Introduction
Prostate cancer is the second leading cause of
male-malignancy-related death in the United States (1). A growing body of data indicates that
the initiation and progression of prostate cancer is influenced by
aging, genetic predisposition, environmental factors such as
toxins, and lifestyle choices such as cigarette smoking. Cigarette
smoking has been identified as the most preventable cause of cancer
morbidity and mortality, yet, 20% of US adult males were reported
to be cigarette smokers in 2010 (2).
The combustion product of cigarettes is an aerosol
containing more than 3,500 chemical compounds, many of which have
been shown to be carcinogens or mutagens. Smoke generated from
burned cigarettes consists of a particulate solid phase (tar) and a
gaseous phase containing volatile organic compounds, free radicals
and other volatile and semi-volatile compounds (3,4). The
water-soluble components of aqueous cigarette tar can produce the
superoxide anion (O2•−) and subsequently
hydrogen peroxide (H2O2) and the reactive
hydroxyl radical (HO•), which can cause oxidative stress
damage to membrane lipids, proteins and DNA (4), contributing to inflammation and
cancer. Active and passive exposure to products of cigarette
combustion promotes angiogenesis and malignancy of the prostate,
lung, esophagus, bladder, pancreas and cervix (5). Cigarette smoking has also been linked
to an elevated risk of advanced stage and high-grade prostate
cancer, both of which are indicative of a poor prognosis (6). Although much is known about the
epidemiology of cigarette smoking, the underlying cellular and
molecular mechanisms responsible for its carcinogenic potential in
prostate cancer remain unclear.
Heme oxygenase (HO) is a microsomal rate-limiting
enzyme involved in the degredation of heme (7,8). The
three mammalian isoforms of heme oxygenase, HO-1, HO-2 and HO-3,
have distinct patterns of tissue-specific expression (9). HO-1, also known as heat shock protein
32 (HSP-32), is highly expressed in the spleen and liver, and at
lower levels in several other mammalian tissues (10,11)
has been implicated in maintaining cellular homeostasis, reducing
oxidative stress damage, attenuating the inflammatory response,
inhibiting apoptosis and regulating proliferation (12). Conversely, HO-1 is also recognized
as an important proangiogenic mediator (13). HO-1 expression is elevated in
various cancer cells (14–17) and tumors (18–20).
Ectopic expression of HO-1 has been shown to increase VEGF
secretion and enhance VEGF-mediated activities such as
proliferation and migration, leading to improved formation and
growth of capillary-like tubular structures (21,22)
as well as tumor angiogenesis in a mouse model of pancreatic cancer
(21–23). Endothelial cells deficient in HO-1
secrete less VEGF than their wild-type counterparts. Reducing HO-1
expression or inhibiting its enzymatic activity impairs
vGPCR-enhanced survival and VEGF-A expression in endothelial cells
(24), and inhibits VEGF
expression in lung carcinoma (25,26).
A number of cohort studies suggest that cigarette
smoking may be associated with prostate cancer (6), however, the molecular mechanism(s)
linking it to prostate cancer remain elusive. Nuclear HO-1 protein
expression has been observed in various tumors (27–29)
including prostate cancer (19).
These studies, however, were reported as clinical and pathological
observations, and failed to investigate role of nuclear HO-1
expression molecularly in prostate cancer. The present study
explored the relationship between cigarette smoke and nuclear
expression of HO-1 and to investigate molecular mechanism(s) by
which cigarette smoke-induced nuclear translocation of HO-1
promoted VEGF secretion in prostate cancer cells. The present study
demonstrated cigarette smoke induced nuclear translocation of HO-1
in prostate cancer cells. Nuclear-directed expression of HO-1
increased transcriptional activity and secretion of VEGF in
prostate cancer cells. The data revealed that cigarette
smoke-mediated translocation of HO-1 was associated with increased
VEGF secretion, and also suggested that exposure to first- and
secondhand products of cigarette combustion were associated with
prostate cancer via nuclear HO-1-modulated VEGF secretion.
Materials and methods
Reagents
Anti-4-hydroxy-2-nonenal (anti-4-HNE) antibody (cat.
24325) was purchased from Percipio Biosciences (Burlingame, CA).
Anti-GAPDH (cat. sc-137179), anti-lamin B1 (cat. Sc-56144) and
anti-GFP (cat. sc-8334) antibodies were purchased from Santa Cruz
Biotechnology (Santa Cruz, CA). Anti-HO-1 antibody (cat.
ADI-SPA-895-F) was purchased from Enzo Life Sciences (Ann Arbor,
MI). Titanium One-Step RT-PCR kit (cat. 639503) was purchased from
Clontech (Mountain View, CA). Human VEGF ELISA kit (cat. DVE00) was
purchased from R&D Systems (Minneapolis, MN). NE-PER Nuclear
and Cytoplasmic Extraction Reagents (cat. 78833) were from Thermo
Scientific (Rockford, IL). CellTiter 96 Aqueous non-radioactive
cell proliferation assay kit (cat. G5421) was purchased from
Promega Corporation (Madison, WI).
Cells and culture conditions
The prostate carcinoma cell line DU145 (cat. HTB-81)
was obtained from the American Type Culture Collection (Manassas,
VA) and cultured in Eagle’s minimum essential medium (Mediatech
Inc., Manassas, VA) supplemented with 10% FBS. The prostate
adenocarcinoma cell line PC3 (cat. CRL-1435) was also obtained from
the American Type Culture Collection and cultured in DMEM F-12
50/50 medium (Mediatech Inc.), supplemented with 10% FBS. COS-7 and
HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium
(Mediatech Inc.) containing 10% FBS. All cells were maintained at
37°C, in a 5% CO2 incubator.
Cigarettes and the preparation of smoke
medium
3R4F reference cigarettes were purchased from the
Reference Cigarette Program, University of Kentucky (Lexington,
KY). Federal Trade Commission Smoking analysis indicated that 3R4F
cigarettes contained 11.0 mg/cigarette (mg/cig) total particulate
matter (TPM), 9.4 mg/cig of tar, 0.73 mg/cig nicotine, and 12
mg/cig carbon monoxide. Smoke media (SM) was generated by
collecting whole smoke from burning one pack (20 cigarettes) of
reference cigarettes into 100 ml of cell culture media. SM was
filtered and stored at −80°C for further use.
Cell proliferation assay
The assay was performed using a CellTiter 96 Aqueous
non-radioactive cell proliferation assay kit per manufacturer’s
instruction. Briefly, 100 μl of DU145 and PC3
(5×104 cells) cell suspension were grown on 96-well
plates incubated at 37°C and 5% CO2. After 24 h, the
culture medium was either refreshed or replaced with serial
dilutions of SM as indicated. The accumulation of formazan was
measured spectroscopically by absorption at 490 nm.
RNA isolation
Isolation of total RNA was performed using TRIzol
Reagent (Life Technologies, Carlsbad, CA) according to the
manufacturer’s instructions. Briefly, cells were seeded on 6-well
plates and treated with SM or standard cell culture media. A total
of 1.0 ml of TRIzol was added followed by 0.2 ml of chloroform
after 15 min. Samples were vigorously inverted by hand for 15 sec,
incubated at room temperature for 3 min and centrifuged at 12,000 ×
g for 15 min at 4°C. After centrifugation, 0.5 ml of isopropanol
was added to the supernatant. After incubation at room temperature
for 10 min, samples were centrifuged at 12,000 × g for 10 min at
4°C. The pellets were washed with 75% ethanol, dissolved in
RNAse-free water and incubated for 10 min at 60°C. RNA
concentration was measured and the samples were stored at
−80°C.
