Introduction
The caspase 8 (CASP8) gene is located on
human chromosome 2q33.1 and plays a vital role in the apoptotic
pathway as an initiator caspase (1). CASP8 is also a crucial factor
involved in the defense system against malignant proliferation and
tumorigenesis (2–5). When CASP8 expression is
disrupted, RIP3-mediated embryonic lethality is observed in 10.5-
to 11.5-day-old embryonic mice, coincident with vascular, cardiac,
and hematopoietic defects (6–9).
RIP3 plays an essential role in the TNF receptor-1 signaling
pathway and can initiate programmed cell necrosis (10). Owing to alternative splicing,
CASP8 produces at least eight different mRNAs (CASP8
a-h) and shows a very complex pattern of isoform expression
(11,12). Different caspase-8 isoforms harbor
distinct functional properties, with some even counteracting the
apoptosis-initiating effects (12–16).
Many studies have explored how CASP8 regulates apoptosis,
but little is known about the transcriptional regulation of
CASP8 and how the widely differing transcripts are
produced.
The first CASP8 promoter was identified in a
neuroblastoma cell line, upstream of exon 1 (17–19).
Based on the complexity of CASP8 transcription and the
experimental conditions of these studies, however, the possible
existence of cryptic or alternate promoters could not be ruled out.
Hence, we still know little about the transcription factors
responsible for regulating this first promoter, and the
transcriptional regulation mechanism of CASP8 remains to be
elucidated.
Owing to the fundamental physiological function of
CASP8 in apoptosis, it is associated with numerous human
diseases, especially cancers (20–24).
A recent meta-analysis of genome-wide association studies for
esophageal squamous cell carcinoma found a susceptibility locus in
2q33.1 encompassing CASP8 and ALS2CR12 (25). Therefore, in the present study we
closely examined the characteristics of this locus. We identified a
second CASP8 promoter located upstream of the caspase-8
isoform G and developed an effective strategy to identify
transcription factors responsible for regulating this newly
identified promoter. A comprehensive understanding of the overall
transcriptional regulation of CASP8 will provide insight
into the mechanisms that contribute to the etiology of cancers and
their responses to treatment.
Materials and methods
Cell culture
The esophageal cancer cell line KYSE510 was grown in
RPMI-1640 medium (Bioroc, China) supplemented with 10% FBS, 100
U/ml penicillin and 100 μg/ml streptomycin in 5%
CO2 at 37°C. The human embryonic kidney cell line HEK293
and the esophageal cancer cell line EC0156 were grown in Minimal
Essential Medium (Bioroc) supplemented with 10% FBS, 100 U/ml
penicillin and 100 μg/ml streptomycin in 5% CO2
at 37°C.
DNA extraction and PCR
Genomic DNA was extracted from KYSE510 cells using
the QIAamp DNA Blood Mini kit (Qiagen, Germany). The promoter
fragment was synthesized with a TaqDNA polymerase mixture (BioTeke,
China). Thermal cycling conditions included activation of the DNA
polymerase at 94°C for 5 min followed by 30 cycles at 94°C for 30
sec, 55–65°C for 30 sec, and 72°C for 30 sec. The specific
oligonucleotide primers used are shown in Table I.
 | Table I.Primers used in PCR and plasmids
construction. |
Table I.
Primers used in PCR and plasmids
construction.
| Name | Sequence
(5′-3′) |
|---|
| Max-forward |
5′-GGGTCTAGGGCTCAGAGCTT-3′ |
| Max-reverse |
5′-CAGTCACCTCTGGAGGCATT-3′ |
| M5N3-forward |
5′-GGGTCTAGGGCTCAGAGCTT-3′ |
| M5N3-reverse |
5′-ACTTGGATCTGCCCTTCTG-3′ |
| N6-forward |
5′-CCTGCAGTTCCTTCTGTGGT-3′ |
| N6-reverse |
5′-ACTTGGATCTGCCCTTCTG-3′ |
| M3N5-forward |
5′-CCTGCAGTTCCTTCTGTGGT-3′ |
| M3N5-reverse |
5′-AATGCCTCCAGAGGTGACTG-3′ |
| M5P1-forward |
5′-GGGTCTAGGGCTCAGAGCTT-3′ |
| M5P1-reverse |
5′-CCCTGTCGGTGGCAAGTAAT-3′ |
| M5P2-forward |
5′-GCCACCGACAGGGGTTATTA-3′ |
| M5P2-reverse |
5′-GCCACCGACAGGGGTTATTA-3′ |
| M5P3-forward |
5′-CAAGCCCTGCTGAATTTGCT-3′ |
| M5P3-reverse |
5′-CAGAAGGGCAGATCCAAGT-3′ |
| C8L-forward |
5′-TCAGGCTTGTCAGGGGGAT-3′ |
| C8L-reverse |
5′-CTGCAGCTACTCCCACCTTC-3′ |
| Isoform
G-forward |
5′-CACAGGTTCTCCTCCTTTTATCTT-3′ |
| Isoform
G-reverse |
5′-TTCAATAACCACCCTGGCTCTTC-3′ |
| GAPDH-forward |
5′-ACAGCAACAGGGTGGTGGAC-3′ |
| GAPDH-reverse |
5′-TTTGAGGGTGCAGCGAACTT-3′ |
|
Bio-Max-forward |
5′-biotin-GGGTCTAGGGCTCAGAGCTT-3′ |
| Bio-N6-forward |
5′-biotin-CCTGCAGTTCCTTCTGTGGT-3′ |
|
Bio-M5P3-forward |
5′-biotin-CAAGCCCTGCTGAATTTGCT-3′ |
|
Bio-M5P2-forward |
5′-biotin-GCCACCGACAGGGGTTATTA-3′ |
|
Bio-C8L-forward |
5′-biotin-TCAGGCTTGTCAGGGGGATA-3′ |
Promoter fragment plasmid
construction
The amplified promoter fragments were cloned into
the pGL3-Basic vector (Promega). The various pGL3-Basic vectors
were then digested with XhoI and HindIII (Takara,
Japan). The promoter fragments were purified using the Wizard SV
Gel and PCR clean up system (Promega), and subsequently ligated
into a promoter less pGL3-Basic luciferase reporter vector. To
ensure the fidelity of the cloned promoter fragments, all final
constructs were sequenced using the vector-specific primers
RVprimer 3: 5′-CTAGCAAAATAGGCTGTCCC-3′ and RVprimer 4:
5′-GACGATAGTCATGCCCCGCG-3′.
