Introduction
Lung cancer is currently the leading cause of cancer
death in men and women, causing more deaths than colon, breast and
prostate cancer combined (1). Lung
cancers are mainly classified into small cell lung carcinoma (SCLC)
and non-small cell lung carcinoma (NSCLC), but lung cancers are
also sub-classified depending upon different characteristics and
origins of progenitor cells (2–4). For
example, neuroendocrine (NE) phenotypes characterize a spectrum of
tumors, including low-grade typical and intermediate-grade atypical
carcinoid, high-grade large-cell NE carcinoma and SCLC (5,6). NE
lung tumors comprise 20–25% of all invasive lung malignancies.
Among those, SCLC is the most common pulmonary NE tumor which
accounts for 15–20% of invasive lung malignancies whereas carcinoid
tumors and large-cell NE carcinoma represent minor fraction of
invasive lung malignancies (5,6).
Currently, no effective treatments are available to cure SCLC and
other NE lung tumors and it is necessary to identify a biological
feature specifically involved in growth and survival of these tumor
cells.
Autophagy is an important cellular recycling process
to overcome limited availability of nutrients, which is mediated by
a constitutive lysosomal degradation pathway (7). Under stress conditions, autophagy is
upregulated to generate resources for the maintenance of essential
cellular functions (8,9). However paradoxically, autophagy can
also trigger cell death under certain conditions. Therefore, a
balance between these two opposing effects of autophagy influences
cellular differentiation, development, homeostasis and the
development of different diseases, including cancer (7). A key step in mediating autophagy is
the formation of autophagosomes, which is mediated by
microtubule-associated protein-1 light chain-3 (LC3), a mammalian
homolog of yeast autophagy-related gene 8 and the LC3 binding
protein, SQSTM1/p62 (10). During
autophagy, the cytoplasmic form of LC3 (LC3-I, 18 kDa) is recruited
to the autophagosomes, where LC3-II (16 kDa) is generated by
site-specific proteolysis and lipidation near to the C-terminus
(11). Therefore, autophagic
activity is measured biochemically as the amount of LC3-II and p62
that accumulates in the absence or presence of lysosomal
activity.
Recent studies have demonstrated the significance of
autophagy in pulmonary epithelial cell proliferation and survival.
For example, it has been demonstrated that increased autophagy
contributes to the pathogenesis of chronic obstructive pulmonary
disease by promoting epithelial cell death (12). It has also been shown that LC3
confers protection against hypoxia-induced pulmonary hypertension
(13). Nevertheless, the
significance of autophagy in lung cancer is yet unclear. Further,
no studies in the context of autophagy in NE lung tumors have been
reported.
Here, we demonstrate that, in a panel of human lung
cancer lines, steady-state levels of the autophagy marker, LC3, are
relatively high in the cell lines expressing neuron-specific
enolase (NSE), a key NE marker in lung tumor (14,15).
We then show that those cell lines are more sensitive to autophagy
inhibitors than non-NE lung tumor cells. We also investigate the
involvement of AKT and mammalian target of rapamycin (mTOR)
pathways, the two known regulators of autophagy (16,17),
in autophagy regulation in these cells.
Materials and methods
Cell culture and reagents
The human lung cancer lines, DMS53, NCI-H209,
NCI-H82, NCI-H69, NCI-H889, NCI-H345, SHP-77, A549, NCI-H23,
NCI-H460, NCI-H1155, NCI-H358, NCI-H727, NCI-H125 and NCI-H1770
(ATCC), were maintained in phenol red-deficient RPMI-1640
(Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine
serum (FBS), 100 U of penicillin and 100 μg of streptomycin
per ml. Bafilomycin A1 and chloroquine were purchased from Sigma
(St. Louis, MO, USA). Torin 1, rapamycin, AKTi and MK-2206 were
purchased from TOCRIS (Minneapolis, MN, USA), Cell Signaling
Technology (Danvers, MA, USA), EMD Chemicals Inc. (Chicago, IL,
USA) and Active Biochem (Maplewood, NJ, USA), respectively.
Small hairpin RNA (shRNA) expression
construct
The lentiviral pLKO.1-shRNA vector targeting mTOR
(Addgene plasmid 8454) was previously described (18). For lentivirus production, 293T
cells were co-transfected with pLKO.1 and packaging vectors, as
previously described (19,20). Viral supernatants were collected
after 48–72 h and mixed with polybrene (Sigma) at 4–8 μg/ml
before use. Viral titer was determined by scoring cells resistant
to puromycin.
Cell survival and cell cycle assays
To determine cell survival rates, cells were seeded
in 24-well plates (Corning, Corning, NY, USA) at a density of 5,000
cells per well. Cell proliferation and death was determined by
counting trypan blue-stained cells using hemocytometer. For cell
cycle analysis, cells were washed with ice-cold 0.2% BSA in PBS,
resuspended in 250 mM sucrose/40 mM citrate buffer (pH 7.6)
containing 0.5% DMSO. Nuclei were prepared, stained with propidium
iodide (21) and analyzed by LSR
II Flow Cytometer (Becton-Dickinson, Franklin Lakes, NJ, USA) with
a gate that selects single nuclei within a normal size range. The
cell cycle parameters from 10,000 gated nuclei were determined and
subsequent analysis was conducted using FCS Express software (De
Novo software, Los Angeles, CA, USA).