RT-PCR Analyses
RNA transcript levels were semi-quantified using the
Titanium One-Step RT-PCR kit (Clontech) according to the
manufacturer’s instructions using the forward
5′-GAGACGGCTTCAAGCTGGTGATG-3′ and reverse
5′-GTTGAGCAGGAACGCAGTCTTGG-3′ primers for HO-1, and the forward
5′-GAAGGTGAAGGTCGGAGTC-3′ and reverse 5′-GAAGATGGTGATGGGATTTC-3′
primers for GAPDH. The conditions were one cycle of 50°C for 1 h
and 94°C for 5 min, followed by 25 cycles of 94°C for 30 sec, 68°C
for 30 sec and 68°C for 60 sec with an extension of 68°C for 2 min.
The RT-PCR products were visualized on 1% agarose gels stained with
ethidium bromide and quantified by Scion Image Software.
Immunoblot analysis
Cells were washed with PBS and lysed directly on ice
with RIPA buffer. The lysates were transferred to a new tube,
solubilized for 1 h at 4°C and clarified by centrifugation at
12,000 rpm for 20 sec at 4°C. Total cell extract protein
concentration was determined by Bradford assay. Equal amounts of
proteins were loaded and electrophoresed on SDS-PAGE gels, blotted
onto PVDF membrane and incubated with anti-HO-1 and anti-GAPDH
antibodies. Blots were washed 3 times and incubated with a
HRP-conjugated IgG antibody. Protein expression was detected on
X-ray films and quantified using Scion Image Software.
Immunohistochemistry
Cell cultures were washed twice with PBS, fixed with
4% paraformaldehyde and treated with 0.5% Triton X-100 in PBS for
30 min. After 3 washes with PBS, cells were treated with PBS
containing 10% goat serum for 2 h and incubated with anti-4HNE
antibodies for 24 h at 4°C. After 3 washes with PBS, the cells were
incubated with FITC- and/or TRITC-conjugated IgG antibodies for 1 h
at room temperature. The cells were then washed, mounted on slides
and imaged using a Zeiss LSM510 Meta confocal microscope.
Generation of DNA constructs
pEGFP-HO-1 (HO-1 fused with the C-terminus to EGFP)
was created by PCR amplification of HO-1 cDNA using the forward
primer 5′-CAGCGAATTC ACCATGGAGCGTCCGCAACCCGACAGC-3′
containing an EcoRI restriction site and the reverse primer
5′-GAT GGATCCCGATGCGGCCGCCATGGCATAAAGCCCTAC-3′ containing a
BamHI site. The product was ligated into the pEGFP-N3 vector
at the EcoRI and BamHI sites. Similarly,
pEGFP-HO-1/NLS (same as pEGFP-HO-1, but containing tandem nuclear
localization signals between HO-1 and EGFP) was created by PCR
amplification of the pNuc-HO-1 template (described below) using the
same forward primer as above and the reverse primer
5′-CTGGGATCCCTACCTTTCTCTTCTTTTTTGGATCTACCTTTCTCTTC-3′
containing a BamHI site. The product was inserted into
pEGFP-N3 at the unique EcoRI and BamHI restriction
sites.
pFlag-HO-1 (HO-1 with an N-terminal FLAG-tag) was
created by PCR amplification of the HO-1 cDNA using the primers
5′-CAAGCTTGAGCGTCCGCAACCCGACAGC-3′ and
5′-CGGATCCTCATTACATGGCATAAAGCCC-3′, and insertion into the
pFlag-CMV4 vector at the HindIII and BamHI sites.
pNuc-HO-1 (HO-1 with tandem nuclear localization at the C-terminus)
was generated by PCR amplification using the primers
5′-TAGTCGACGAGCGTCCGCAACCC GAC-3′ and
5′-TAGCGGCCGCCATGGCATAAAGCCCT-3′ and insertion into the
pCMV/myc/nuc vector at SalI and NotI restriction
sites. Expression in HEK293T cells was confirmed by immunoblot
analysis using an anti-HO-1 antibody.
Luciferase assay
HEK293T or COS7 cells were co-transfected with
combinations of plasmids as indicated in the figure legends using
Lipofectamine reagent (Life Technologies). Cell lysates were
prepared and luciferase and β-galactosidase activities were
quantified using a Luciferase assay kit (Promega) according to the
manufacturer’s instructions. The effect of the transfected proteins
on promoter transcriptional activity was assessed by measuring
luciferase activity normalized to β-galactosidase activity.
Statistics
Statistical significance was determined using the
Student’s t-test and one-way ANOVA. Data represent the mean ± SD of
independent experiments, with a p<0.05 considered statistically
significant.
Results
Effect of cigarette smoke on growth of
prostate cancer cells
Many epidemiological research studies have shown
that cigarette smoking is linked to aggravation of cancer
progression. Here, we examined the concentration of cigarette smoke
medium which supported the growth of prostate cancer cells.
Cigarette smoke medium (SM) was prepared as described in Materials
and methods. Concentrations of total particulate materials (TPM),
tar, nicotine (Nico) and carbon monoxide (CO) in SM were estimated
from the Federal Trade Commission Smoking analysis as described in
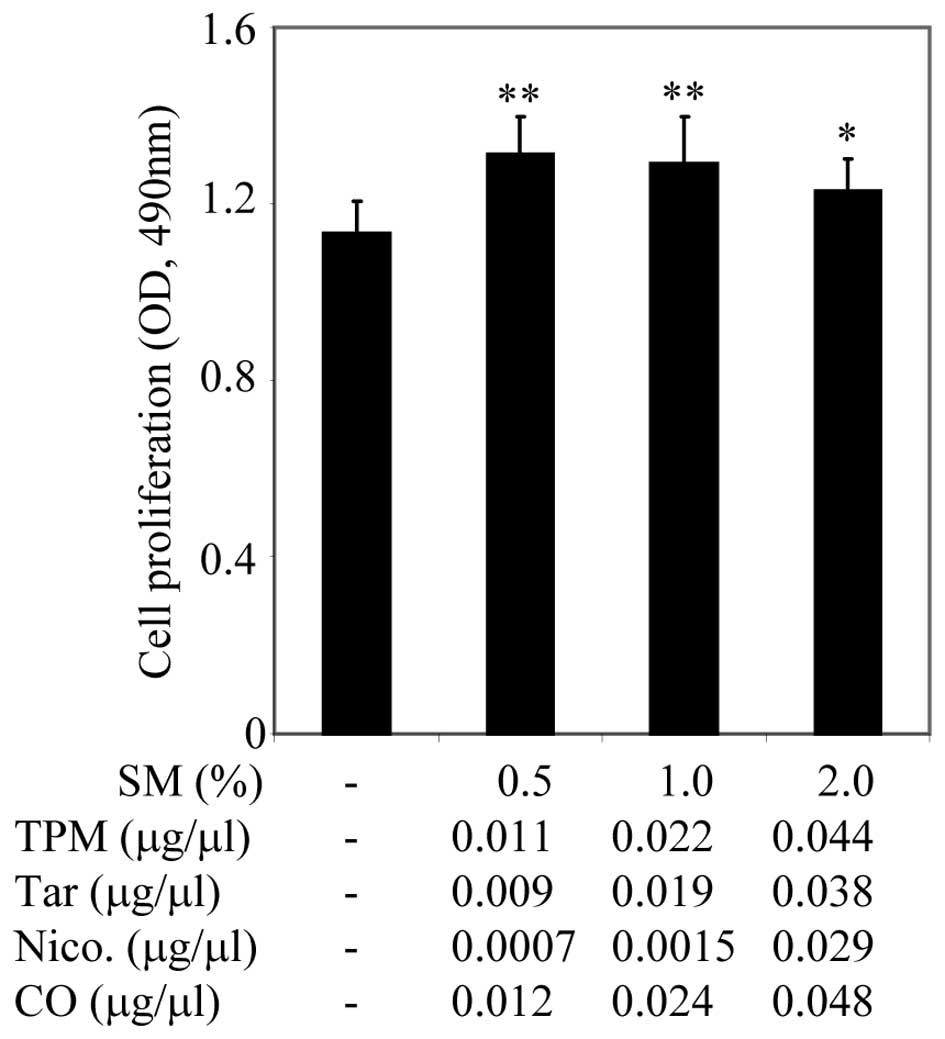
Materials and methods and indicated in Fig. 1. PC3 cells were plated onto 96-well
plates containing 10% FBS culture medium. After 24 h, the cell
culture medium was replaced with culture medium containing 0.5%
FBS. After a further 24 h, cell cultures were treated with serial
dilutions of SM ranging from 0.5–2% (v/v), for 48 h, as indicated
in Fig. 1. Cell proliferation was
assessed using the CellTiter Non-Radioactive Cell Proliferation
Assay, known as MTS. Treatment with 0.5 and 1% SM significantly
promoted growth of PC3 cells, however, 2% SM did not appear to have
a significant inhibitory effect on cell proliferation (Fig. 1). SM (4, 10 and 20%) inhibited cell
growth (data not shown). SM supported the growth of PC3 cells in a
dose-dependent manner, in particular, SM (0.5%). SM (0.5%) in cell
culture media was used for further studies.