Transient transfection and signal
detection
For the dual lucif-erase reporter assay, KYSE510
cells were seeded in a 6-well plate at a density of
2×105 cells per well for at least 20 h prior to
transfection. The constructed plasmids and the Renilla
luciferase internal control plasmid (pRL-TK) were transfected into
the cells using Lipofectamine (Invitrogen). After 24 h, the cells
were treated with the lysis buffer from the Dual-Luciferase
Reporter Assay System (Promega). The signals were measured using an
automatic microplate reader (Synergy H1, BioTek). For knockdown of
the transcriptional activator protein PURα, the cells were
transfected with siRNAs (5′-CCACCUAUCGCAACUCCAUTT-3′ and 5′-AUGGAGU
UGCGAUAGGUGGTT-3′) for 24 h prior to transfection with the
constructed promoter and control plasmids for 24 h. The sequences
negative control siRNAs are 5′-UUCUCCGAA CGUGUCACGUTT-3′ and
5′-ACGUGACACGUUCGGA GAATT-3′.
DNA-protein affinity purification
Nuclear extracts were obtained from KYSE510 cells
using the ProteoExtract Subcellular Proteome Extraction kit
(Calbiochem, EMD Biosciences Inc., Germany). The protein
concentration of the nuclear extract was determined by Bradford
method. The primers used to amplify the promoter and non-promoter
sequence DNA fragments were labeled with biotin at the 5′ terminus.
Streptavidin magnetic beads (Invitrogen) were washed three times
with phosphate-buffered saline (PBS) before use. For affinity
purification, 3 μg of each biotin-labeled DNA fragment was
incubated with 30 μl of the magnetic-bead slurry for 20 min.
Unbound DNA fragments were removed, and 500 μg of nuclear
protein extracts was added to the streptavidin bead-biotin-labeled
DNA fragments and incubated at 4°C overnight. The non-promoter
control DNA fragment was used to decrease the abundance of
non-specific DNA-binding proteins, such as those that bind
histones, in the nuclear extracts. The bead-DNA-protein complex was
washed with TBS (50 mM Tris, 300 mM NaCl) three times, and the
proteins were eluted using 2% SDS. The eluted proteins were
subjected to SDS-PAGE, and visualized by silver staining. Protein
bands were excised and identified by in-gel trypsin digestion with
subsequent analysis by MS (Q Exactive Orbitrap, Thermo Scientific).
Mascot version 2.3.01 (Matrix Science Inc.) was used to analyze the
data and search the databases.
Western blot analysis
The elution products from affinity purification or
cell lysates were denatured in SDS-PAGE sample buffer containing
0.5 M Tris-HCl (pH 6.8), 2% SDS, 10% DTT, 10% glycerol and 0.01%
bromophenol blue, boiled for 5 min, and then analyzed by SDS-PAGE
followed by transfer to a PVDF membrane (Millipore, Germany). Each
membrane was blocked for 1 h in PBS containing 10% nonfat milk.
After blocking, each membrane was incubated overnight with rabbit
polyclonal anti-E2F-1, mouse monoclonal anti-PURα, or rabbit
polyclonal anti-RNA polymerase II (Pol II) (Santa Cruz
Biotechnology Inc.). After washing with TBS containing 20% (w/v)
Tween-20, each membrane was incubated with horseradish
peroxidase-conjugated secondary antibody for 2 h and visualized
with the SuperSignal West Femto Maximum Sensitivity Substrate
(Thermo Fisher Scientific).
Immunoprecipitation assay
KYSE510 cells cultured in 10-cm dishes to 90%
confluency were washed with ice-cold PBS and lysed for 30 min on
ice in lysis buffer containing 1% (w/v) Triton X-100, 0.15 M NaCl,
and 30 mM Tris-HCl (pH 7.5) with protease inhibitors (Roche,
Germany). Lysates were sonicated and centrifuged at 10,000 × g at
4°C for 15 min. The protein concentration was determined by
Bradford assay. For immuno precipitation, 1 mg of the resulting
extract was incubated at 4°C overnight with anti-PURα or anti-E2F-1
and Dynabeads Protein G (Invitrogen). Immunoprecipitates were
washed three times with lysis buffer and the beads were directly
boiled in 1% SDS-PAGE loading buffer.
Immunofluorescence under confocal
microscopy
For immuno fluorescence, KYSE510 cells were fixed in
10% (w/v) paraformaldehyde on poly-L-lysine-coated slides for 30
min at room temperature and washed three times with PBS (pH 7.4).
The cells were blocked with PBS (pH 7.5) supplemented with 1% (w/v)
BSA and 0.1% (w/v) Triton X-100 for 30 min at room temperature.
Washed cells were incubated for 30 min at room temperature with
primary rabbit anti-human E2F-1 and mouse anti-human PURα (Santa
Cruz Biotechnology Inc.). Then the cells were incubated in the dark
for 60 min with Alexa Fluor 488-conjugated goat anti-rabbit and
Alexa Fluor 594-conjugated goat anti-mouse (Life Technologies).
Cells were stained with DAPI and examined using fluorescence
confocal microscopy (Leica Tcs SP2, Germany).
Quantitative RT-PCR
The total mRNA was extracted using TRIzol
(Invitrogen). After the quality control was examined, mRNA was
transformed to cDNA by reverse transcriptase kit (Tiangen). The
quantitative PCR was completed using SYBR Green Master (ROX)
(Roche), and the system included SYBR Green Master (ROX) (2X) 10.0
μl, PCR forward primer (10 μM) 0.6 μl, PCR
reverse primer (10 μM) 0.6 μl, Template cDNA 2.0
μl, ddH2O ≤20.0 μl. The reaction was
conducted under ABI PRISM® 7500. The quality control and
Ct values of the reaction were analyzed using SDS software.
ENCODE database analysis
ENCODE is a DNA elements encyclopaedia in human cell
lines (26). We mainly used the
ChIP-seq, histone methylation and DNA methylation data. The human
cell lines represented in Fig. 1
from ENCODE include GM12878 (lymphoblastoid cells), H1-hESC
(embryonic stem cells), K562 (bone marrow), HeLa S3 (cervix
adenocarcinoma epithelial cells), HEP G2 (hepatocellular carcinoma
cells), HUVEC (umbilical vein endothelial cells), A549 (lung
carcinoma cells), IMR90 (lung fibroblasts), MCF-7 (breast cancer
epithelial cells), and HESC (embryonic stem cells).
Statistical analysis
The data presented are the mean ± standard
deviation. To examine differences between two groups in the
luciferase reporter assay, t-tests were applied using SPSS version
17.0 (IBM software). Mann-Whitney U test was used to compare the
mRNA expression of two groups in the RT-PCR experiments. P-values
<0.05 were considered statistically significant.
