiratory infection (2).
The skin lesions in Sweet’s syndrome are painful,
erythematous and papular or nodular. The lesions may coalesce and
progress to form plaques over a period of days to weeks. These skin
lesions are seen in nearly half of patients with
malignancy-associated Sweet’s syndrome and 36% of patients with
drug-induced Sweet’s syndrome (5).
Unique characteristics have been observed in the
skin lesions seen in malignancy-associated Sweet’s syndrome,
sometimes as bullous, ulcerated lesions, with morphologic features
of pyoderma gangrenosum (41,47).
Cutaneous pathergy, for instance, in post mastectomy lymphedema has
also been observed to incite Sweet’s syndrome skin lesions
(18).
Manifestations of Sweet’s syndrome have also
occurred in the ears, eyes, central nervous system, mouth, bone,
muscle, heart, lung, liver, intestines, kidneys and hematologic
system (6). Lungs are the most
common extracutaneous site. Symptoms include mild dyspnea to acute
respiratory distress syndrome and imaging demonstrates diffuse
ground-glass opacities or consolidation. Bronchoscopy shows
erythematous pustules with ulcerations in the tracheobronchial
tree. Bronchoalveolar lavage is characterized by neutrophilic
predominance with negative cultures (48).
Anemia has been reported in 82–83% of patients with
malignancy-associated Sweet’s syndrome and is found in 100% of
patients with drug-induced Sweet’s syndrome but is an exceedingly
infrequent finding in patients with idiopathic or classical Sweet’s
syndrome (5). Additionally,
abnormal platelet count is infrequently found in idiopathic or
classical Sweet’s syndrome but has been reported to be present in
68% of patients with hematologic malignancy and Sweet’s syndrome
(4). Abnormal platelet counts have
been reported in half of patients with drug-induced Sweet’s
syndrome and Sweet’s syndrome with solid malignancy (5). Peripheral leukocytosis is frequently
observed in Sweet’s syndrome, however, this is not always the case.
Some of the patients with malignancy-associated Sweet’s syndrome
may have neutropenia as described above (36).
Differential diagnosis and diagnostic work
up
Sweet’s syndrome is characterized by neutrophilic
dermatosis or neutrophilic panniculitis, therefore any conditions
that present similarly histologically need to be considered in the
differential diagnosis of the disease (2,49).
As described earlier, leukemia cutis is a condition that can occur
either separately or concurrently with Sweet’s syndrome. The
primary means of differentiation is that the dermal infiltrate
associated with leukemia cutis will consist of neoplastic immature
leukocytes in contrast to the mature neutrophils seen in Sweet’s
syndrome (2,49). Chloromas also have some overlap
with leukemia cutis. Occasionally, hematological malignancy can
predispose individuals to infection which include abscess
formation, cellulitis and panniculitis thus producing challenges in
diagnosis of Sweet’s syndrome (50). Behcet’s disease and pyoderma
gangrenosum can both be confused with Sweet’s syndrome due to
histological findings and an association with hematological
neoplasias (7). Other etiologies
that are commonly confused with Sweet’s syndrome include
leukocytoclastic vasculitis, periarteritis nodosa and granuloma
faciale (3). Several other
disorders of neutrophilic dermatosis including bowel (intestinal)
bypass syndrome, erythema elevatum diutinum, halogenoderma,
neutrophilic eccrine hidradenitis and rheumatoid neutrophilic
dermatitis should be considered (49).
A lesional skin biopsy for routine histopathologic
evaluation is a useful procedure to confirm a clinically suspected
diagnosis of Sweet’s syndrome. It may also be prudent to submit
lesional tissue for bacterial, fungal, mycobacterial and possibly
viral cultures (2).
Treatment
The treatment of choice in malignancy-associated
Sweet’s syndrome is treatment of the underlying malignancy, which
can result in complete resolution of the individual’s Sweet’s
syndrome (2). However, patients
usually require the course of systemic corticosteroids 1 mg/kg for
3–4 weeks. Surgical intervention have also occasionally promoted
resolution of Sweet’s syndrome when the dermatosis is associated
with solid tumors (2).
There are no specific guidelines for the treatment
of malignancy-associated Sweet’s syndrome, therefore hematologists
and oncologists treat Sweet’s syndrome same as classical Sweet’s
syndrome. Sweet’s syndrome caused by anticancer agents sometimes
involves withdrawal or temporary discontinuation of anticancer
agents, use of systemic corticosteroids, and/or rechallenge with
either the same anticancer agents or different agents.