Cigarette smoke induced expression of
HO-1 in prostate cancer cells
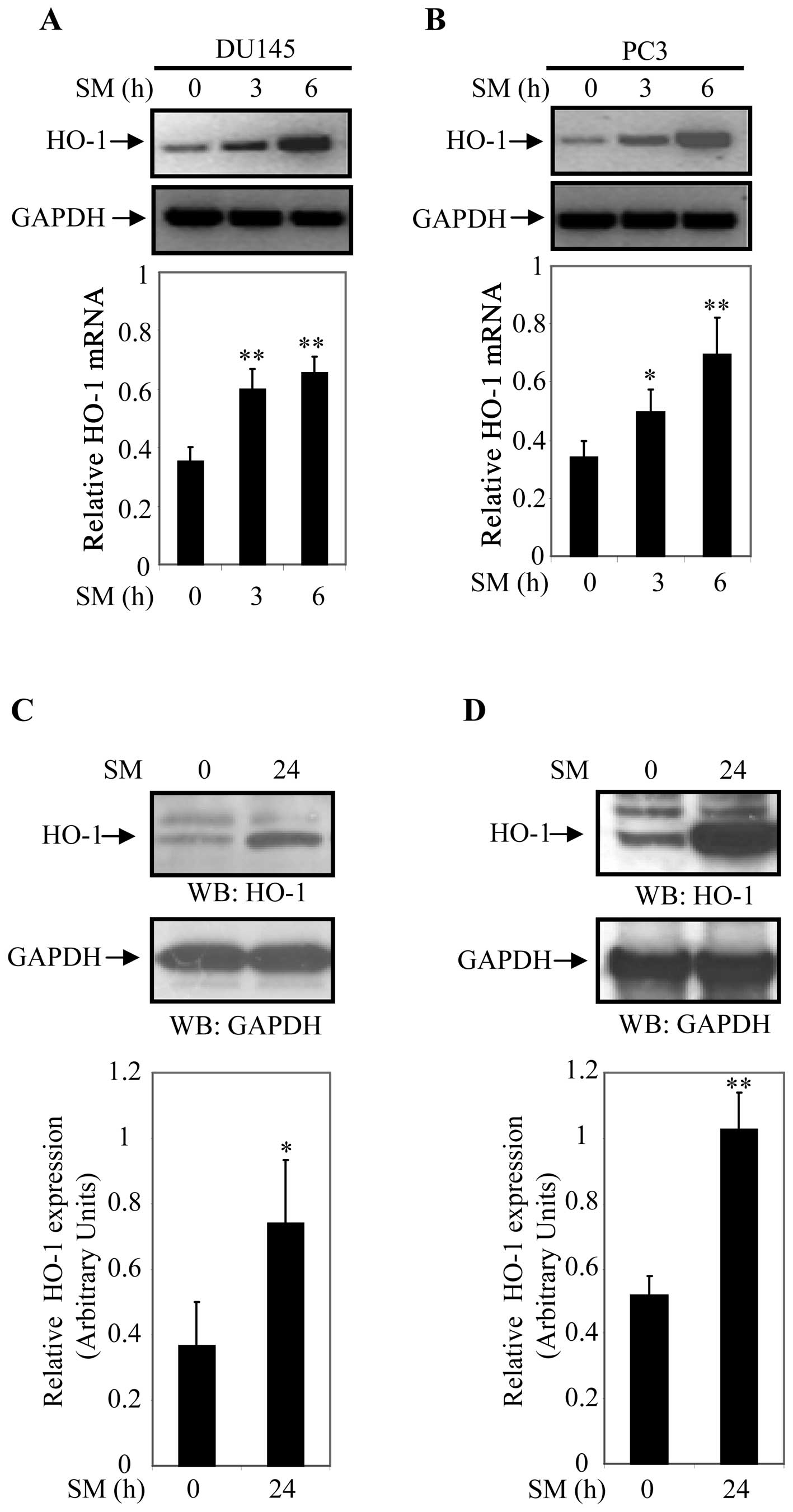
Next, we examined whether SM induces expression of
HO-1 in DU145 and PC3 cells. For this study, cells were treated for
0, 3 and 6 h with 0.5% SM. Steady state transcript levels of HO-1
and GAPDH (internal control) were assessed by semi-quantitative PCR
analysis. HO-1 transcript levels increased in 0.5% SM-treated DU145
and PC3 cells in a time-dependent manner (Fig. 2A and B). HO-1 mRNA levels were 1.7-
and 1.9-fold higher in DU145 and 1.5- and 2.0-fold higher in PC3
cells after 3 and 6 h of 0.5% SM treatment, respectively (Fig. 2A and B). These results suggested
that HO-1 plays a central role in the response to SM exposure in
prostate cancer cells.
To further analyze the effect of SM on activation of
HO-1, DU145 and PC3 cells were treated with SM for 24 h and
expression of HO-1 was determined by western blot analyses. Cell
extracts of DU145 and PC3 cells treated with 0.5% SM for 0 and 24 h
were prepared and protein expression was determined by western blot
analysis using an anti-HO-1 antibody. Expression of HO-1
(normalized to GAPDH) was significantly higher in SM treated cells
compared to controls (Fig. 2C and
D). HO-1 expression was 2.1-fold (p=0.047) higher in DU145
cells and 2.0-fold (p=0.002) higher in PC3 cells after 24 h of 0.5%
SM treatment compared to controls (0 h of treatment). Relative
levels of HO-1 expression were determined based on data derived
from three independent experiments (Fig. 2C and D). These finding indicated
that 0.5% SM induced steady-state HO-1 mRNA and protein levels in
DU145 and PC3 cells.
Cigarette smoke induced VEGF secretion in
prostate cancer cells
VEGF has been implicated in tumor progression and
expression of VEGF has been reported in a number of cell lines and
clinical specimens derived from a broad range of cancers (30–33).