Results
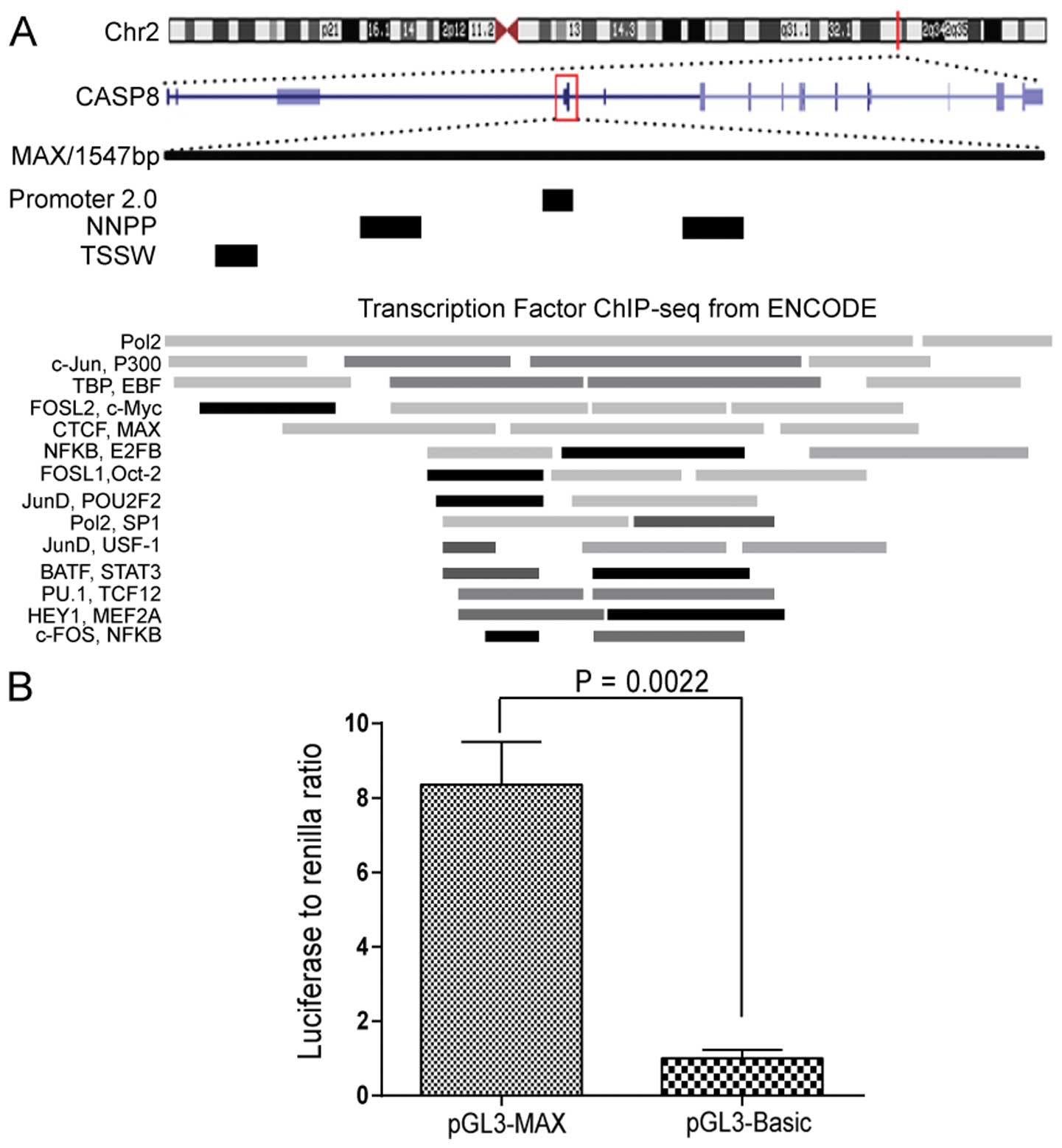
A fragment within CASP8 on chromosome 2
shows transcriptional activity
Analysis of data from ChIP coupled with deep
sequencing (ChIP-seq) in the Human Encyclopedia of DNA elements
(ENCODE) database revealed a region on human chromosome 2q33.1 that
contained binding sites for numerous transcription factors and thus
might be involved in transcriptional regulation of CASP8.
This region is located within chr2: 202,122,236 to 202,123,227 and
25 kb downstream of the previously described CASP8 promoter.
Transcript of caspase-8 isoform G is adjacent to this region. To
examine whether this region has promoter characteristics, we
analyzed this fragment (1547 bp, termed MAX) using three promoter
prediction programs; four potential promoter sequences were
identified (Fig. 1A). To confirm
that the fragment is transcriptionally active, we introduced MAX
into a promoter-deficit luciferase reporter vector (pGL3-Basic).
When transfected into KYSE510 cells, the pGL3-MAX construct
resulted in considerably higher luciferase activity than the
pGL3-Basic vector alone (P=0.0022, Fig. 1B).
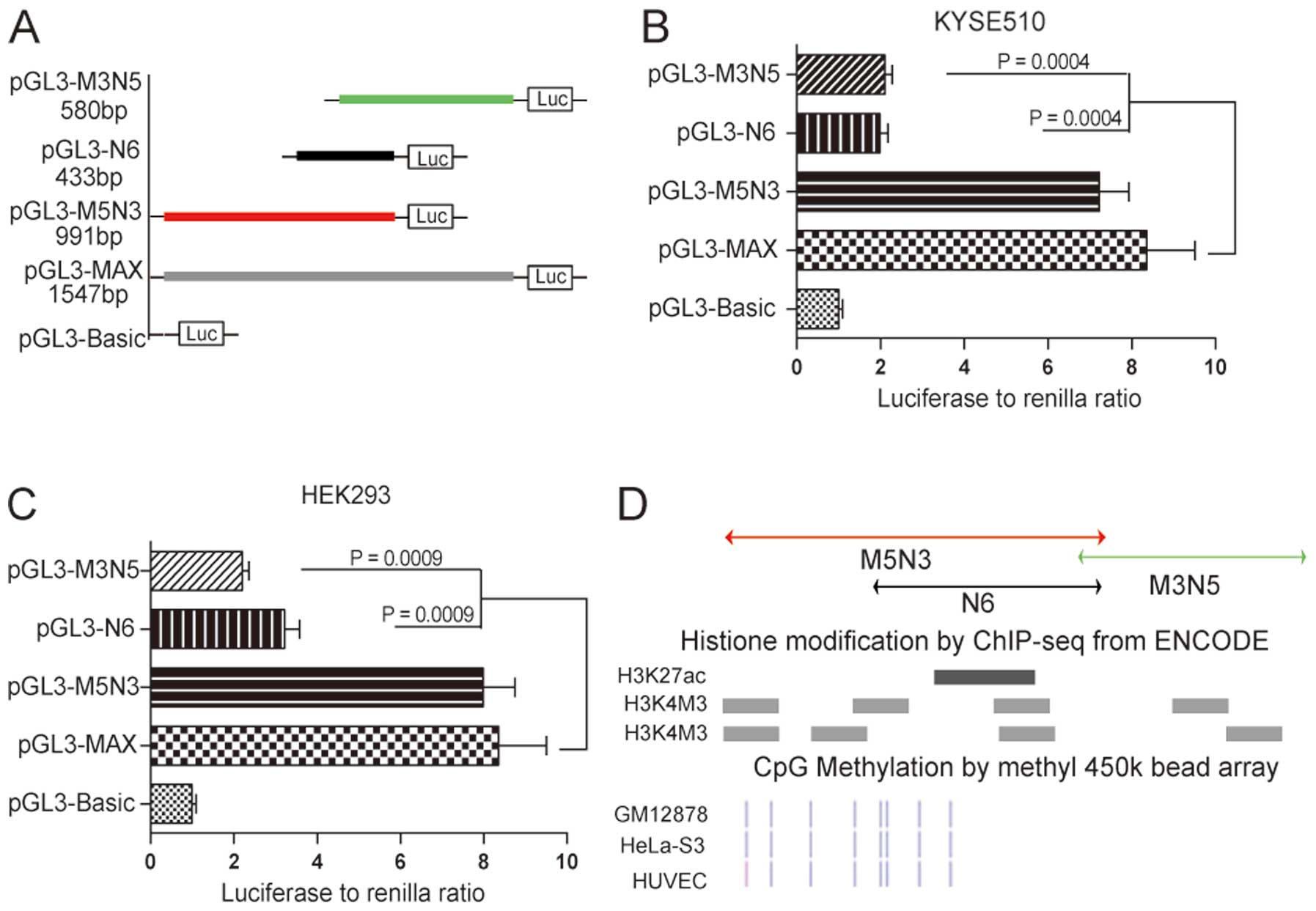
Core promoter of the fragment is
restricted to the region with transcription-related epigenetic
modifications
To identify the core promoter region within the
1547-bp MAX fragment, we first constructed reporter vectors
containing three different imbricating truncations of MAX that were
transfected into KYSE510 cells for the luciferase activity assay
(Fig. 2A). The construct
containing the 5′-terminus of MAX (pGL3-M5N3) retained luciferase
activity comparable to MAX, whereas constructs containing
3′-terminal fragments (pGL3-N6 and pGL3-M3N5) led to statistically
significant decreases in transcriptional activity (76.2%, P=0.0004,
and 74.8%, P=0.0004, respectively; Fig. 2B). These results suggested that the
region contained in pGL3-M5N3 encompassed the core of this novel
CASP8 promoter. Data obtained with KYSE510 cells was
validated in HEK293 cells, in which we observed similar results for
pGL3-M5N3, pGL3-N6 and pGL3-M3N5 (Fig.
2C).
We also examined potential epigenetic modifications
of these three truncated fragments by analyzing these regions in
the ENCODE database. Histone modifications, including acetylation
of lysine 27 in histone H3 and trimethylation of lysine 4 in
histone H3, are indicative of actively transcribed promoters. There
was significant enrichment of these two kinds of histone
modifications within fragment M5N3 compared with fragments M3N5 and
N6 (Fig. 2D). CpG methylation
status is also related to the degree of transcriptional activation
of a DNA region (27). All of the
CpG sites in M3N5 were methylated according to the ENCODE database,
whereas the M5N3 region contained many unmethylated or partially
methylated CpG sites, which also suggested that fragment M5N3 was
more transcriptionally active than M3N5 and N6 (Fig. 2D). We therefore concluded that
fragment M5N3 encompasses the core sequence of this novel
CASP8 promoter.
The pGL3-M5N3 construct (991 bp) only decreased the