First line agents
Systemic corticosteroids are the mainstay of therapy
for Sweet’s syndrome. Prednisone, at a dosage of 1 mg/kg/day
(usually ranging from 30 to 60 mg/day), may be given as a single
morning dose. After a clinical response is observed, prednisone can
be lowered by 10 mg/day within a period of 4–6 weeks. However, some
patients may require 2 to 3 months of treatment or intravenous
therapy (2). Intravenous pulse
administration of methylprednisolone sodium succinate (≤1000
mg/day) over one or more hours, daily for 3–5 days, may be utilized
for patients whose condition is refractory to other therapies
(51). The dose is followed by
tapering oral dose of corticosteroid or another immunosuppressant
agent (2,50). The use of systemic corticosteroids
should prompt evaluation of every patient for possible prophylactic
proton-pump inhibitor therapy.
Localized manifestations of Sweet’s syndrome may be
treated with high-potency topical corticosteroids including
intra-lesional corticosteroids (such as triamcinolone acetonide at
a dose ranging from 3 to 10 mg/ml) as a single injection or as
multiple sequential treatments if necessary (49). However, potential exacerbation of
the lesions through pathergy should be considered before initiating
intralesional therapy (50). Also,
high-potency topical corticosteroids (such as clobetasol propionate
0.05% in either a cream, ointment, gel or foam vehicle) can be
applied to the Sweet’s syndrome skin lesions (49,50).
Other treatments include potassium iodide which is
more affective in vasculitis and hypothyroidism-associated Sweet’s
syndrome (51). Potassium iodide
has been used (53) in solid
malignancy-associated Sweet’s syndrome with response. Patients who
are treated with potassium iodide have improved within 48 h and
cutaneous lesions clear within one week in most cases (52). In some cases the drug was withdrawn
after only 2 weeks and no recurrence was seen (53,54).
Potassium iodide can be administered orally as 300 mg
enteric-coated tablets, 3 times each day (for a daily dose of 900
mg). However, severe vasculitis is a concern after potassium iodide
administration (55).
Several larger studies have shown colchicine at a
dose of 0.5 mg three times each day is an effective agent for the
successful management of patients with Sweet’s syndrome (55,56).
One report presented twenty patients with Sweet’s syndrome of whom
90% responded to colchicine therapy from 10 to 21 days (56).
Second line agents
Dapsone and cyclosporine can be given in combination
with or without steroids in patients that do not respond to first
line therapy (54). The oral dose
of cyclosporine ranges from 2 to 10 mg/kg/day. Rapid response
within one week is usually recorded with cyclosporine. However,
tapering is difficult in some cases. Therefore, patients who
receive cyclosporine 10 mg/kg/day, should be tapered by 2 mg/kg/day
every 2 days and discontinued on day 21 (57). Dapsone has been used in combination
with oral steroids. The initial oral dose of dapsone ranges from
100 to 200 mg per day (55).
Other second line stand-alone agents are
indomethacin 150 mg orally for 7 days followed by 100 mg orally for
14 days have been shown with good response (58). Clofazimine has been used in
patients who failed treatment with methylprednisolone for chronic
or relapsing Sweet’s syndrome with ‘almost complete remission’ in
six patients. Clofazimine was dosed daily as 200 mg for 4 weeks
followed by 4 additional weeks at 100 mg per day (4).
Other systemic drugs in isolated case reports have
also been shown to be effective for the treatment of Sweet’s
syndrome include chlorambucil and cyclophosphosphamide,
antimetabolites, immunoglobulins, interferon-α, tumor necrosis
factor and the anti-angiogenic agents infliximab and thalidomide
(2).
Conclusions
In this report we have reviewed pathophysiology,
diagnosis, manifestations for malignancy-associated Sweet’s
syndrome, various new anticancer agents associated with Sweet’s
syndrome and experiences in the management of this rare
syndrome.
Unfortunately, little is known about pathogenesis of
Sweet’s syndrome. The syndrome may occur as a paraneoplastic
manifestation for a cancer or may be the first sign of malignancy.
Reappearance of lesions may herald relapse in patients with
malignancy-associated Sweet’s syndrome. In some cases, anticancer
agents cause Sweet’s syndrome for unclear pathogenesis and unknown
significance.
As the story of Sweet’s syndrome pathogenesis
continues to be unraveled, we may begin to learn risk factors that
predispose to this disease and better understand how to manage
it.