Here we examined whether exposure to SM stimulated VEGF secretion
in prostate cancer cells. DU145 and PC3 cells were grown in culture
medium supplemented with 10% FBS. After 24 h, cells were refreshed
with culture medium containing 0.5% FBS, and then treated with 0.5%
SM for 0 and 24 h. Cell culture supernatants were collected after 0
and 24 h, and VEGF secretion was assessed by ELISA per the
manufacturer’s instructions. VEGF increased 1.65-fold (p=0.0002) in
DU145 cells (Fig. 3A) and
4.38-fold (p=0.0002) in PC3 cells (Fig. 3B) after 24 h treatment with SM.
These results suggested that SM induced VEGF secretion in prostate
cancer cells.
Cigarette smoke induced nuclear
translocation of HO-1 in prostate cancer cells
Recent studies reported that nuclear localization of
HO-1 was associated with prostate cancers (19), and head and neck squamous cell
carcinomas (27). Treatment of
DU145 and PC3 cells with SM induced mRNA and protein levels of HO-1
as compared to control counterparts (Fig. 2). We examined whether SM induced
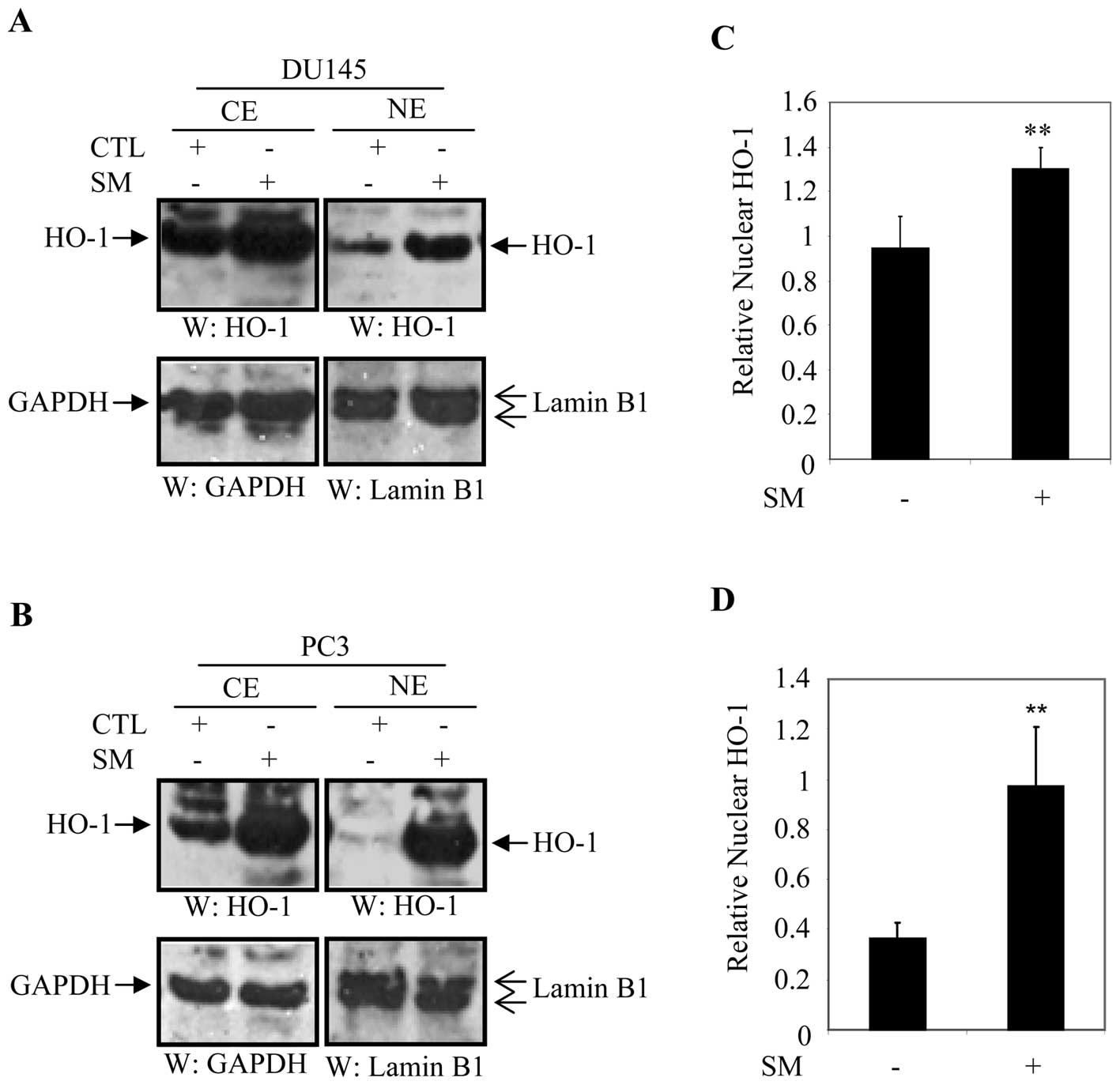
nuclear translocation of HO-1 in DU145 and PC3 cells. Cellular
fractionation was performed to determine the cellular distribution
of HO-1 in SM-treated and untreated cells. DU145 and PC3 cells were
treated with either culture medium or 0.5% SM and cellular
fractions were blotted with anti-HO-1, anti-GAPDH (cytoplasmic
maker) or anti-lamin B1 (nuclear marker) antibody after 24 h. The
level of cytoplasmic HO-1 was significantly higher in SM-treated
DU145 and PC3 cells compared to their control counterparts
(Fig. 4A and B).
We also observed that levels of HO-1 in the nuclear
fraction significantly increased in DU145 and PC3 cells treated
with SM compared to untreated controls (Fig. 4A and B). For quantification
purposes, expression of nuclear HO-1 was normalized to that of
lamin B1 and expressed in arbitrary units. Data were averaged from
three independent experiments. Relative levels of nuclear HO-1 were
1.37-fold (p=0.001) higher in DU145 cells and 2.65-fold (p=0.01)
higher in PC3 cells treated with 0.5% SM compared to controls
(Fig. 4C and D).
Nuclear-directed expression of HO-1 in
HEK293 cells
SM increased cytoplasmic expression and nuclear
translocation of HO-1 in prostate cancer cells (Fig. 4). SM-mediated expression of HO-1
also correlated with an increase in VEGF secretion (Fig. 3). These observations prompted us to
investigate the role of cytoplasmic and nuclear HO-1 in the
regulation of VEGF. For this study, we generated two constructs by
fusing HO-1 and HO-1 with C-terminal nuclear localization signals
(NLS) to the N-terminus of EGFP. The generated constructs were
designated pEGFP-HO-1 and pEGFP-HO-1/NLS. HEK293 cells were
transfected and stained with F-actin (cytoplasmic marker) and DAPI
(nuclear marker). As anticipated, cells transfected with pEGFP-HO-1
expressed HO-1 exclusively in the cytosol (Fig. 5A), whereas cells transfected with
pEGFP-HO-1/NLS expressed HO-1/NLS exclusively in the nucleus
(Fig. 5B), demonstrating that a
carboxyl terminal NLS could efficiently mediate nuclear expression
of HO-1 in vitro (Fig. 5B).
Exclusive expression of HO-1/NLS in the nucleus can therefore be
used to examine the function of nuclear HO-1 in vitro, thus
mimicking endogenous HO-1 in prostate cancer tissues as shown
previously (19,33). Furthermore, this system can be used
to compare function of cytoplasmic and nuclear HO-1 in VEGF
transcriptional activation and secretion.