luciferase activity in KYSE510 cells by 13.7% relative to the full
MAX fragment (Fig. 2B). We
continued to explore the parts of this fragment that were most
vital for maintaining transcriptional function. Three imbricating
truncations of M5N3 were introduced into pGL3-Basic (pGL3-M5P1, 359
bp; pGL3-M5P2, 404 bp; pGL3-M5P3, 313 bp) and transfected into
KYSE510 cells (Fig. 3A). Comparing
the activities of these three constructs to that of pGL3-M5N3, we
found that pGL3-M5P1 and pGL3-M5P2 decreased the transcriptional
activity significantly (P<0.0001), whereas pGL3-M5P3 retained
comparable activity (Fig. 3B). The
M5P3 sequence (Fig. 3C), which
likely contains the essential core of this newly identified
promoter, has been submitted to GenBank under accession number
KF765385.
DNA-protein affinity purification
combined with LC-MS/MS identifies PURα as a specific transcription
factor for the newly identified promoter
To identify which transcription factors contribute
to the activity of new promoter, we developed a strategy to
separate, enrich and identify the nuclear proteins that
specifically bind to fragment M5N3 (Fig. 4A). The biotin-labeled fragment
(BM5N3) was conjugated to streptavidin-coupled magnetic beads and
then incubated with nuclear proteins extracted from KYSE510 cells.
To eliminate non-specific DNA-binding proteins, such as those
binding histones, we pre-incubated the nuclear proteins with a
non-promoter control fragment from a conserved exon of most
CASP8 transcripts in which ENCODE ChIP-seq data suggested
there are no potential transcription factor-binding sites (BC8L;
Fig. 4B). The bound proteins were
eluted and examined by SDS-PAGE. Silver staining of the gels
revealed several bands that were enriched in the BM5N3 elutions
compared with the BC8L elution. Most notably, when the nuclear
extracts were pre-incubated twice with the control fragment
(BM5N3-2), a greatly enriched band was evident in the region in
which most transcription factors are distributed (35–55 kDa;
Fig. 4B).
To identify the proteins in the specific enriched
bands in the promoter fragment elutions (Fig. 4B), we extracted the bands and
performed LC-MS/MS. Based on the MS/MS spectra of the peptides, we
identified proteins using Mascot software. After excluding proteins
that were also present in the corresponding control bands, we
retained the 12 promoter-specific proteins listed in Table II. The selected proteins were
analyzed in terms of three parameters, peptide score, molecular
mass, and subcellular localization. We excluded four proteins
(KLP6, MUCL1, GNAS and FBN3) based on the fact that their actual
known molecular mass fell outside the 35- to 55-kDa gel region that
we extracted. Two other proteins (PDHA1 and CLEC14A) were excluded
because of their low peptide scores. Of the remaining six proteins,
the transcriptional activator protein PURα showed the appropriate
subcellular localization and known function and thus was chosen as
the candidate transcription factor to be validated.
| Table II.Proteins identified by LC-MS/MS
specific to promoter fragment. |
Table II.