Immunoblot analysis
Cells harvested at various times were lysed and
analyzed for protein concentration using the BCA reagent (Pierce,
Rockford, IL, USA), as previously described (22). Protein (50–100 μg) was resolved by
SDS-PAGE, transferred to a polyvinylidene difluoride membrane
filter (Bio-Rad, Hercules, CA, USA) and stained with Fast Green
reagent (Thermo Fisher Scientific, Waltham, MA, USA). Membrane
filters were then blocked in 0.1 M Tris (pH 7.5)/0.9% NaCl/0.05%
Tween-20/5% non-fat dry milk and incubated with appropriate
antibodies. Antibodies were diluted as follows: NSE, 1:2,500; PARP,
1:1000 (Thermo Fisher Scientific); AKT, 1:2,500; phospho-AKT
(Ser473), 1:5,000; phospho-GSK-3β (Ser9), 1:2,500; phospho-ERK1/2
(Thr202/Tyr204), 1:2,500; GAPDH, 1:5,000; phospho-p70S6K1 (Thr389),
1:2,000; phospho-mTOR (Ser2481), 1:2,000; phospho-4E-BP1 (S65),
1:1,000; phospho-S6 (S235/236), 1:1,000 (Cell Signaling
Technology); p62, 1:2,000 (Santa Cruz Biotech, Santa Cruz, CA,
USA); LC3B, 1:2,000 (MBL International, Woburn, MA, USA). The
Supersignal West Pico and Femto chemiluminescence kits (Pierce)
were used for visualization of the signal. Immunoblots were scanned
and analyzed using Image Lab (Bio-Rad).
Results
The levels of LC3-I are upregulated in NE
lung tumor cell lines
To profile steady-state levels of autophagy in
different lung tumor types, we analyzed LC3 levels in 15 human lung
tumor cell lines, including 7 SCLC and 8 NSCLC cell lines. In this
panel, cell lines were also examined for NSE expression as a marker
of NE phenotype. All SCLC cell lines except for DMS53 expressed
high levels of NSE whereas among the NSCLC cell lines, only the
known NE lines, NCI-H1155 and NCI-H1770 (23), expressed similarly high levels of
NSE (Fig. 1).
In this cell line panel, we detected relatively high
LC3-I levels in strong correlation with high NSE levels (Fig. 1). Because LC3-II levels were not
clearly detected under the culture condition using 10% FBS
(Fig. 1, left panel), we also
analyzed cells maintained in the media containing 0.1% FBS, which
would increase the cellular demand for autophagy (Fig. 1, right panel). Indeed, the
formation of LC3-II became prominent in most cell lines under this
serum-starved culture condition (Fig.
1, right panel). However, this condition did not increase
LC3-II formation in the NSE expressing cell lines, NCI-H209,
NCI-H69, NCI-H1155 and NCI-H1770 cells (Fig. 1, right panel) and very
