1. Introduction
Prostate cancer is the most common non-cutaneous
malignancy in males causing one in three tumor-associated deaths in
Germany and the second most common cause of cancer-related death in
the United States (1) (Krebs in
Deutschland; www.krebsdaten.de). Androgen deprivation
therapy is the cornerstone of the urooncological management of
advanced hormone-sensitive prostate cancer (2). However, patients inevitably enter a
castration resistant stage in which tumor progression occurs
despite androgen deprivation. These metastatic castration resistant
prostate cancer (mCRPC) patients have, until recently, received
chemotherapy (docetaxel, mitoxantrone) as first line therapy
(3,4). However, the introduction of novel and
highly potent anti-androgens such as abiraterone or enzalutamide
has led to a shift in the clinical practice and these agents are
now also being used in chemo-naive mCRPC patients (5,6).
Nevertheless, treatment of mCRPC remains a major challenge due to
the heterogeneity of the disease and the inevitable development of
therapy resistance. It is therefore of importance to better
understand the biology of prostate cancer progression in order to
develop improved therapeutic strategies.
Recent advances in comprehensive genomic profiling
have identified four major signaling nodes that are most frequently
altered in prostate cancer: i) the androgen receptor (AR) signaling
pathway; ii) the PI3K pathway; iii) the Ras/Raf/MEK/ERK pathway;
and iv) the retinoblastoma protein (pRB) signaling pathway
(7). These pathways were altered
at high frequencies in both primary tumors and metastatic samples
in an exemplary study (7). The AR
was identified as the most commonly mutated gene in prostate cancer
with 56–100% of tumors examined containing mutated AR. Loss of
PTEN, a negative regulator of the PI3K pathway, is a hallmark of
prostate cancer and occurs in 42–100% of the tumors analyzed. The
Ras/Raf/MEK/ERK pathway was altered in 43–90% of tumors and the pRB
signaling pathway was altered in 34–74% of prostate cancers
(7).
Importantly, extensive crosstalk and redundancy
exists between these signaling pathways leading to the hypothesis
that therapy resistance may readily develop when only one of the
four pathways is targeted (7–9).
This suggests that developing either novel therapeutic agents, or
therapeutic regimes, that target a broader spectrum of pathway
components may improve the clinical benefit of systemic therapy
(Table I). In this review, we
highlight the most commonly altered signaling pathways in advanced
prostate cancer which need to be taken into account for the
development of such new rational therapeutic strategies (Fig. 1).