Proteins identified by LC-MS/MS
specific to promoter fragment.
| Uniprot | Score | Mass | Protein | Gene |
|---|
| P25311 | 68 | 34237 |
Zinc-α-2-glycoprotein | AZGP1 |
| Q00577 | 55 | 34889 | Transcriptional
activator protein Pur-α | PURA |
| Q9P0J7 | 36 | 41919 | E3
ubiquitin-protein ligase KCMF1 | KCMF1 |
| P15328 | 35 | 29799 | Folate receptor
α | FOLR1 |
| P04406 | 29 | 36030 |
Glyceraldehyde-3-phosphate
dehydrogenase | GAPDH |
| Q8NBX0 | 27 | 47121 | Saccharopine
dehydrogenase-like oxidoreductase | SCCPDH |
| B7ZC32 | 25 | 108185 | Kinesin-like
protein KLP6 | KLP6 |
| Q96DR8 | 24 | 9034 | Mucin-like protein
1 | MUCL1 |
| Q5JWF2 | 22 | 110956 | Guanine
nucleotide-binding protein G(s) subunit α | GNAS |
| P08559 | 22 | 43268 | Pyruvate
dehydrogenase E1 component subunit α | PDHA1 |
| Q86T13 | 17 | 51603 | C-type lectin
domain family 14 member A | CLEC14A |
| Q75N90 | 15 | 300149 | Fibrillin-3 | FBN3 |
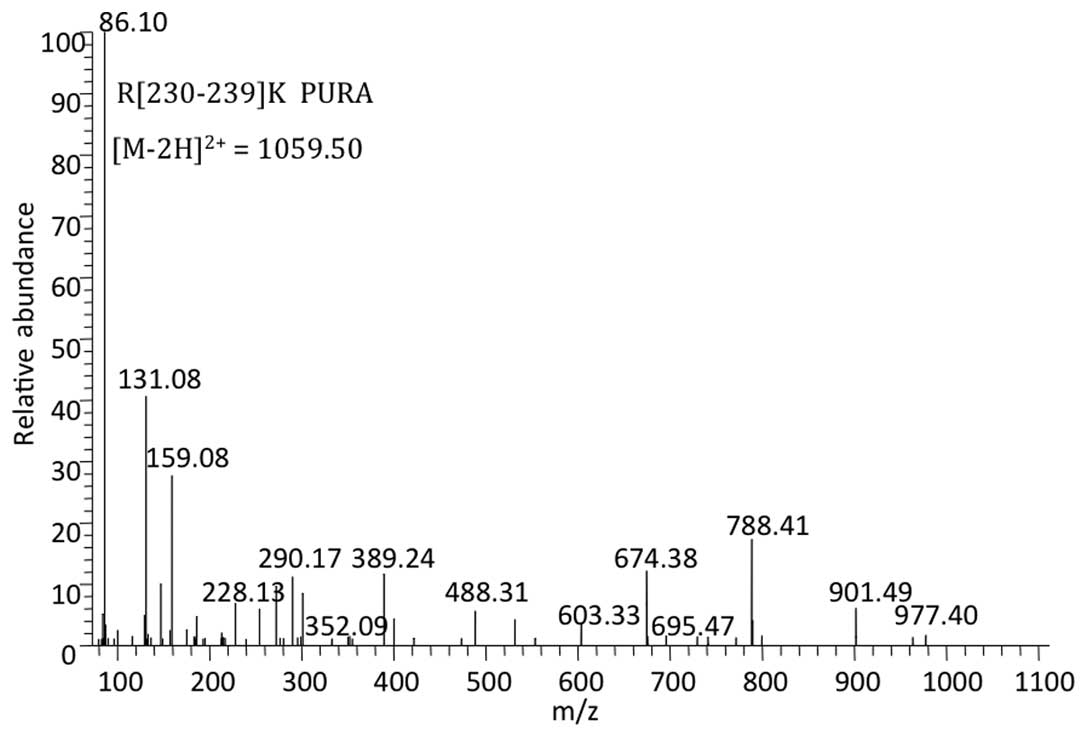
The MS/MS spectrum showed a peptide with mass
1059.50 Da reconstituted from the doubly charged ion of 530.76 m/z.
The sequence obtained from this spectrum revealed that the peptide
originated from arginine 230 to lysine 239 [R(230–239)K] of PURα
[National Center for Biotechnology Information (NCBI) gene
identifier 5813] and was unique to PURα (Fig. 5). Mascot software calculated the
probability (P-value) of the identified peptide being a random
event and transformed the P-value to a peptide score (the peptide
score was 20 when P=0.05). The peptide score of the unique peptide
from PURα was 41 (P=0.00074), indicating the reliability of the
result.
PURα may exert its function by
interacting with E2F-1
To validate the MS/MS results, we first repeated the
DNA-protein affinity purification using just the core promoter
region M5P3. Silver staining of the electrophoresed elution
products showed a significantly enriched band at ∼40 kDa, which was
identical to the band isolated with the longer M5N3 fragment
(Fig. 6A). Western blotting of the
eluted products confirmed the presence of PURα. We also examined
E2F-1 in the western blotting (Fig.
6B) because PURα and E2F family members are known to
inter-regulate (28). To determine
if PURα and E2F-1 physically interact, we performed reciprocal
immunoprecipitation assays. The results showed that PURα and E2F-1
directly or indirectly interacted (Fig. 6C and D). Pol II is responsible for
synthesizing messenger RNA in eukaryotes and is an essential part
of the transcriptional machinery (29,30).
A Pol II signal was also detected in the PURα immunoprecipitates
(Fig. 6D), suggesting that PURα
and E2F-1 are likely components of the transcriptional complex.
Immunohistochemistry with PURα and E2F-1 antibodies and confocal
microscopy confirmed the colocalization of these proteins in the
nucleus of KYSE510 cells (Fig.
6E).