Nuclear localization of HO-1 promoted
transcriptional activity of VEGF
Next, we generated two constructs of HO-1,
pFlag-HO-1 (HO-1 with N-terminal FLAG-tag) and pNuc-HO-1/NLS (HO-1
with C-terminal NLS). We then assessed whether cytoplasmic or/and
nuclear expression of HO-1 enhanced transcriptional activity of
VEGF. HEK293 cells were co-transfected with the VEGF promoter
(pVEGF) and either pFlag-HO-1 (cytoplasmic HO-1) or pNuc-HO-1/NLS
(nuclear HO-1) in a dose-dependent manner (Fig. 6A). Cell extracts were prepared
after 24 h, and luciferase activity normalized to β-Gal activity
was assayed to measure VEGF promoter activity. The expression
levels of HO-1 were determined by western blot analysis with
anti-HO-1 antibody. Membranes were stripped and reblotted with an
anti-GAPDH antibody to ensure equal loading. Relative VEGF promoter
activity was approximately 6-fold higher in cells transfected with
pNuc-HO-1 and approximately 3-fold higher in cells transfected with
pFlag-HO-1 compared to their control counterparts (Fig. 6A).
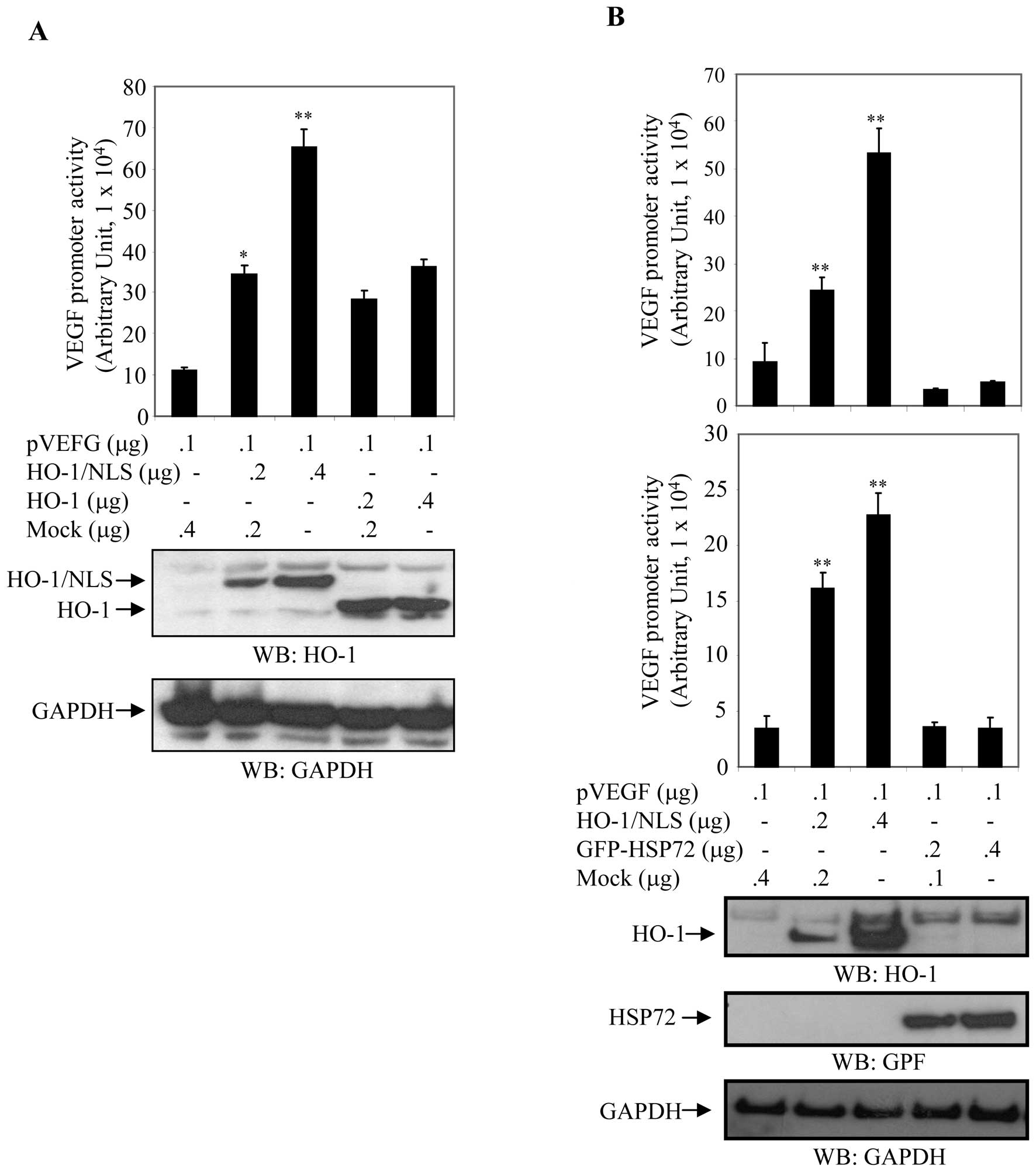 | Figure 6Nuclear localization of HO-1 promoted
transcriptional activity of VEGF. (A) Differential activation of
VEGF transcriptional activity by cytoplasmic and nuclear HO-1.
HEK293 cells were co-transfected with the VEGF promoter and HO-1 or
HO-1/NLS in a dose-dependent fashion, as indicated. After 24 h,
VEGF promoter activity (luciferase activity) was measured and
normalized to β-Gal activity. Relative VEGF promoter activity
derived from three experiments was expressed in arbitrary units.
Cell extracts were also blotted with anti-HO-1 and anti-GAPDH
antibodies. Columns, mean; bars, SD; *p<0.05;
**p<0.01. pVEGF, VEGF promoter; HO-1, plasmid
expressing HO-1 in cytosol; HO-1/NLS, plasmid expressing nuclear
HO-1; NLS, nuclear localization signal. (B) Differential activation
of VEGF transcriptional activity by heat shock proteins. HEK293 and
COS7 cells were co-transfected with the VEGF promoter and
pNuc-HO-1/NLS or EGFP-HSP72 in a dose-dependent manner as
indicated. After 24 h, luciferase activity was measured and
normalized to β-galactosidase (β-Gal) activity to quantify VEGF
promoter activity. Relative VEGF promoter activity was expressed in
arbitrary units. Micrograph is representative of three independent
experiments. Cell extracts were also blotted with anti-HO-1,
anti-GFP and anti-GAPDH antibodies. GAPDH served as internal
control. Columns, mean; bars, SD; *p<0.05;
**p<0.01. HO-1, also known as heat shock protein 32;
and HSP72, heat shock protein 72; NLS, nuclear localization signal.
Upper panel: HEK293; lower panel: COS7. |
HEK293 and COS7 cells were cotransfected with the
VEGF promoter (pVEGF) and either pNuc-HO-1/NLS [HO-1, also known as
Heat Shock Protein 32 (10)] or
pEGFP-HSP72 (HSP72, heat shock protein 72) as a control in a
dose-dependent manner. Cell extracts were prepared after 24 h and a
luciferase assay was performed to measure VEGF promoter activity.
Luciferase activity was normalized and expressed in arbitrary units
relative to β-Gal activity. Expression levels of HO-1 and HSP72
were determined by western blot analysis with anti-HO-1 and
anti-GFP antibodies. Blotting with anti-GAPDH antibody demonstrated
equal loading of cell extracts. Relative luciferase activity was
upregulated in a dose-dependent manner in cells transfected with
HO-1/NLS, whereas luciferase activity was relatively unchanged in
cells transfected with GFP-HSP72 (Fig.
6B). VEGF promoter activity was 2.60-fold (p<0.001) and
5.7-fold (p<0.001) higher in HEK293T cells transfected 0.2 and
0.4 μg of pNuc-HO-1/NLS, respectively (Fig. 6B), and 4.61-fold (p<0.001) and
6.47-fold (p<0.001) higher when COS7 cells which were similarly
cotransfected. In contrast, transfection of HEK293 and COS7 cells
with GFP-HSP72 had no statistically significant effect on
luciferase activity (Fig. 6B).