intriguingly, this phenomenon was correlated with relatively high
basal levels of AKT phosphorylation at Ser473, an indication of AKT
activation (24) (Fig. 1).
In this cell line panel, we also examined the mTOR
and ERK1/2 pathways, which are often deregulated in cancer.
However, neither ERK1/2 activity, as indicated by phosphorylation
of their activation loop (Thr202/Tyr204 of ERK1 and Thr183/Tyr185
of ERK2), nor activity of the mTOR pathway, as indicated by
phosphorylation of 4E binding protein 1 (4E-BP) and the ribosomal
protein S6, were significantly correlated with the low LC3-II
levels (Fig. 1, right panel).
4E-BP is phosphorylated by mTOR and S6 is a substrate of S6 kinase
1 (S6K1), which is also phosphorylated by mTOR (25). These data demonstrate a strong
correlation between LC3 and NSE levels, suggesting a potential
significance of autophagy in NE lung tumor cells. These data also
suggested a potential involvement of AKT in autophagy regulation in
certain NE lung tumor types.
NE lung tumor cells are more sensitive to
autophagy inhibitors than non-NE lung tumor cells
To determine the significance of autophagy in NE
lung tumor cells, we examined the effects of inhibition of
steady-state autophagy in NCI-H69, NCI-H209 and NCI-H1155 cells
using bafilomycin A1 and chloroquine, the lysosomotropic agents
that inhibit lysosomal degradation of autophagosome. As expected,
these cells exhibited highly increased LC3-II and p62 levels within
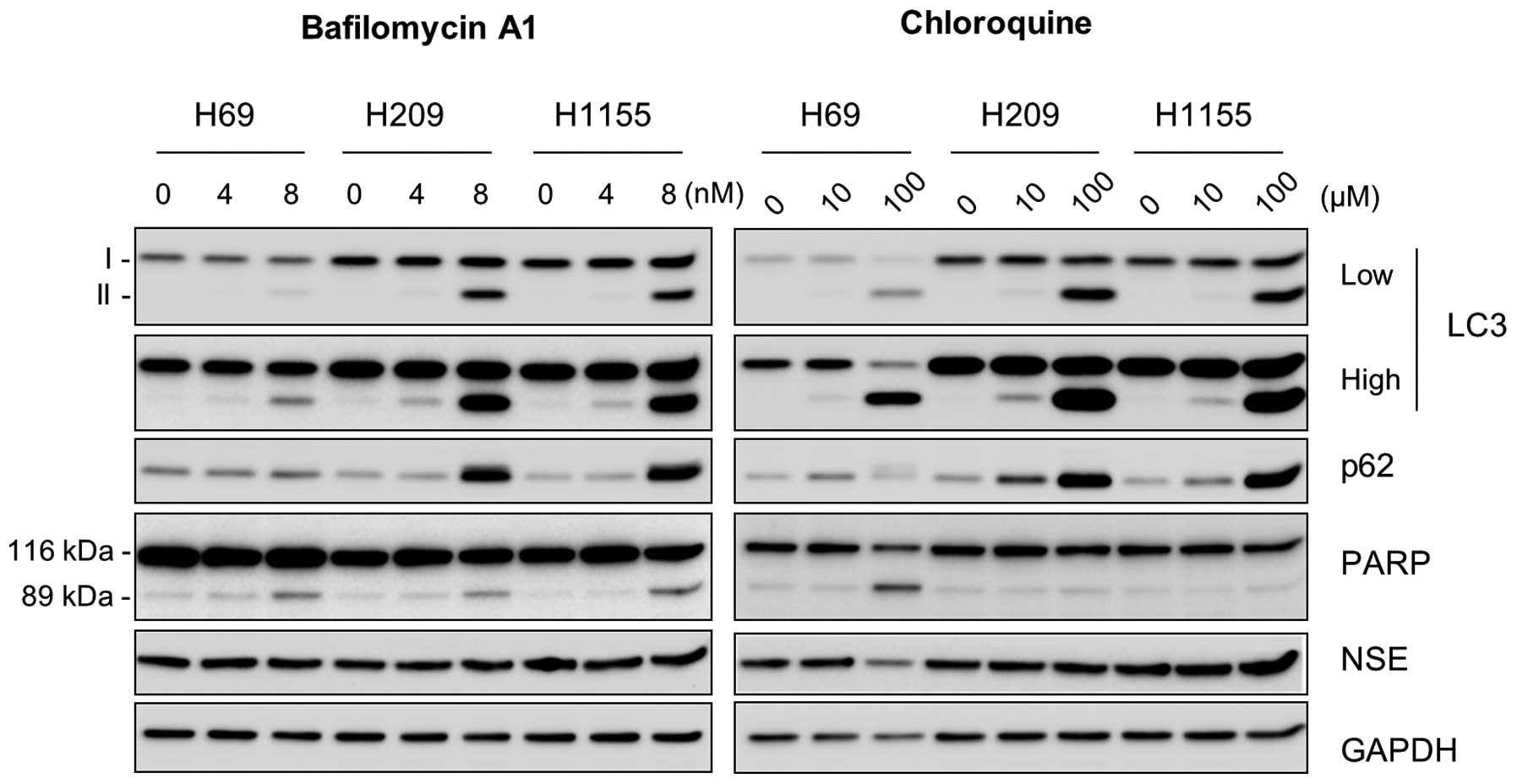
48 h in response to the inhibitor treatment (Fig. 2), which indicates the accumulation
of these proteins due to delayed autophagy. Of note, bafilomycin A1
significantly increased cleavage of poly(ADP-ribose) polymerase
(PARP), an indication of caspase-dependent apoptosis, in all these
cell lines, although chloroquine increased PARP cleavage only in
NCI-H69 cells (Fig. 2). In
contrast, NSE levels were unaffected under these conditions except
that chloroquine decreased NSE levels in NCI-H69 cells (Fig. 2, right panel). These data suggest
the importance of autophagy for survival, but not for NE phenotype,
of NE lung tumor cells.
Subsequently, we analyzed NCI-H69, NCI-H209 and
NCI-H1155 cells in comparison with the non-NE lung tumor cell
lines, NCI-H23, NCI-H460 and NCI-H727 for their sensitivity to
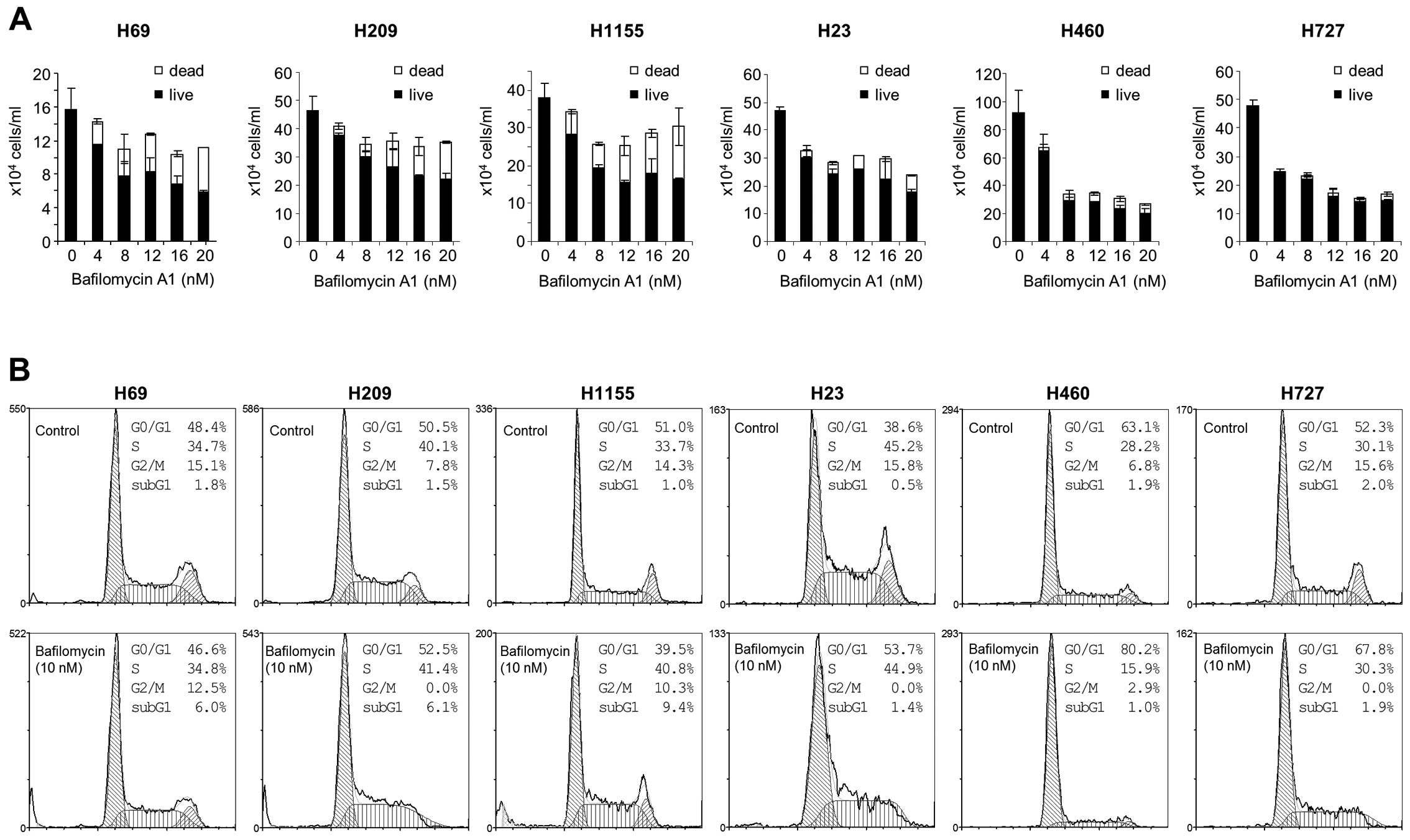
different doses of bafilomycin A1. We found that bafilomycin A1
could effectively suppress proliferation of these tumor lines