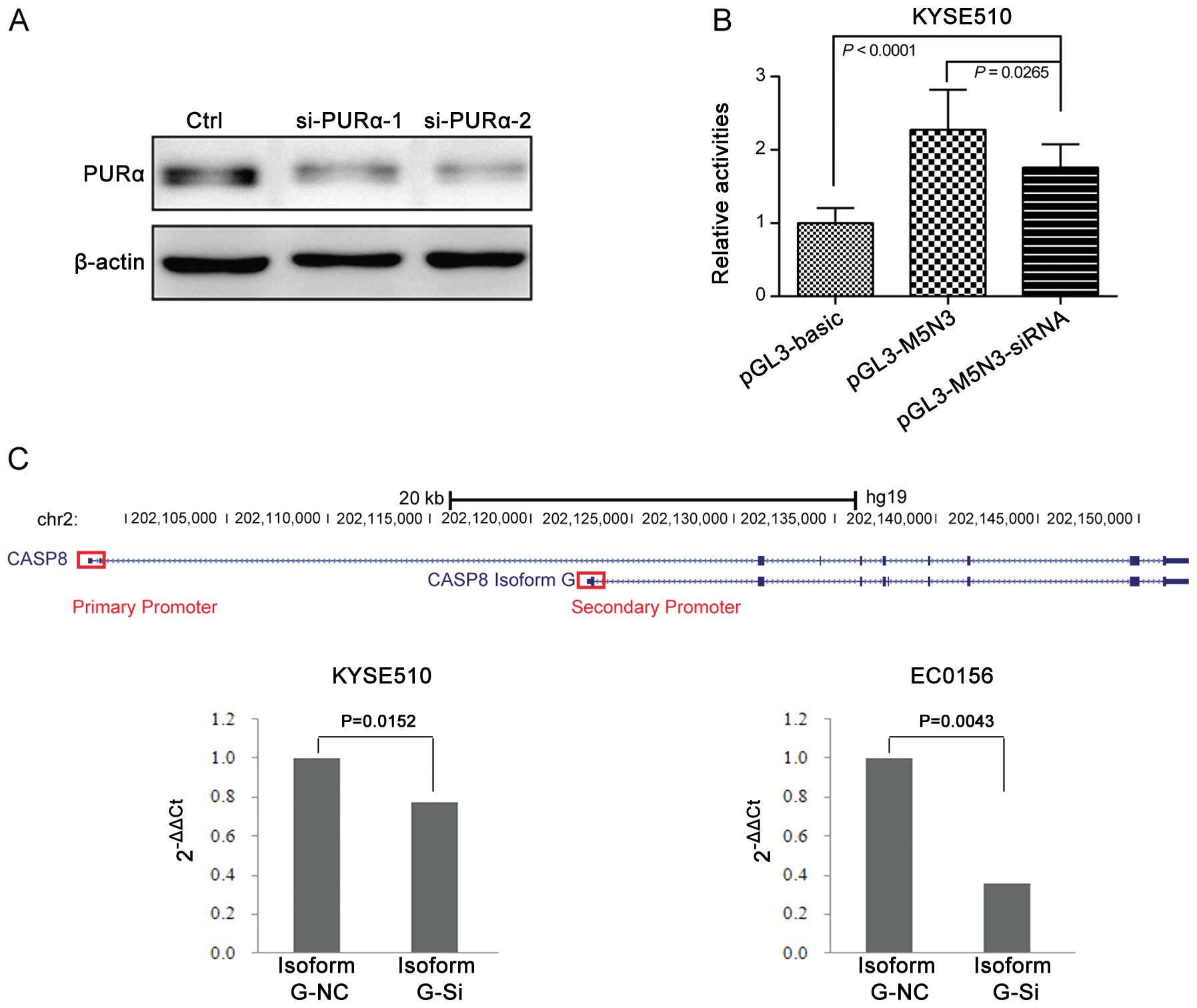
Knockdown of PURα attenuates the
transcriptional activity of the novel promoter and the mRNA
expression of CASP8 isoform G
To evaluate the function of PURα and its
contribution to the transcriptional machinery, we knocked down PURα
in KYSE510 cells using siRNA-mediated RNAi and then transfected the
pGL3-M5N3 construction to these cells and measured promoter
activity. Western blotting confirmed that the siRNA decreased PURα
protein expression (Fig. 7A). The
pGL3-M5N3 promoter activity was still higher than the
promoter-deficient pGL3-Basic plasmid in the PURα knockdown strain
(P<0.0001) but was significantly lower than for pGL3-M5N3
without PURα knockdown (P=0.0265; Fig.
7B). Based on the results of transcriptional activity, we
continued to test whether PURα could influence the mRNA expression
of gene CASP8. The secondary promoter locates surround the
5′-UTR of isoform G, so we designed primers specific to isoform G.
We observed significant down regulation of mRNA expression of
isoform G in KYSE510 cells after knowing down PURα (P=0.0152;
Fig. 7C). We validated this result
in another esophageal cell line (EC0156; P=0.0043; Fig. 7C). These results point to PURα as
responsible for the transcriptional activity of the secondary
promoter and it is able to promote the mRNA expression of isoform
G.
Discussion
Transcriptional regulation is a complex and dynamic
process involving concerted modulation of transcription initiation,
alternative splicing and post-transcriptional modifications
(31–33). Complex transcriptional units can
produce multiple mature mRNAs by a variety of mechanisms, including
alternative splicing or use of alternative promoters or alternative
start sites around a single promoter (34,35).
Mammalian promoters have been identified at various unexpected
positions in the genome, such as in intergenic regions far from
known genes, in the 3′-UTRs of known protein-coding genes, in
coding exons, and in introns (36–38).
Many human genes have secondary or alternative promoters (39–43).
At least eight different CASP8 transcripts
have been identified. This variety of transcripts can be explained,
in part, by transcription from the known promoter of CASP8
(17–19). Of note, a six-nucleotide
insertion-deletion polymorphismin this promoter may be associated
with susceptibility to multiple cancers and can influence
expression of certain CASP8 isoforms (44). However, results are contradictory
about the association of this polymorphism with disease (45–54).
We believe that this contradiction stems, in part, from the
existence of the second promoter that we identified for
CASP8, which suggests more complex transcriptional
regulation than previously thought. The ENCODE project aims to
identify all functional elements in the human genome sequence
(26,55). Based on this database, some new
features and mechanisms of transcriptional systems have been
characterized at the overall genome level (56,57).
To further uncover the regulatory mechanisms underlying individual
genes, we suggest analysis of the ENCODE data for specific region,
as was done in this study. The ENCODE database provides an
excellent platform to identify potential unknown DNA elements, like
secondary promoters.
Identification of transcription factors associated
with a specific gene has often relied on analysis of binding sites
for known candidate factors and on ChIP-seq techniques, neither of
which can identify novel factors (58). In this study, we developed a
strategy that incorporated MS to identify proteins that
specifically bind a selected DNA fragment. This strategy could
easily be applied to the identification of binding proteins for
many other DNA elements and thus could greatly expand the depth of
research on transcriptional regulation.
PURα is a member of the PUR family of proteins, it
can bind to both DNA (either single- or double-stranded) and RNA,
and functions in the initiation of DNA replication, regulation of
transcription and mRNA translation (59). PURα is associated with many types
of neoplasias and brain development (60). As a single-stranded nucleic
acid-binding protein, PURα has DNA helix-destabilizing activity,
which is consistent with the requirement for duplex DNA unwinding
during initiation of transcription and replication (61). The fact that PURα knockdown
decreased the activity of the secondary promoter fragment suggests
that this protein is directly involved in CASP8
transcription. The RT-PCR results further confirmed that PURα was
responsible for the mRNA expression of CASP8, especially
certain transcripts such as isoform G. Isoform G is the longest
isoform of caspase-8 (538 amino acids), also known as
procaspase-8L. Apart from the well known apoptosis role, caspase-8
also has some nonapoptotic functions such as regulation of
proliferation and differentiation of B cells and NK cells (62,63),
and these functions might be exerted by a certain isoform. PURα
interacts with many transcription factors, including E2F-1
(28). We found E2F-1 at the
second CASP8 promoter and confirmed the physical interaction
between PURα and E2F-1 and also revealed a relationship between
PURα and Pol II.
In summary, we identified a secondary promoter of
CASP8 in the 5′-UTR and exon 1 of isoform G. Through
affinity purification combined with MS, we identified PURα as a
promoter-specific transcription factors that appears to function
together with E2F-1. The presence of this functional secondary
promoter in CASP8 suggests a complex pattern of gene
regulation and may also explain some of the contradictory results
obtained in previous studies of CASP8. Further research on
the complicated regulation mechanism of CASP8 will provide a
greater understanding of programmed cell death.