These results showed that nuclear HO-1 significantly increased
transcriptional activity of the VEGF promoter in a dose-dependent
manner, while HSP72 had an insignificant effect on the activity of
the VEGF promoter (Fig. 6B). These
findings suggested that nuclear expression of HO-1 plays an
important role in the transcriptional activity of VEGF.
Ectopic expression of nuclear HO-1
promoted VEGF secretion in prostate cancer cells
Given that ectopic expression of nuclear HO-1
increased VEGF promoter activity (Fig.
6), we sought to further examine the effect of nuclear HO-1 on
VEGF secretion using ELISA. For this analysis, PC3 cells were used
because of their high transfection efficiency. PC3 cells were
transfected with mock, pFlag-HO-1 or pNuc-HO-1/NLS. Cell cultures
were washed and replaced with fresh medium containing 0.5% FBS
within 24 h. Cell culture supernatants were collected and analyzed
for VEGF using ELISA. Ectopic expression of HO-1/NLS significantly
enhanced VEGF secretion compared to cells transfected with HO-1
(Fig. 7A). Next, cell cultures
were replaced with fresh medium containing 0.5% FBS and then
treated with SM for 24 h. Supernatants were collected and VEGF
concentration was measured by ELISA. A significant increase in VEGF
secretion was observed in cells transfected with HO-1/NLS compared
to cells transfected with mock or HO-1 (Fig. 7B). These data suggested that
nuclear HO-1 was involved in promoting VEGF secretion.
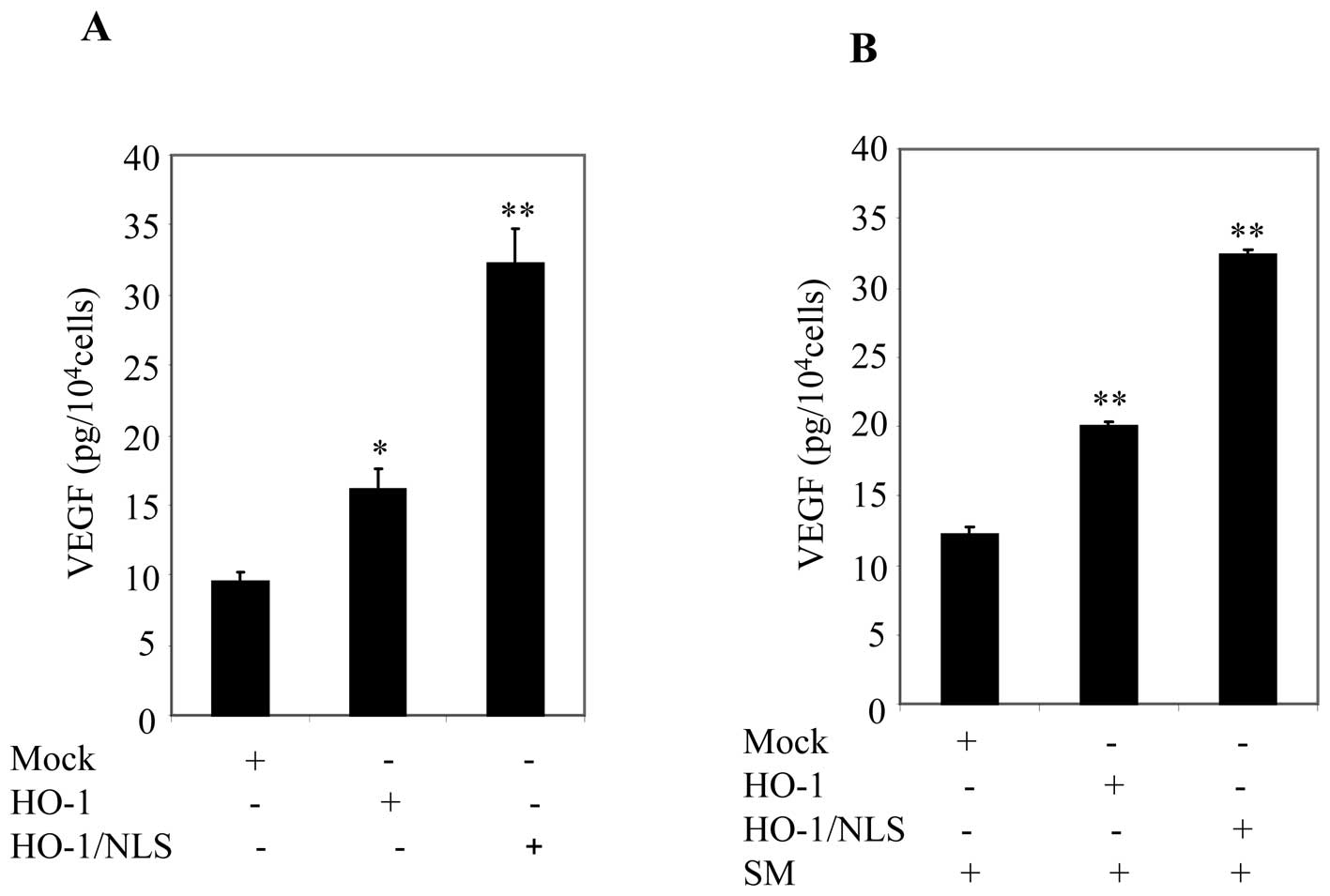 | Figure 7Ectopic expression of nuclear HO-1
promoted VEGF secretion in prostate cancer cells. (A) PC3 cells
were transfected with mock, HO-1 or HO-1/NLS. After 24 h, cells
were starved with cell culture medium containing 0.5% FBS. After 24
h, supernatants were collected, and VEGF secretions were measured
by using ELISA. Columns, mean; bars, SD; *p<0.05;
**p<0.01. (B) Cell culture, in (A), then were
replenished with cell culture medium containing 0.5% FBS plus SM.
After 24 h, supernatants were collected and VEGF secretions were
measured by using ELISA. Columns, mean; bars, SD;
*p<0.05; **p<0.01. |
Discussion
Cigarette smoking represents one of the most serious
problems for public health, and at present accounts for 6 million
deaths annually worldwide (34).
Although the relevance of this epidemic is known, the molecular
mechanism(s) underlying its toxicity and carcinogenic potential
remain elusive. Cigarette smoking has been linked to cancers of the
lung, breast and brain (34), but
while some research studies have shown that cigarette smoking is
not associated with the incidence of prostate cancer, other reports
suggested that current or recent cigarette smoking is linked to an
elevated risk of mortality, advanced stage or high-grade disease
(6). Cigarette smoking may
therefore be involved in the progression rather than the initiation
of prostate cancer. A number of studies demonstrated that
angiogenesis is associated with the progression of prostate cancer
(31,32). Previous reports detected higher
levels of HO-1 protein in various tumor tissues compared to normal
tissue (33,35–40),
and was associated with tumor progression of head and neck squamous
cell carcinomas (27). The
angiogenic cytokine VEGF has a central role in tumor angiogenesis
by binding and activating the receptors, VEGFR1 and VEGFR2. The
VEGF/VEGFR axis promotes endothelial cell differentiation, cell
growth, tubular formation and migration (41–45).
It has also been reported that HO-1 is an important proangiogenic
mediator, which further supports tumor progression (12). The present study explores the
relationship between cigarette smoke, HO-1 expression and VEGF
secretion in prostate cancer cells.