Due to lack of our understanding of pathophysiology
of Sweet’s syndrome and in the absence of evidence based
literature, many physicians regardless of the underlying disease
associated with this entity, have successfully used corticosteroids
in these patients. The duration of remission is variable between
recurrent episodes of the dermatosis. Occasionally, patients
require broad spectrum antibiotics if skin lesions are secondarily
infected.
We believe that malignancy-associated Sweet’s
syndrome and anticancer medications-associated Sweet’s syndrome
behave differently. However, when patients are receiving
antineoplastic agents, it is difficult to define underlying cause
of Sweet’s syndrome and its true significance. Future studies
should focus on understanding the pathophysiology of this disease
and its association with new anticancer drugs along with risk
factors so that prompt treatment without interruption of cancer
management can be instituted.
Acknowledgements
We would like to thank Dr Jeff North
for providing the figure of Sweet’s syndrome in a patient treated
at our institution.
References
|
1.
|
Sweet RD: An acute febrile neutrophilic
dermatosis. Br J Dermatol. 76:349–356. 1964. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Cohen PR: Sweet’s syndrome: a
comprehensive review of an acute febrile neutrophilic dermatosis.
Orphanet J Rare Dis. 2:342007.
|
|
3.
|
Su WPD and Liu H-NH: Diagnostic criteria
for Sweet’s syndrome. Cutis. 37:167–174. 1986.
|
|
4.
|
von den Driesch P: Sweet’s syndrome (acute
febrile neutrophilic dermatosis). J Am Acad Dermatol. 31:535–556.
1994.
|
|
5.
|
Cohen PR and Kurzrock R: Sweet’s syndrome
and cancer. Clin Dermatol. 11:149–157. 1993.
|
|
6.
|
Paydas S: Sweet’s syndrome: a revisit for
hematologists and oncologists. Crit Rev Oncol Hematol. Oct
6–2012.(Epub ahead of print). View Article : Google Scholar
|
|
7.
|
Cohen PR, Talpaz M and Kurzrock R:
Malignancy-associated Sweet’s syndrome: review of the world
literature. J Clin Oncol. 6:1887–1897. 1988.
|
|
8.
|
Haverstock C, Libecco JF, Sadeghi P, et
al: Tender erythematous plaques in a woman with acute myelogenous
leukemia. Arch Dermatol. 142:235–240. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Disel U, Paydas S, Yavuz S, et al:
Bilateral ear Sweet’s syndrome in a case with relapse acute
myeloblastic leukemia. Leuk Res. 30:3642006.
|
|
10.
|
Cohen PR: Cutaneous paraneoplastic
syndromes. Am Fam Physician. 50:1273–1282. 1994.PubMed/NCBI
|
|
11.
|
Kurzrock R and Cohen PR: Mucocutaneous
paraneoplastic manifestations of hematologic malignancies. Am J
Med. 99:207–216. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Kurzrock R and Cohen PR: Cutaneous
paraneoplastic syndromes in solid tumors. Am J Med. 99:662–671.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Cohen PR: Cutaneous manifestations of
internal malignancy. Section V, Dermatology. Current Practice of
Medicine. Callen JP and Bone RC: Current Medicine, Philadelphia,
PA, Inc; 2(V): pp. 19.1–19.3. 1996
|
|
14.
|
Cohen PR and Kurzrock R: Paraneoplastic
syndromes of the skin. Current Diagnosis 9. Conn RB, Borer WZ and
Snyder JW: W.B. Saunders Co; Philadelphia, PA: pp. 1199–1206.
1997
|
|
15.
|
Morgan KW, Callen JP and Kentucky L:
Sweet’s syndrome in acute myelogenous leukemia presenting as
periorbital cellulitis with an infiltrate of leukemic cells. J Am
Acad Dermatol. 45:590–595. 2001.
|
|
16.
|
Caughman W, Stern R and Haynes H:
Neutrophilic dermatosis of myeloproliferative disorders: atypical
forms of pyoderma gangrenosum and Sweet’s syndrome associated with
myeloproliferative disorders. J Am Acad Dermatol. 9:751–758.
1983.PubMed/NCBI
|
|
17.
|
Bamelis M, Boyden B, Sente F, et al:
Sweet’s syndrome and acute myelogenous leukemia in a patient who
presented with a sudden massive swelling of the tongue.
Dermatology. 190:335–337. 1995.
|
|
18.
|
García-Río I, Pérez-Gala S, Aragüés M, et
al: Sweet’s syndrome on the area of postmastectomy lymphoedema. J
Eur Acad Dermatol Venereol. 20:401–405. 2006.
|
|
19.
|
Matta M and Kurban AK: Sweet’s syndrome:
systemic association. Cutis. 12:561–565. 1973.
|
|
20.
|
Elsner P, Hartmann AA and Lechner W:
Sweet’s syndrome associated with Yersinia enterocolitica infection.