Acknowledgements
This study was supported by grants of
the High-tech R&D Program (2012AA020206, 2012AA02A503 and
2013AA041201), State Key Projects for Basic Research (2011CB910703)
and NSFC (81372591, 81372385, 81321091) of China. We thank Dr
Yutaka Shimada at Hyogo College of Medicine for providing the KYSE
510 cells. The authors greatly appreciate Nan Zhao, Kun Jia, Bo
Han, Lanping Zhou and Fang Liu for their technical support, and
thank Dr Yulin Sun for helpful discussions and comments.
References
|
1.
|
Shi Y: Mechanisms of caspase activation
and inhibition during apoptosis. Mol Cell. 9:459–470. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Raff M: Cell suicide for beginners.
Nature. 396:119–122. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Thompson CB: Apoptosis in the pathogenesis
and treatment of disease. Science. 267:1456–1462. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Hengartner MO: The biochemistry of
apoptosis. Nature. 407:770–776. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Li H, Zhu H, Xu CJ and Yuan J: Cleavage of
BID by caspase 8 mediates the mitochondrial damage in the Fas
pathway of apoptosis. Cell. 94:491–501. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Varfolomeev EE, Schuchmann M, Luria V, et
al: Targeted disruption of the mouse Caspase 8 gene ablates cell
death induction by the TNF receptors, Fas/Apo1, and DR3 and is
lethal prenatally. Immunity. 9:267–276. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Kang TB, Ben-Moshe T, Varfolomeev EE, et
al: Caspase-8 serves both apoptotic and nonapoptotic roles. J
Immunol. 173:2976–2984. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Kang TB, Oh GS, Scandella E, et al:
Mutation of a self-processing site in caspase-8 compromises its
apoptotic but not its nonapoptotic functions in bacterial
artificial chromosometransgenic mice. J Immunol. 181:2522–2532.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Sakamaki K, Inoue T, Asano M, et al: Ex
vivo whole-embryo culture of caspase-8-deficient embryos normalize
their aberrant phenotypes in the developing neural tube and heart.
Cell Death Differ. 9:1196–1206. 2002. View Article : Google Scholar
|
|
10.
|
Galluzzi L, Kepp O and Kroemer G: RIP
kinases initiate programmed necrosis. J Mol Cell Biol. 1:8–10.
2009. View Article : Google Scholar
|
|
11.
|
Boldin MP, Goncharov TM, Goltsev YV and
Wallach D: Involvement of MACH, a novel MORT1/FADD-interacting
protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell.
85:803–815. 1996. View Article : Google Scholar
|
|
12.
|
Scaffidi C, Medema JP, Krammer PH and
Peter ME: FLICE is predominantly expressed as two functionally
active isoforms, caspase-8/a and caspase-8/b. J Biol Chem.
272:26953–26958. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Horiuchi T, Himeji D, Tsukamoto H,
Harashima S, Hashimura C and Hayashi K: Dominant expression of a
novel splice variant of caspase-8 in human peripheral blood
lymphocytes. Biochem Biophys Res Commun. 272:877–881. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Himeji D, Horiuchi T, Tsukamoto H, Hayashi
K, Watanabe T and Harada M: Characterization of caspase-8L: a novel
isoform of caspase-8 that behaves as an inhibitor of the caspase
cascade. Blood. 99:4070–4078. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Miller MA, Karacay B, Zhu X, O’Dorisio MS
and Sandler AD: Caspase 8L, a novel inhibitory isoform of caspase
8, is associated with undifferentiated neuroblastoma. Apoptosis.
11:15–24. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Mohr A, Zwacka RM, Jarmy G, et al:
Caspase-8L expression protects CD34+hematopoietic
progenitor cells and leukemic cells from CD95-mediated apoptosis.
Oncogene. 24:2421–2429. 2005.PubMed/NCBI
|
|
17.
|
Casciano I, De Ambrosis A, Croce M, et al:
Expression of the caspase-8 gene in neuroblastoma cells is
regulated through an essential interferon-sensitive response
element (ISRE). Cell Death Differ. 11:131–134. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Casciano I, Banelli B, Croce M, et al:
Caspase-8 gene expression in neuroblastoma. Ann NY Acad Sci.
1028:157–167. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Teitz T, Wei T, Valentine MB, et al:
Caspase 8 is deleted or silenced preferentially in childhood
neuroblastomas with amplification of MYCN. Nat Med. 6:529–535.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Kim HS, Lee JW, Soung YH, et al:
Inactivating mutations of caspase-8 gene in colorectal carcinomas.
Gastroenterology. 125:708–715. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Mandruzzato S, Brasseur F, Andry G, Boon T
and van der Bruggen P: A CASP-8 mutation recognized by cytolytic T
lymphocytes on a human head and neck carcinoma. J Exp Med.
186:785–793. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Soung YH, Lee JW, Kim SY, et al: CASPASE-8
gene is inactivated by somatic mutations in gastric carcinomas.
Cancer Res. 65:815–821. 2005.PubMed/NCBI
|
|
23.
|
Takita J, Yang HW, Chen YY, et al: Allelic
imbalance on chromosome 2q and alterations of the caspase 8 gene in
neuroblastoma. Oncogene. 20:4424–4432. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Hopkins-Donaldson S, Ziegler A, Kurtz S,
et al: Silencing of death receptor and caspase-8 expression in
small cell lung carcinoma cell lines and tumors by DNA methylation.
Cell Death Differ. 10:356–364. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Abnet CC, Wang Z, Song X, et al: Genotypic
variants at 2q33 and risk of esophageal squamous cell carcinoma in
China: a meta-analysis of genome-wide association studies. Hum Mol
Genet. 21:2132–2141. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Maher B: ENCODE: The human encyclopaedia.
Nature. 489:46–48. 2012. View Article : Google Scholar
|
|
27.
|
Vaillant I and Paszkowski J: Role of
histone and DNA methylation in gene regulation. Curr Opin Plant
Biol. 10:528–533. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Darbinian N, White MK, Gallia GL, Amini S,
Rappaport J and Khalili K: Interaction between the pura and E2F-1
transcription factors. Anticancer Res. 24:2585–2594.
2004.PubMed/NCBI
|
|
29.
|
Kadonaga JT: Regulation of RNA polymerase
II transcription by sequence-specific DNA binding factors. Cell.
116:247–257. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Oei SL, Griesenbeck J, Schweiger M and
Ziegler M: Regulation of RNA polymerase II-dependent transcription
by poly(ADP-ribosyl)ation of transcription factors. J Biol Chem.
273:31644–31647. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Coulon A, Chow CC, Singer RH and Larson
DR: Eukaryotic transcriptional dynamics: from single molecules to
cell populations. Nat Rev Genet. 14:572–584. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Lenhard B, Sandelin A and Carninci P:
Metazoan promoters: emerging characteristics and insights into
transcriptional regulation. Nat Rev Genet. 13:233–245.