regardless of NE phenotypes (Fig.
3A). However intriguingly, the mechanisms of growth inhibition
appeared quite different between NE and non-NE lung tumor lines.
Bafilomycin A1 treatment increased cell death more significantly in
NCI-H69, NCI-H209 and NCI-H1155 cells than in NCI-H23, NCI-H460 and
NCI-H727 cells, as determined by trypan blue staining (Fig. 3A). Consistent with this,
bafilomycin A1 significantly increased sub-G1 phase cell
populations in NCI-H69, NCI-H209 and NCI-H1155 cells whereas the
drug induced G0/G1 phase arrest, but did not increase sub-G1
population, in NCI-H23, NCI-H460 and NCI-H727 cells (Fig. 3B). Increases in sub-G1 phase
population indicate onset of programmed cell death and thus, accord
with increased trypan blue staining (Fig. 3A) and PARP cleavage (Fig. 2, left panel) in bafilomycin
A1-treated NCI-H69, NCI-H209 and NCI-H1155 cultures. These data
suggest that autophagy inhibition can induce different growth
inhibitory effects in different lung tumor types, i.e.,
cytotoxicity in NE lung tumor types versus cytostasis in non-NE
lung tumor types.
AKT and mTOR pathways regulate autophagy
in certain NE lung tumor types in an opposing context
The correlation between low LC3-II levels and high
AKT phosphorylation in NCI-H69, NCI-H209 and NCI-H1155 cells
(Fig. 1) led us to investigate the
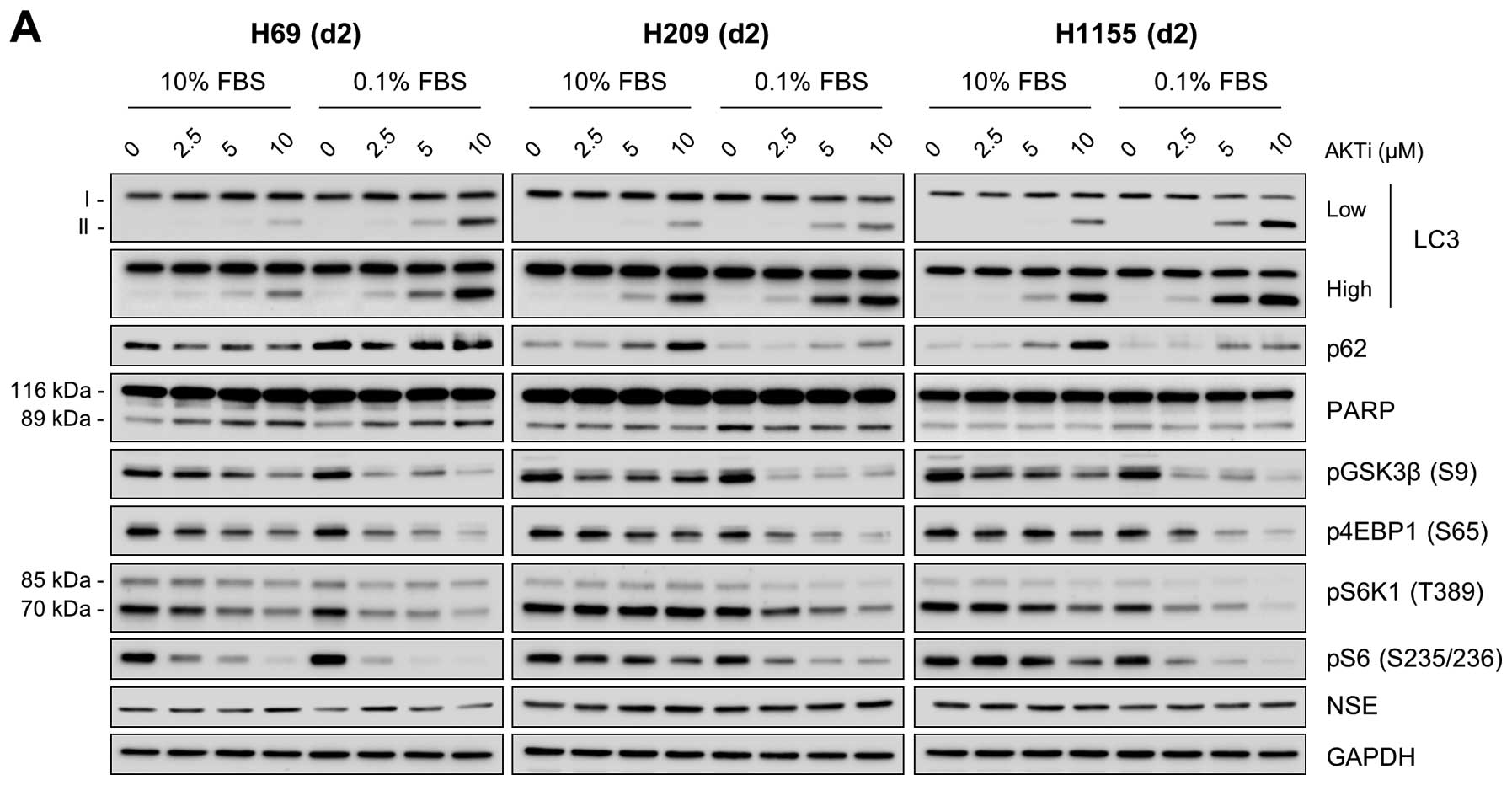
role of AKT in autophagy regulation in these cells. For this, we
examined the effects of AKT inhibition in these cells using the two
structurally-unrelated AKT specific inhibitors, AKTi and MK-2206.
We found that inhibition of AKT activity, as indicated by decreased
phosphorylation of its substrate GSK3β, substantially increased
LC3-II levels in these cells regardless of the culture conditions,
i.e., 10% versus 1% FBS (Fig. 4).
Along with this, p62 levels were also increased in these cells,
which was more significant in cells maintained using 10% FBS
(Fig. 4). However, NSE expression
was not affected by these inhibitors, suggesting that AKT may not
affect NE phenotypes of these cells. These effects of AKT
inhibition are consistent with the effects of bafilomycin A1 and
chloroquine on these NE lung tumor cell lines, suggesting a role
for AKT in autophagy regulation in certain NE lung tumor types.
Since mTOR complex (mTORC) 1 is well known for its
antagonizing effect on autophagy (16,17)
and because high mTOR pathway activity was also detected in