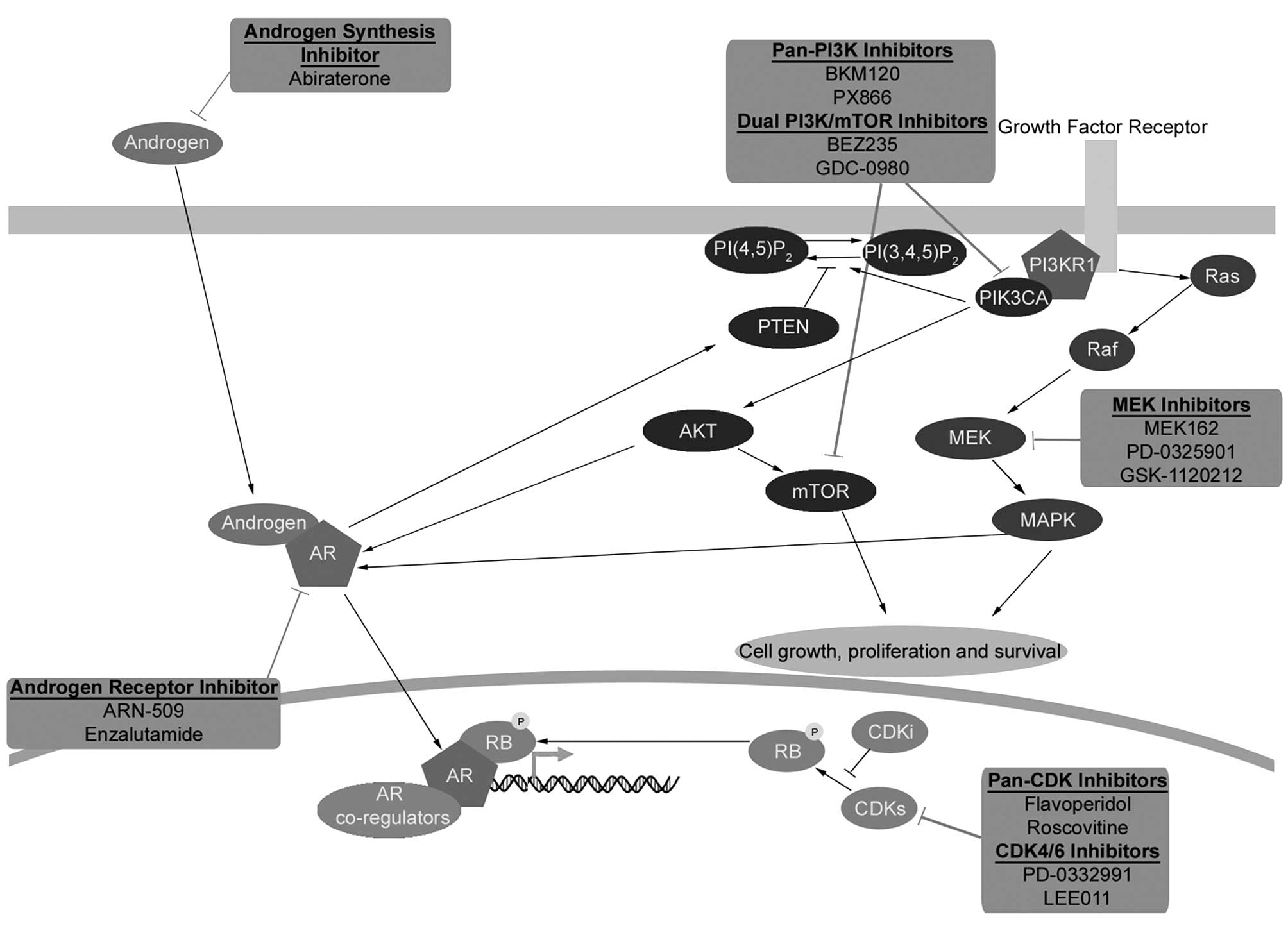 | Figure 1AR, PI3K, Ras/Raf/MEK/ERK and pRB
pathway crosstalk and pharmacological inhibition. Recent advances
in genomic profiling have identified four major signaling pathways
that are most frequently altered in prostate cancer: i) the
androgen receptor (AR) signaling pathway; ii) the PI3K pathway;
iii) the Ras/Raf/MEK/ERK pathway; and iv) the retinoblastoma (pRB)
signaling pathway. Extensive crosstalk and redundancy exists
between these signaling pathways suggesting that devising a
therapeutic regime that targets more than one of these pathways may
provide a clinical advantage. Androgen signaling is mediated
through AR, a ligand-activated transcription factor that is the
main therapeutic target of first-line therapies for advanced
prostate cancer. The inactive form of the AR resides within the
cytoplasm, binding of androgens to the receptor leads to its
nuclear translocation and transcription of androgen-dependent
genes. Inhibition of the AR receptor can lead to activation of
other signaling pathways such as the PI3K pathway through pathway
crosstalk. The PI3K pathway is the second most frequently
deregulated signaling pathway in prostate cancer behind only the AR
signaling pathway. The PI3K pathway is normally activated following
growth factor stimulation of receptor tyrosine kinases ultimately
resulting in the activation of AKT, the central effector of many
downstream signaling pathways regulating protein synthesis, cell
cycle, cell death, cell growth and cell survival. AKT activation
can be reversed by PTEN, which negatively regulates PI3K pathway
activation. Another important target of the PI3K signaling pathway
is the serine/threonine kinase mTOR (mammalian target of
rapamycin). The Ras/Raf/MEK/ERK signaling pathway regulates
fundamental cellular processes, including proliferation,
differentiation and cell survival and is frequently activated in
several cancer types, including prostate cancer. The PI3K and
Ras/Raf/MEK/ERK pathways have been shown to interact extensively
and frequent co-activation of these two pathways has been observed
in prostate cancer. Ras/Raf/MEK/ERK signaling is stimulated by
growth factor receptor activation of the small GTPase Ras, which in
turn activates the protein kinase Raf leading to the activation of
the dual specificity MAPK kinase (MEK1/MEK2), and finally
phosphorylation of extracellular signal-regulated kinases
(ERK1/ERK2). Ras/Raf/MEK/ERK pathway can sustain the transcription
of androgen responsive genes, and therefore prostate cancer cell
proliferation, even when the AR is inhibited. Retinoblastoma (RB1)
is a tumor suppressor gene with somatic alterations in multiple
cancers whose gene product (pRB) inhibits cell cycle progression.