Dermatologica. 173:85–89. 1986.
|
|
21.
|
Arun B, Berberian B, Azumi N, et al:
Sweet’s syndrome during treatment with all-trans retinoic acid in a
patient with acute promyelocytic leukemia. Leuk Lymphoma.
31:613–615. 1998.
|
|
22.
|
Paydas S, Sahin B and Zorludemir S:
Sweet’s syndrome accompanying leukaemia: seven cases and review of
the literature. Leuk Res. 24:83–86. 2000.
|
|
23.
|
Arbetter KR, Hubbard KW, Markovic SN, et
al: Case of granulocytic colony-stimulating factor-induced Sweet’s
syndrome. Am J Hematol. 61:126–129. 1999.PubMed/NCBI
|
|
24.
|
Giasuddin AS, El-Orfi AH, Ziu MM and
El-Barnawi NY: Sweet’s syndrome: is the pathogenesis mediated by
helper T cell type 1 cytokines. J Am Acad Dermatol. 39:940–943.
1998.
|
|
25.
|
Tuerlinckx D, Bodart E, Despontin K, et
al: Sweet’s syndrome with arthritis in a 8-month-old boy. J
Rheumatol. 26:440–442. 1999.
|
|
26.
|
Kawakami T, Ohashi S, Kawa Y, et al:
Elevated serum granulocyte colony-stimulating factor levels in
patients with active phase of Sweet syndrome and patients with
active Behcet disease: implication in neutrophil apoptosis
dysfunction. Arch Dermatol. 140:570–574. 2004. View Article : Google Scholar
|
|
27.
|
Oiso N, Watanabe K and Kawada A:
Granulocyte colony-stimulating factor-induced Sweet syndrome in a
healthy donor. Br J Haematol. 135:1482006. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Shinojima Y, Toma Y and Terui T: Sweet
syndrome associated with intrahepatic cholangiocarcinoma producing
granulocyte colony-stimulating factor. Br J Haematol.
154:1103–1104. 2006.
|
|
29.
|
Requena L, Kutzner H, Plamedo G, et al:
Histiocytoid Sweet syndrome: a dermal infiltration of immature
neutrophilic granulocytes. Arch Dermatol. 141:834–842. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Walker DC and Cohen PR:
Trimethoprim-sulfamethoxazole associated acute febrile neutrophilic
dermatosis: case report and review of drug-induced Sweet’s
syndrome. J Am Acad Dermatol. 34:918–923. 1996.PubMed/NCBI
|
|
31.
|
Cohen PR and Kurzrock R: Sweet’s syndrome.
A neutrophilic dermatosis classically associated with acute onset
and fever. Clin Dermatol. 18:265–282. 2000.
|
|
32.
|
Bidyasar S, Montoya M, Suleman K, et al:
Sweet syndrome associated with granulocyte colony-stimulating
factor. J Clin Oncol. 26:4355–4356. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Llamas-Velasco M, García-Martín P,
Sánchez-Pérez J, et al: Sweet’s syndrome with subcutaneous
involvement associated with pegfilgrastim treatment: first reported
case. J Cutan Pathol. 40:46–49. 2013.
|
|
34.
|
Van Regenmortel N, Van de Voorde K, De
Raeve H, et al: Bortezomib-induced Sweet’s syndrome. Haematologica.
90:ECR432005.
|
|
35.
|
Tanguy-Schmidt A, Avenel-Audran M, Croué
A, et al: Bortezomib-induced acute neutrophilic dermatosis. Ann
Dermatol Venereol. 136:443–446. 2009. View Article : Google Scholar
|
|
36.
|
Alencar C, Abramowitz M, Parekh S, et al:
Atypical presentations of Sweet’s syndrome in patients with MDS/AML
receiving combinations of hypomethylating agents with histone
deacetylase inhibitors. Am J Hematol. 84:688–689. 2009.
|
|
37.
|
Trickett HB, Cumpston A and Craig M:
Azacitidine-associated Sweet’s syndrome. Am J Health Syst Pharm.
69:869–871. 2012.PubMed/NCBI
|
|
38.
|
Deininger MW, O’Brien SG, Ford JM, et al:
Practical management of patients with chronic myeloid leukemia
receiving imatinib. J Clin Oncol. 21:1637–1647. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Liu D, Seiter K, Mathews T, et al: Sweet’s
syndrome with CML cell infiltration of the skin in a patient with
chronic-phase CML while taking Imatinib Mesylate. Leuk Res.