NCI-H69, NCI-H209 and NCI-H1155 cells (Fig. 1), we next investigated the effects
of mTOR inhibitors, torin 1 and rapamycin, on LC3 and p62 in these
cells. Whereas torin 1 inhibits both mTORC1 and mTORC2, rapamycin
inhibits only mTORC1 (26). Both
inhibitors effectively inhibited phosphorylation of S6K1 and its
substrate S6, although torin 1 inhibited 4E-BP1 phosphorylation
more effectively than rapamycin (Fig.
5), indicating significantly reduced mTOR activity in these
cells. Under these conditions, p62 levels were significantly
decreased in all three cell lines (Fig. 5). No significant increases in
LC3-II levels were detected, although torin 1-treated NCI-H69 cells
exhibited mild increases, which were still lower than the levels
increased by the AKT inhibitors (Fig.
5; compare with Fig. 4). These
changes are consistent with the known effects of mTORC1 inhibition
to trigger autophagy, leading to p62 depletion (16,17).
These opposite effects on p62 of the AKT and mTOR inhibitors
suggest that the mTOR and AKT pathways have opposing roles in
autophagy regulation in certain NE lung tumor types.
 | Figure 5.mTOR inhibitors, torin 1 and
rapamycin, consistently affect autophagy markers, but not AKT
activity, in NE lung tumor cells. Cells were treated with
increasing doses of the mTOR inhibitors, torin 1 (A) and rapamycin
(B), in RPMI-1640 containing 10% FBS for 2 days. LC3 processing,
p62 accumulation, PARP cleavage, AKT phosphorylation and mTOR
pathway activity (indicated by phosphorylation of 4E-BP, S6K1 and
S6) were analyzed by western blotting of total cell lysates. GAPDH
was the control for protein loading. |
AKT and mTOR pathways crosstalk in
certain NE lung tumor types
AKT can regulate mTORC1 activity via phosphorylation
of tuberous sclerosis complex 2 or PRAS40 (27–29).
Conversely, mTOR can also regulate AKT activity via mTORC2-mediated
phosphorylation of Ser473 in the hydrophobic motif of AKT (18). Given the coincident activation of
AKT and mTOR pathways in NCI-H69, NCI-H209 and NCI-H1155 cells, we
determined whether these two pathways can crosstalk in these cells
by examining the effects of AKT and mTOR inhibitors on the
surrogate markers of each pathway.
AKTi and MK-2206 treatments mildly but consistently
decreased phosphorylation of 4E-BP1 and S6K1 with the decreases
being more significant under the low serum culture condition
(Fig. 4), suggesting that AKT can
affect activity of the mTOR pathway in these NE lung tumor cells.