In response to oxidative stress, cells have evolved
multiple protective mechanisms to neutralize and clear toxic
molecules and restore cellular redox homeostasis. Induction of HO-1
is a fundamental cellular defense process against oxidative stress
caused by environmental stimuli. Cells lacking HO-1 are susceptible
to free radical damage and oxidative injury, which results in high
levels of endothelial damage and prolonged inflammation (10,11).
Treatment with SM induced HO-1 mRNA expression and increased HO-1
protein in prostate cancer cells (Fig.
2). Induction of HO-1 may therefore provide the first line of
cellular defense of prostate cancer cells against the oxidative
stimulus of cigarette smoke. Thus, an increase in HO-1 levels may
be required for survival of prostate cancer cells. This result is
consistent with previous analyses of HO-1 expression in different
types of cancer. Overexpression of HO-1 has been reported in
lymphosarcoma (35), brain tumors
(36), renal carcinoma (37), hepatoma (38), Kaposi sarcoma (39), pancreatic cancer (40) and chronic myeloid leukemia
(41). Cigarette smoking has been
shown to be associated with an elevated risk of mortality or
advanced stage prostate cancer, but not with incidence of prostate
cancer (6). These lines of
evidence strongly suggest that SM-mediated induction of HO-1 may be
associated with the progression of prostate cancer.
HO-1 has been reported to promote VEGF secretion and
facilitate VEGF-mediated activities such as promoting the
efficiency of cell proliferation and migration and improving the
formation of capillary-like tubular structures and capillary
outgrowth (14,15). Treatment of prostate cancer cells
with SM induced expression of HO-1 (Fig. 2) and enhanced VEGF secretion in
both DU145 and PC3 cells (Fig. 3).
Ectopic expression of HO-1 (also known as heat shock protein 32)
and another heat shock protein, HSP72 were tested for their ability
to induce VEGF transcriptional activity. While HSP72 failed to
increase transcriptional activity of VEGF, HO-1 induced significant
transcriptional activity of VEGF (Fig.
6) and VEGF secretion (Fig.
7). These data suggested that cigarette smoke may therefore
promote the progression of prostate cancer through HO-1-modulated
VEGF increase.
Previous studies reported that nuclear translocation
of HO-1 was associated with prostate cancer (19), and its nuclear expression had a
strong correlation with the grade of differentiation of oral
squamous cell carcinomas (28,29)
and the tumor progression of head and neck squamous cell carcinomas
(27). Together with our finding
that SM induced nuclear translocation in prostate cancer, this
suggested that SM-mediated nuclear localization of HO-1 is
contributable to the progression of prostate cancer. Given that
nuclear localization may be associated with the progression of
prostate cancer, we asked whether nuclear HO-1 was involved in the
promotion of VEGF secretion in prostate cancer. To address this
question we attempted to express HO-1 exclusively in nuclei of
cells by tagging it with tandem nuclear localization signals.
Interestingly, ectopic expression of nuclear HO-1 had the potential
to significantly induce VEGF transcriptional activity and
secretion, whereas cytoplasmic HO-1 did not (Fig. 6). Taken together, these findings
strongly suggest that nuclear localization of HO-1 induced by
cigarette smoke plays a central role in VEGF secretion and may
contribute to tumor angiogenesis and the progression of prostate
cancer.
Our study revealed the mechanism by which cigarette
smoking is associated with prostate cancer through nuclear HO-1 and
VEGF regulation. This study also provides new insights into the
involvement of cigarette smoke and HO-1 in prostate cancer.
Therapies targeting nuclear HO-1 may therefore represent a novel
approach for the treatment of prostate cancer.
Acknowledgements
This work was supported by grants from
Flight Attendant Medical Research Institute (S.S.) and National
Institutes of Health [NIH RO1-HL092811 (S.T.)]. We would like to
thank Dr Jerome Groopman, Chief of Division of Experimental
Medicine, for facilitating this study. We also appreciate Dr
Farheen Arshad and Dr Paula Kuzontkoski for their assistance.
References
|
1
|
Siegel R, Ward E, Brawley O and Jemal A:
Cancer statistics, 2011: the impact of eliminating socioeconomic
and racial disparities on premature cancer deaths. CA Cancer J
Clin. 61:212–236. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Danaei G, Ding EL, Mozaffarian D, Taylor
B, Rehm J, Murray CJ and Ezzati M: The preventable causes of death
in the United States: comparative risk assessment of dietary,
lifestyle, and metabolic risk factors. PLoS Med. 6:e10000582009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pryor WA and Stone K: Oxidants in
cigarette smoke. Radicals, hydrogen peroxide, peroxynitrate, and
peroxynitrite. Ann NY Acad Sci. 686:12–27. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hecht SS: Tobacco smoke carcinogens and
lung cancer. J Natl Cancer Inst. 91:1194–1210. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Portal-Nuñez S, Shankavaram UT, Rao M,
Datrice N, Atay S, Aparicio M, Camphausen KA, Fernández-Salguero
PM, Chang H, Lin P, Schrump DS, Garantziotis S, Cuttitta F and
Zudaire E: Aryl hydrocarbon receptor-induced adrenomedullin
mediates cigarette smoke carcinogenicity in humans and mice. Cancer
Res. 72:5790–5800. 2012.PubMed/NCBI
|
|
6
|
Zu K and Giovannucci E: Smoking and
aggressive prostate cancer: a review of the epidemiologic evidence.
Cancer Causes Control. 20:1799–1810. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tenhunen R, Marver HS and Schmid R: The
enzymatic conversion of heme to bilirubin by microsomal heme
oxygenase. Proc Natl Acad Sci USA. 61:748–755. 1968. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kikuchi G, Yoshida T and Noguchi M: Heme
oxygenase and heme degradation. Biochem Biophys Res Commun.
338:558–567. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Prawan A, Kundu JK and Surh YJ: Molecular
basis of heme oxygenase-1 induction: implications for
chemoprevention and chemoprotection. Antioxid Redox Signal.
7:1688–1703. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Maines MD and Abrahamsson PA: Expression
of heme oxygenase-1 (HSP32) in human prostate: normal,
hyperplastic, and tumor tissue distribution. Urology. 47:727–733.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Maines MD and Gibbs PE: 30 some years of
heme oxygenase: from a ‘molecular wrecking ball’ to a ‘mesmerizing’
trigger of cellular events. Biochem Biophys Res Commun.
338:568–577. 2005.PubMed/NCBI
|
|
12
|
Jozkowicz A, Was H and Dulak J: Heme
oxygenase-1 in tumors: is it a false friend? Antioxid Redox Signal.
9:2099–2117. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cherrington JM, Strawn LM and Shawver LK:
New paradigms for the treatment of cancer: the role of
anti-angiogenesis agents. Adv Cancer Res. 79:1–38. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Busserolles J, Megias J, Terencio MC and
Alcaraz MJ: Heme oxygenase-1 inhibits apoptosis in Caco-2 cells via
activation of Akt pathway. Int J Biochem Cell Biol. 38:1510–1517.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen X, Ding YW, Yang G, Bondoc F, Lee MJ
and Yang CS: Oxidative damage in an esophageal adenocarcinoma model
with rats. Carcinogenesis. 21:257–263. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu ZM, Chen GG, Ng EK, Leung WK, Sung JJ
and Chung SC: Upregulation of heme oxygenase-1 and p21 confers
resistance to apoptosis in human gastric cancer cells. Oncogene.