28(Suppl 1): S61–S63. 2004.
|
|
40.
|
Ayirookuzhi SJ, Ma L, Ramshesh P, et al:
Imatinib-induced Sweet syndrome in a patient with chronic myeloid
leukemia. Arch Dermatol. 141:368–370. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Hoverson AR, Davis MD, Weenig RH, et al:
Neutrophilic dermatosis (Sweet syndrome) of the hands associated
with lenalidomide. Arch Dermatol. 142:1070–1071. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Thieu KP, Rosenbach M, Xu X, et al:
Neutrophilic dermatosis complicating lenalidomide therapy. J Am
Acad Dermatol. 61:709–710. 2009. View Article : Google Scholar
|
|
43.
|
Tageja N, Giorgadze T and Zonder J:
Dermatological complications following initiation of lenalidomide
in a patient with chronic lymphocytic leukaemia. Intern Med J.
41:286–288. 2011. View Article : Google Scholar
|
|
44.
|
Aue G, Njuguna N, Tian X, et al:
Lenalidomide induced upregulation of CD80 on tumor cells correlates
with T-cell activation, the rapid onset of a cytokine release
syndrome and leukemic cell clearance in chronic lymphocytic
leukemia. Haematologica. 94:1266–1273. 2009. View Article : Google Scholar
|
|
45.
|
Yan ZS, Li DP, Jiang EL, et al:
Development of Sweet syndrome in an acute promyelocyte leukemia
patient during treatment with all-trans retinoic acid - case report
and literature review. Zhonghua Xue Ye Xue Za Zhi. 28:462–465.
2007.PubMed/NCBI
|
|
46.
|
Dubois C, Schlageter MH, de Gentile A, et
al: Modulation of IL-8, IL-1 beta, and G-CSF secretion by all-trans
retinoic acid in acute promyelocytic leukemia. Leukemia.
8:1750–1757. 1994.PubMed/NCBI
|
|
47.
|
Bielsa S, Baradad M, Marti RM, et al:
Sweet’s syndrome with bullous lesions. Actas Dermosifiliogr.
96:315–316. 2005.
|
|
48.
|
Shugarman IL, Schmit JM, Sbicca JA, et al:
Easily missed extracutaenous manifestation of malignancy associated
Sweet’s syndrome: systemic inflammatory response syndrome. J Clin
Oncol. 29:e702–705. 2011.PubMed/NCBI
|
|
49.
|
Cohen PR: Neutrophilic dermatoses: a
review of current treatment options. Am J Clin Dermatol.
10:301–312. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Cohen PR and Kurzrock R: Sweet’s syndrome:
a review of current treatment options. Am J Clin Dermatol.
3:117–131. 2002.
|
|
51.
|
Spillman DH, Carabajal MG, Errecaborde MS,
et al: Acute febrile neutrophilic dermatosis (Sweet’s syndrome).
In: Book of Abstracts. 18th World Congress of Dermatology.
Dermatology Progress and Perspectives; June 12–18; New York City.
abs. 147. 1992
|
|
52.
|
Cohen PR, Holder WR, Tucker SB, Kono S and
Kurzrock R: Sweet syndrome in patients with solid tumors. Cancer.
72:2723–2731. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
53.
|
Horio T, Imamura S, Danno K, et al:
Treatment of acute neutrophilic dermatosis (Sweet’s syndrome) with
potassium iodide. Dermatologica. 160:341–347. 1980.
|
|
54.
|
Burrall B: Sweet’s syndrome (acute febrile
neutrophilic dermatosis). Dermatol Online J. 5:81999.
|
|
55.
|
Bourke JF, Keohane S, Long CC, et al:
Sweet’s syndrome and malignancy in the U.K. Br J Dermatol.
137:609–613. 1997.
|
|
56.
|
Maillard H, Leclech C, Peria P, et al:
Colchicine for Sweet’s syndrome. A study of 20 cases. Br J
Dermatol. 140:565–566. 1999.
|
|
57.
|
von den Driesch P, Steffan C, Zobe A, et
al: Sweet’s syndrome-therapy with cyclosporin. Clin Exp Dermatol.
19:274–277. 1994.
|
|
58.
|
Jeanfils S, Joly P, Young P, et al:
Indomethacin treatment of eighteen patients with Sweet’s syndrome.
J Am Acad Dermatol. 36:436–439. 1997.PubMed/NCBI
|