Whereas torin 1 inhibited AKT phosphorylation in these cell lines
(Fig. 5A), rapamycin rather
increased AKT phosphorylation in NCI-H209 and NCI-H1155 cells,
albeit not in NCI-H69 (Fig. 5B),
suggesting that mTORC1 and mTORC2 may antagonistically regulate AKT
in certain NE lung tumors.
The effect of torin 1, which inhibits both mTORC1
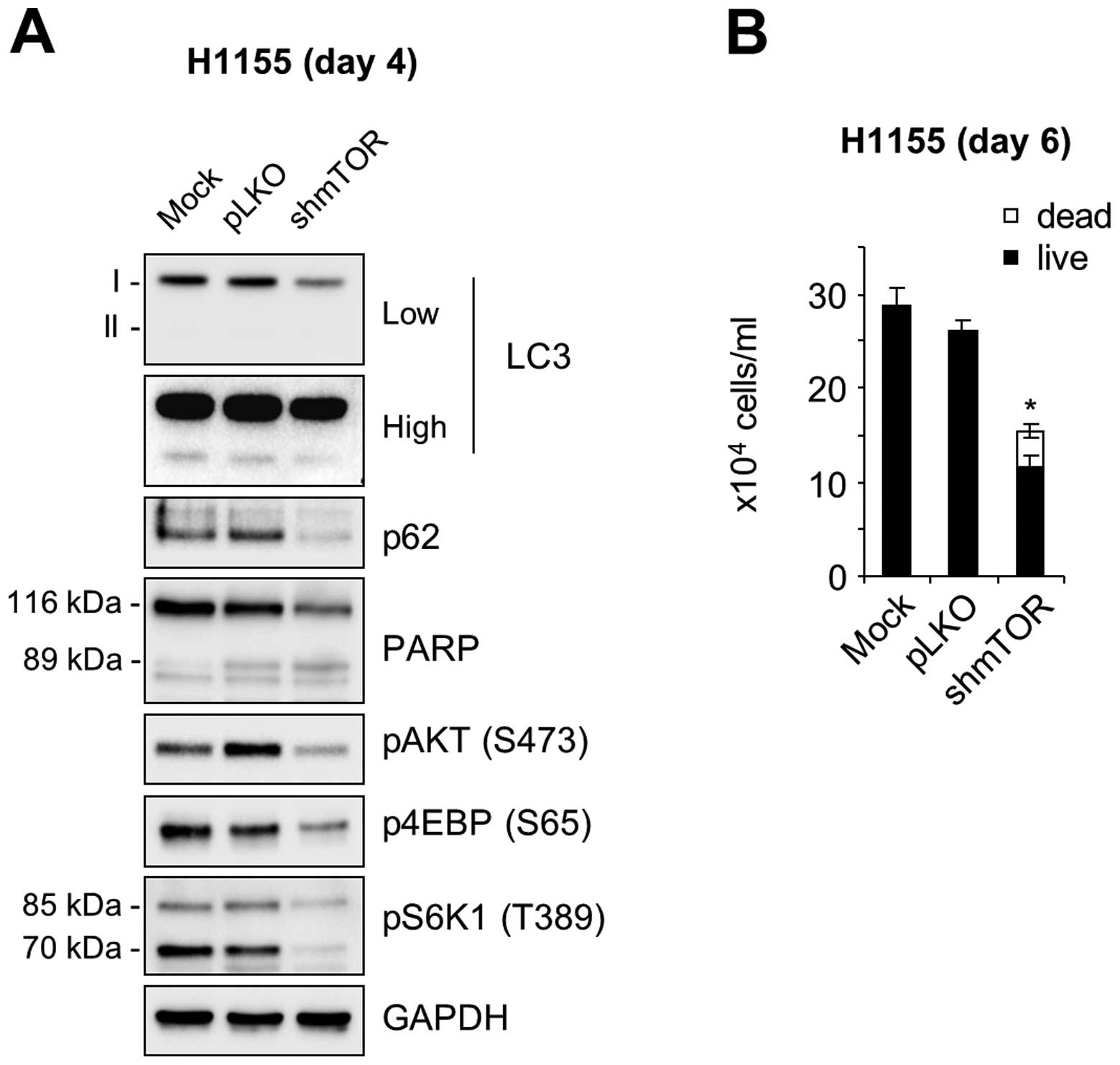
and mTORC2 activity, was confirmed by a lentiviral shRNA construct
that was previously used to specifically knockdown mTOR activity in
cells (18). Consistent with the
effects of torin 1 and rapamycin, mTOR knockdown also reduced the
levels of p62, as determined in NCI-H1155 cells (Fig. 6A). Under this condition, AKT
phosphorylation was significantly decreased (Fig. 6A), which is consistent with the
effect of torin 1. Of note, mTOR knockdown significantly increased
cell death and suppressed cell proliferation in NCI-H1155 cultures
(Fig. 6B). These data therefore
suggest that the AKT and mTOR pathway can antagonistically regulate
autophagy in certain NE lung tumor cells and that crosstalk between
these pathways may contribute to the regulation of autophagy and
cell survival in the tumors.
Discussion
This study demonstrates that human NE lung tumor
cell lines maintain relatively high LC3 levels and sensitivity to
autophagy inhibition when compared with non-NE lung cancer types,
suggesting that autophagy may have important roles in NE lung
tumors.