2012.PubMed/NCBI
|
|
33.
|
Kornblihtt AR, Schor IE, Allo M, Dujardin
G, Petrillo E and Munoz MJ: Alternative splicing: a pivotal step
between eukaryotic transcription and translation. Nat Rev Mol Cell
Biol. 14:153–165. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Ayoubi TA and Van De Ven WJ: Regulation of
gene expression by alternative promoters. FASEB J. 10:453–460.
1996.PubMed/NCBI
|
|
35.
|
Landry JR, Mager DL and Wilhelm BT:
Complex controls: the role of alternative promoters in mammalian
genomes. Trends Genet. 19:640–648. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Carninci P, Kasukawa T, Katayama S, et al:
The transcriptional landscape of the mammalian genome. Science.
309:1559–1563. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Katayama S, Tomaru Y, Kasukawa T, et al:
Antisense transcription in the mammalian transcriptome. Science.
309:1564–1566. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Carninci P, Sandelin A, Lenhard B, et al:
Genome-wide analysis of mammalian promoter architecture and
evolution. Nat Genet. 38:626–635. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Yoo EJ, Cooke NE and Liebhaber SA:
Identification of a secondary promoter within the human B cell
receptor component gene hCD79b. J Biol Chem. 288:18353–18365. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Laszkiewicz A, Cebrat M, Miazek A and
Kisielow P: Complexity of transcriptional regulation within the Rag
locus: identification of a second Nwc promoter region within the
Rag2 intron. Immunogenetics. 63:183–187. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Pinte S, Guerardel C, Deltour-Balerdi S,
Godwin AK and Leprince D: Identification of a second G-C-rich
promoter conserved in the human, murine and rat tumor suppressor
genes HIC1. Oncogene. 23:4023–4031. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Rini D and Calabi F: Identification and
comparative analysis of a second runx3 promoter. Gene. 273:13–22.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Burton EA, Tinsley JM, Holzfeind PJ,
Rodrigues NR and Davies KE: A second promoter provides an
alternative target for therapeutic up-regulation of utrophin in
Duchenne muscular dystrophy. Proc Natl Acad Sci USA.
96:14025–14030. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Sun T, Gao Y, Tan W, et al: A
six-nucleotide insertion-deletion polymorphism in the CASP8
promoter is associated with susceptibility to multiple cancers. Nat
Genet. 39:605–613. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Haiman CA, Garcia RR, Kolonel LN,
Henderson BE, Wu AH and Le Marchand L: A promoter polymorphism in
the CASP8 gene is not associated with cancer risk. Nat Genet.
40:259–260. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Frank B, Rigas SH, Bermejo JL, et al: The
CASP8 -652 6N del promoter polymorphism and breast cancer risk: a
multicenter study. Breast Cancer Res Treat. 111:139–144. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Cybulski C, Wokolorczyk D, Gliniewicz B,
et al: A six-nucleotide deletion in the CASP8 promoter is not
associated with a susceptibility to breast and prostate cancers in
the Polish population. Breast Cancer Res Treat. 112:367–368. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Pittman AM, Broderick P, Sullivan K, et
al: CASP8 variants D302H and -652 6N ins/del do not influence the
risk of colorectal cancer in the United Kingdom population. Br J
Cancer. 98:1434–1436. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Xiao MS, Chang L, Li WL, et al: Genetic
polymorphisms of the CASP8 gene promoter may not be associated with
colorectal cancer in Han Chinese from Southwest China. PLoS One.
8:e675772013. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
de Martino M, Haitel A, Schatzl G,
Klingler HC and Klatte T: The CASP8 -652 6N insertion/deletion
promoter polymorphism is associated with renal cell carcinoma risk
and metastasis. J Urol. 190:717–722. 2013.PubMed/NCBI
|
|
51.
|
Malik MA, Zargar SA and Mittal B: A
six-nucleotide deletion polymorphism in the casp8 promoter is
associated with reduced risk of esophageal and gastric cancers in
Kashmir valley. Indian J Hum Genet. 17:152–156. 2011. View Article : Google Scholar
|
|
52.
|
Li C, Lu J, Liu Z, et al: The
six-nucleotide deletion/insertion variant in the CASP8 promoter
region is inversely associated with risk of squamous cell carcinoma
of the head and neck. Cancer Prev Res. 3:246–253. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53.
|
Wang M and Zhang Z, Tian Y, Shao J and
Zhang Z: A six-nucleotide insertion-deletion polymorphism in the
CASP8 promoter associated with risk and progression of bladder
cancer. Clin Cancer Res. 15:2567–2572. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
54.
|
Ni C, Ye Y, Wang M, et al: A
six-nucleotide insertion-deletion polymorphism in the CASP8
promoter is associated with risk of coal workers’ pneumoconiosis. J
Toxicol Environ Health A. 72:712–716. 2009.PubMed/NCBI
|
|
55.
|
Qu H and Fang X: A brief review on the
Human Encyclopedia of DNA Elements (ENCODE) project. Genomics
Proteomics Bioinformatics. 11:135–141. 2013. View Article : Google Scholar
|
|
56.
|
Gerstein MB, Kundaje A, Hariharan M, et
al: Architecture of the human regulatory network derived from
ENCODE data. Nature. 489:91–100. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57.
|
Bajic VB, Brent MR, Brown RH, et al:
Performance assessment of promoter predictions on ENCODE regions in
the EGASP experiment. Genome Biol. 7(Suppl 1): S31–13. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
58.
|
Bardet AF, Steinmann J, Bafna S, Knoblich
JA, Zeitlinger J and Stark A: Identification of transcription
factor binding sites from ChIP-seq data at high resolution.
Bioinformatics. 29:2705–2713. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
59.
|
Johnson EM: The Pur protein family: clues
to function from recent studies on cancer and AIDS. Anticancer Res.
23:2093–2100. 2003.PubMed/NCBI
|
|
60.
|
Johnson EM, Daniel DC and Gordon J: The
pur protein family: genetic and structural features in development
and disease. J Cell Physiol. 228:930–937. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
61.
|
Darbinian N, Gallia GL and Khalili K:
Helix-destabilizing properties of the human single-stranded DNA-
and RNA-binding protein Puralpha. J Cell Biochem. 80:589–595. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
62.
|
Algeciras-Schimnich A, Barnhart BC and
Peter ME: Apoptosis-independent functions of killer caspases. Curr
Opin Cell Biol. 14:721–726. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
63.
|
Launay S, Hermine O, Fontenay M, Kroemer
G, Solary E and Garrido C: Vital functions for lethal caspases.
Oncogene. 24:5137–5148. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
64.
|
Knudsen S: Promoter2.0: for the
recognition of PolII promoter sequences. Bioinformatics.
15:356–361. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
65.
|
Reese MG: Application of a time-delay
neural network to promoter annotation in the Drosophila
melanogaster genome. Comput Chem. 26:51–56. 2001. View Article : Google Scholar
|
|
66.
|
Solovyev V and Salamov A: The Gene-Finder
computer tools for analysis of human and model organisms genome
sequences. Proc Int Conf Intell Syst Mol Biol. 5:294–302.
1997.PubMed/NCBI
|