23:503–513. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nishie A, Ono M, Shono T, Fukushi J,
Otsubo M, Onoue H, Ito Y, Inamura T, Ikezaki K, Fukui M, Iwaki T
and Kuwano M: Macrophage infiltration and heme oxygenase-1
expression correlate with angiogenesis in human gliomas. Clin
Cancer Res. 5:1107–1113. 1999.PubMed/NCBI
|
|
18
|
Fang J, Sawa T, Akaike T, Akuta T, Sahoo
SK, Khaled G, Hamada A and Maeda H: In vivo antitumor activity of
pegylated zinc protoporphyrin: targeted inhibition of heme
oxygenase in solid tumor. Cancer Res. 63:3567–3574. 2003.PubMed/NCBI
|
|
19
|
Sacca P, Meiss R, Casas G, Mazza O, Calvo
JC, Navone N and Vazquez E: Nuclear translocation of haeme
oxygenase-1 is associated to prostate cancer. Br J Cancer.
97:1683–1689. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tanaka S, Akaike T, Fang J, Beppu T, Ogawa
M, Tamura F, Miyamoto Y and Maeda H: Antiapoptotic effect of haem
oxygenase-1 induced by nitric oxide in experimental solid tumour.
Br J Cancer. 88:902–909. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Duckers HJ, Boehm M, True AL, Yet SF, San
H, Park JL, Clinton Webb R, Lee ME, Nabel GJ and Nabel EG: Heme
oxygenase-1 protects against vascular constriction and
proliferation. Nat Med. 7:693–698. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jazwa A, Loboda A, Golda S, Cisowski J,
Szelag M, Zagorska A, Sroczynska P, Drukala J, Jozkowicz A and
Dulak J: Effect of heme and heme oxygenase-1 on vascular
endothelial growth factor synthesis and angiogenic potency of human
keratinocytes. Free Radic Biol Med. 40:1250–1263. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sunamura M, Duda DG, Ghattas MH, Lozonschi
L, Motoi F, Yamauchi J, Matsuno S, Shibahara S and Abraham NG: Heme
oxygenase-1 accelerates tumor angiogenesis of human pancreatic
cancer. Angiogenesis. 6:15–24. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Marinissen MJ, Tanos T, Bolos M, de
Sagarra MR, Coso OA and Cuadrado A: Inhibition of heme oxygenase-1
interferes with the transforming activity of the Kaposi sarcoma
herpes-virus-encoded G protein-coupled receptor. J Biol Chem.
281:11332–11346. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cisowski J, Loboda A, Jozkowicz A, Chen S,
Agarwal A and Dulak J: Role of heme oxygenase-1 in hydrogen
peroxide-induced VEGF synthesis: effect of HO-1 knockout. Biochem
Biophys Res Commun. 326:670–676. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hirai K, Sasahira T, Ohmori H, Fujii K and
Kuniyasu H: Inhibition of heme oxygenase-1 by zinc protoporphyrin
IX reduces tumor growth of LL/2 lung cancer in C57BL mice. Int J
Cancer. 120:500–505. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gandini NA, Fermento ME, Salomón DG,
Blasco J, Patel V, Gutkind JS, Molinolo AA, Facchinetti MM and
Curino AC: Nuclear localization of heme oxygenase-1 is associated
with tumor progression of head and neck squamous cell carcinomas.
Exp Mol Pathol. 93:237–245. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tsuji MH, Yanagawa T, Iwasa S, Tabuchi K,
Onizawa K, Bannai S, Toyooka H and Yoshida H: Heme oxygenase-1
expression in oral squamous cell carcinoma as involved in lymph
node metastasis. Cancer Lett. 138:53–59. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lin Q, Weis S, Yang G, Weng YH, Helston R,
Rish K, Smith A, Bordner J, Polte T, Gaunitz F and Dennery PA: Heme
oxygenase-1 protein localizes to the nucleus and activates
transcription factors important in oxidative stress. J Biol Chem.
282:20621–20633. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Walsh K, Sriprasad S, Hopster D, Codd J
and Mulvin D: Distribution of vascular endothelial growth factor
(VEGF) in prostate disease. Prostate Cancer Prostatic Dis.
5:119–122. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sasco AJ, Secretan MB and Straif K:
Tobacco smoking and cancer: a brief review of recent
epidemiological evidence. Lung Cancer. 2:S3–S9. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Aragon-Ching JB: Active surveillance for
prostate cancer: has the time finally come? J Clin Oncol.
28:e265–e266. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Aragon-Ching JB, Madan RA and Dahut WL:
Angiogenesis inhibition in prostate cancer: current uses and future
promises. J Oncol. 2010:3618362010.PubMed/NCBI
|
|
34
|
World Health Organization: Tobacco 2011
[cited]. Available from: http://www.who.int/mediacentre/factsheets/fs339/en/index.html.
|
|
35
|
Schacter BA and Kurz P: Alterations in
hepatic and splenic microsomal electron transport system
components, drug metabolism, heme oxygenase activity, and
cytochrome P-450 turnover in Murphy-Sturm lymphosarcoma-bearing
rats. Cancer Res. 42:3557–3564. 1982.
|
|
36
|
Deininger MH, Meyermann R, Trautmann K,
Duffner F, Grote EH, Wickboldt J and Schluesener HJ: Heme oxygenase
(HO)-1 expressing macrophages/microglial cells accumulate during
oligodendroglioma progression. Brain Res. 882:1–8. 2000. View Article : Google Scholar
|
|
37
|
Goodman AI, Choudhury M, da Silva JL,
Schwartzman ML and Abraham NG: Overexpression of the heme oxygenase
gene in renal cell carcinoma. Proc Soc Exp Biol Med. 214:54–61.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Doi K, Akaike T, Fujii S, Tanaka S, Ikebe
N, Beppu T, Shibahara S, Ogawa M and Maeda H: Induction of haem
oxygenase-1 nitric oxide and ischaemia in experimental solid
tumours and implications for tumour growth. Br J Cancer.
80:1945–1954. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
McAllister SC, Hansen SG, Ruhl RA, Raggo
CM, DeFilippis VR, Greenspan D, Früh K and Moses AV: Kaposi
sarcoma-associated herpesvirus (KSHV) induces heme oxygenase-1
expression and activity in KSHV-infected endothelial cells. Blood.
103:3465–3473. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Berberat PO, Dambrauskas Z, Gulbinas A,
Giese T, Giese N, Künzli B, Autschbach F, Meuer S, Büchler MW and
Friess H: Inhibition of heme oxygenase-1 increases responsiveness
of pancreatic cancer cells to anticancer treatment. Clin Cancer
Res. 11:3790–3798. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mayerhofer M, Florian S, Krauth MT,
Aichberger KJ, Bilban M, Marculescu R, Printz D, Fritsch G, Wagner
O, Selzer E, Sperr WR, Valent P and Sillaber C: Identification of
heme oxygenase-1 as a novel BCR/ABL-dependent survival factor in
chronic myeloid leukemia. Cancer Res. 64:3148–3154. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ferrara N, Mass RD, Campa C and Kim R:
Targeting VEGF-A to treat cancer and age-related macular
degeneration. Annu Rev Med. 58:491–504. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shibuya M: Vascular permeability/vascular
endothelial growth factor. Angiogenesis: An Integrative Approach
from Science of Medicine. Folkman J and Figg WD: Springer; New
York, NY: pp. 89–98. 2008, View Article : Google Scholar
|
|
44
|
Li X, Claesson-Welsh L and Shibuya M: VEGF
receptor signal transduction. Methods Enzymol. 443:261–284. 2008.
View Article : Google Scholar
|
|
45
|
Kim KJ, Li B, Winer J, Armanini M, Gillett
N, Phillips HS and Ferrara N: Inhibition of vascular endothelial
growth factor-induced angiogenesis suppresses tumour growth in
vivo. Nature. 362:841–844. 1993. View Article : Google Scholar : PubMed/NCBI
|