The striking correlation between LC3 and NSE levels
in lung tumor cells leads to a question why LC3 levels are high in
NE lung tumors and what advantages it confers to the tumor type. It
was previously proposed that autophagy in lung epithelium indicates
an adaptive response to stress-induced injury, which is caused by
hypoxia, oxidants, inflammation, ischemia-reperfusion, endoplasmic
reticulum stress, pharmaceuticals, or cigarette smoke (30). Particularly, chronic exposure to
cigarette smoke or cigarette smoke extract increased autophagy in
mouse lungs and in pulmonary epithelial cells (12). Further, knockdown of cigarette
smoke-induced autophagy mediators inhibited apoptosis in
vitro, suggesting that autophagy has a role in regulating lung
epithelial cell survival (12).
Since SCLC is mainly caused by tobacco smoke (2), it is conceivable that the relatively
high LC3 levels in SCLC cells may reflect an etiological alteration
attributed to tobacco smoke. Additional explanations are also
available. A recent study suggests that LC3 confers protection
against hypoxia-induced pulmonary hypertension by inhibiting
proliferation of pulmonary artery wall cells (13). A similar mechanism may underlie the
progression of NE lung tumors. For example, LC3 may confer an
advantage for tumor cell survival under a hypoxic condition, which
is often associated with the development of solid tumors.
The relatively high sensitivity of the tested NE
lung tumor cell lines to autophagy inhibition may present a
potential clinical significance. Currently, no effective treatments
are available to cure SCLC or other NE lung tumors. Conventionally,
SCLC is initially treated by combination chemotherapy using
cisplatin or carboplatin plus etoposide with an option to include
radiation therapy, which results in overall high response rates
(60–80%) (4). However, tumors
relapse within months after the initial therapy and topotecan is
the only approved agent for recurrent or progressive SCLC (31,32).
Accordingly, SCLC patients have a very poor survival of <5% at 5
years (33). Atypical carcinoids
and large cell NE carcinomas also pose clinical problems because
the optimal therapy for them is not available (5,6).
Therefore, there is a significant demand for the development of new
therapeutic strategies for SCLC and other NE lung tumors. Autophagy
inhibition may be useful to suppress NE lung tumors because
autophagy inhibition induces programmed cell death in NE lung tumor
cells. Of note, recent studies show that a number of different
chemotherapeutic agents induce autophagic alteration as a mechanism
underlying their therapeutic effects (34). Indeed, chloroquine has been
evaluated in multiple clinical studies of different cancers
(34). Our results suggest a
careful consideration of these therapeutic modalities in NE lung
cancer. In addition, since our study suggests that AKT and mTOR
pathways are among the key signaling pathways that regulate
autophagy in certain NE lung tumors, it may be possible to target
these kinases to disrupt the balance of autophagy in the
tumors.
Intriguingly, it has been suggested that NSE
expression is associated with the degree of tumor malignancy and,
thus, NSE has been proposed as a marker for staging and monitoring
of NE lung tumor (14,35,36).
Therefore, the strong correlation between NSE and LC3 levels may
indicate the possibility that an autophagic alteration underlies NE
lung tumor malignancy and that LC3 is a potential prognostic
biomarker. Distinct alterations in metabolism and signal
transduction might lead to unique biological and clinical features
of lung cancer and identification of these alterations could
contribute to the development of novel therapeutic strategies. Our
study suggests that autophagy may be a unique feature
characterizing NE lung tumors.
Abbreviations:
|
4E-BP
|
4E binding protein 1;
|
|
GAPDH
|
glyceraldehyde 3-phosphate
dehydrogenase;
|
|
LC3
|
microtubule-associated protein-1 light
chain-3;
|
|
mTOR
|
mammalian target of rapamycin;
|
|
mTORC
|
mTOR complex;
|
|
NE
|
neuroendocrine;
|
|
NSE
|
neuron-specific enolase;
|
|
NSCLC
|
non-small cell lung carcinoma;
|
|
S6
|
ribosomal protein S6;
|
|
S6K1
|
S6 kinase 1;
|
|
SCLC
|
small cell lung carcinoma;
|
Acknowledgements
We thank Dr Barry Nelkin at Johns
Hopkins Medical Institute for cell lines and for critical review of
this manuscript. This study was supported by FAMRI Young
Investigator Award (062438), American Cancer Society
(RSGM-10-189-01-TBE) and National Cancer Institute (R01CA138441) to
J.P.
References
|
1.
|
Siegel R, DeSantis C, Virgo K, et al:
Cancer treatment and survivorship statistics, 2012. CA Cancer J
Clin. 62:220–241. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Herbst RS, Heymach JV and Lippman SM: Lung
cancer. N Engl J Med. 359:1367–1380. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
O’Byrne KJ, Gatzemeier U, Bondarenko I, et
al: Molecular biomarkers in non-small-cell lung cancer: a
retrospective analysis of data from the phase 3 FLEX study. Lancet
Oncol. 12:795–805. 2011.PubMed/NCBI
|
|
4.
|
Pietanza MC and Rudin CM: Novel
therapeutic approaches for small cell lung cancer: the future has
arrived. Curr Probl Cancer. 36:156–173. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Travis WD: Advances in neuroendocrine lung
tumors. Ann Oncol. 21(Suppl 7): vii65–vii71. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Swarts DR, Ramaekers FC and Speel EJ:
Molecular and cellular biology of neuroendocrine lung tumors:
evidence for separate biological entities. Biochim Biophys Acta.
1826:255–271. 2012.PubMed/NCBI
|
|
7.
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Mathew R and White E: Autophagy in
tumorigenesis and energy metabolism: friend by day, foe by night.
Curr Opin Genet Dev. 21:113–119. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Mizushima N and Komatsu M: Autophagy:
renovation of cells and tissues. Cell. 147:728–741. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Klionsky DJ, Codogno P, Cuervo AM, et al:
A comprehensive glossary of autophagy-related molecules and
processes. Autophagy. 6:438–448. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Tanida I, Ueno T and Kominami E: LC3 and
autophagy. Methods Mol Biol. 445:77–88. 2008. View Article : Google Scholar
|
|
12.
|
Chen ZH, Kim HP, Sciurba FC, et al: Egr-1
regulates autophagy in cigarette smoke-induced chronic obstructive
pulmonary disease. PLoS One. 3:e33162008. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Lahm T and Petrache I: LC3 as a potential
therapeutic target in hypoxia-induced pulmonary hypertension.
Autophagy. 8:1146–1147. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Righi L, Volante M, Rapa I, Scagliotti GV
and Papotti M: Neuroendocrine tumours of the lung. A review of
relevant pathological and molecular data. Virchows Arch. 451(Suppl
1): S51–S59. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Taneja TK and Sharma S: Markers of small
cell lung cancer. World J Surg Oncol. 2:102004. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Lum JJ, DeBerardinis RJ and Thompson CB:
Autophagy in metazoans: cell survival in the land of plenty. Nat
Rev Mol Cell Biol. 6:439–448. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Maiuri MC, Tasdemir E, Criollo A, et al:
Control of autophagy by oncogenes and tumor suppressor genes. Cell
Death Differ. 16:87–93. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Sarbassov DD, Guertin DA, Ali SM and
Sabatini DM: Phosphorylation and regulation of Akt/PKB by the
rictor-mTOR complex. Science. 307:1098–1101. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Mostoslavsky G, Fabian AJ, Rooney S, Alt
FW and Mulligan RC: Complete correction of murine Artemis
immunodeficiency by lentiviral vector-mediated gene transfer. Proc
Natl Acad Sci USA. 103:16406–16411. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Rubinson DA, Dillon CP, Kwiatkowski AV, et
al: A lentivirus-based system to functionally silence genes in
primary mammalian cells, stem cells and transgenic mice by RNA
interference. Nat Genet. 33:401–406. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Vindelov LL, Christensen IJ and Nissen NI:
A detergent-trypsin method for the preparation of nuclei for flow
cytometric DNA analysis. Cytometry. 3:323–327. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Hong SK, Yoon S, Moelling C, Arthan D and
Park JI: Noncatalytic function of ERK1/2 can promote
Raf/MEK/ERK-mediated growth arrest signaling. J Biol Chem.
284:33006–33018. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Phelps RM, Johnson BE, Ihde DC, et al:
NCI-Navy Medical Oncology Branch cell line data base. J Cell
Biochem. (Suppl 24): 32–91. 1996. View Article : Google Scholar
|
|
24.
|
Hers I, Vincent EE and Tavare JM: Akt
signalling in health and disease. Cell Signal. 23:1515–1527. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Dazert E and Hall MN: mTOR signaling in
disease. Curr Opin Cell Biol. 23:744–755. 2011. View Article : Google Scholar
|
|
26.
|
Thoreen CC, Kang SA, Chang JW, et al: An
ATP-competitive mammalian target of rapamycin inhibitor reveals
rapamycin-resistant functions of mTORC1. J Biol Chem.
284:8023–8032. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Laplante M and Sabatini DM: mTOR signaling
in growth control and disease. Cell. 149:274–293. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Potter CJ, Pedraza LG and Xu T: Akt
regulates growth by directly phosphorylating Tsc2. Nat Cell Biol.
4:658–665. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Vander Haar E, Lee SI, Bandhakavi S,
Griffin TJ and Kim DH: Insulin signalling to mTOR mediated by the
Akt/PKB substrate PRAS40. Nat Cell Biol. 9:316–323. 2007.PubMed/NCBI
|
|
30.
|
Ryter SW and Choi AM: Autophagy in the
lung. Proc Am Thorac Soc. 7:13–21. 2010. View Article : Google Scholar
|
|
31.
|
Eckardt JR, von Pawel J, Pujol JL, et al:
Phase III study of oral compared with intravenous topotecan as
second-line therapy in small-cell lung cancer. J Clin Oncol.
25:2086–2092. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
O’Brien ME, Ciuleanu TE, Tsekov H, et al:
Phase III trial comparing supportive care alone with supportive
care with oral topotecan in patients with relapsed small-cell lung
cancer. J Clin Oncol. 24:5441–5447. 2006.PubMed/NCBI
|
|
33.
|
Merrill RM, Henson DE and Barnes M:
Conditional survival among patients with carcinoma of the lung.
Chest. 116:697–703. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Maes H, Rubio N, Garg AD and Agostinis P:
Autophagy: shaping the tumor microenvironment and therapeutic
response. Trends Mol Med. 19:428–446. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Pujol JL, Quantin X, Jacot W, Boher JM,
Grenier J and Lamy PJ: Neuroendocrine and cytokeratin serum markers
as prognostic determinants of small cell lung cancer. Lung Cancer.
39:131–138. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Giovanella L, Piantanida R, Ceriani L, et
al: Immunoassay of neuron-specific enolase (NSE) and serum
fragments of cytokeratin 19 (CYFRA 21.1) as tumor markers in small
cell lung cancer: clinical evaluation and biological hypothesis.
Int J Biol Markers. 12:22–26. 1997.
|