pRB activity is predominately controlled by cell cycle regulated
cyclin-dependent kinase (CDK) activity. When CDK activity is
inhibited, pRB remains in a hypophosphorylated and active state
repressing E2F-regulated gene transcription and inhibiting cell
cycle progression. Inactivation of pRB prevents cells from
restraining cellular proliferation and leads to the aberrant
expression of E2F-responsive genes. Dysregulation of E2F binding
motif-containing genes is a hallmark of CRPC. Pharmacological
inhibitors of the different signaling pathways are shown. |
 | Table IInhibitors of the AR, PI3K,
Ras/Raf/MEK/ERK and RB pathway. |
Table I
Inhibitors of the AR, PI3K,
Ras/Raf/MEK/ERK and RB pathway.
| Agent | Pathway
inhibition |
|---|
| Abiraterone | AR |
| Enzalutamide | AR |
| ARN 509 | AR |
| CFG920 | AR |
| Ortenerol
(TAK700) | AR |
| BKM120 | PI3K |
| PX866 | PI3K |
| BEZ235 | PI3K/mTOR |
| GDC-0980 | PI3K/mTOR |
| MK2206 | AKT |
| GDC-0068 | AKT |
| Everolimus | TORC1 |
| Temsirolimus | TORC1 |
| PD98059 | MEK |
| U0126 | MEK |
| PD184352
(CI-1040) | MEK |
| PD0325901 | MEK |
| Selumetinib | MEK |
| RDEA119 | MEK |
| SL327 | MEK |
| Pimasertib
(AS-703026) | MEK |
| BIX 02188 | MEK |
| BIX 02189 | MEK |
| AZD8330 | MEK |
| TAK-733 | MEK |
| Trametinib
(GSK1120212) | MEK |
| AZD 5438 | CDKs |
| SCH 727965 | CDKs |
| Seliciclib | CDKs |
| Flavopiridol | CDKs |
| PD0332991 | CDK4/6 |
| LEE011 | CDK4/6 |
2. Androgen receptor signaling
Androgen signaling is mediated through the androgen
receptor (AR), a ligand-activated transcription factor that is the
main therapeutic target of first-line therapies for advanced
prostate cancer. The inactive form of the AR resides within the
cytoplasm. Binding of androgens to the receptor leads to receptor
activation and translocation into the nucleus, where the
transcription of androgen-dependent genes is induced (10). Androgen-deprivation therapy (or
chemical castration) is an effective first-line therapy for
metastatic prostate cancer, but despite good initial responses,
castrate-resistant prostate cancer commonly develops leading to
tumors that are insensitive to androgen ablation (11).
Castration resistance can develop through several
mechanisms, most commonly by gene amplification, activating
mutations of the AR, or expression of AR splice variants which
promotes maintenance of AR activity and signaling capabilities at
castrate hormone levels (7,12–15).
Interestingly, AR gene amplification has been detected in 28% of
CRPCs after androgen ablation, but has not been observed in tumor
samples without prior exposure to androgen deprivation (12). Receptor hypersensitivity to
androgens can also occur in CRPC through increased receptor
stability, enhanced nuclear localization and overexpression of
nuclear co-activators (16,17).
Furthermore, AR point mutations have been demonstrated to confer AR
promiscuity leading to an activation by non-androgen ligands such
as progesterone and estradiol (18,19).
These observations suggest a selective pressure to maintain
AR-mediated signaling in CRPC despite androgen deprivation
conditions.
Based on this fact, CRPC remains to a certain extent
sensitive to second-generation AR antagonists and hormone synthesis
blockers, which includes drugs that target AR signaling at the
androgen production and conversion level and at the receptor level
such as abiraterone or enzalutamid (20). However, it has been shown that
inhibition of the AR receptor can lead to activation of other
signaling pathways via crosstalk, such as the PI3K pathway. For
example, AR blockade can lead to a reduced expression of the
AR-responsive immunophilin FBPK5, a chaperone for the AKT
phosphatase PHLPP, and ultimately an increased level of
phosphorylated AKT, a downstream target of PI3K (11). Further, non-ligand mediated
activation of AR signaling has been shown to be induced through
crosstalk with other oncogenic pathways such as the
mitogen-activated protein kinase (MAPK) pathway (21,22).
3. Pharmacological inhibition of androgen
receptor signaling action
Abiraterone
Chemical castration leads to decreased production of
testicular androgens but adrenal glands and even prostate cancer
tissue can continue to produce androgens, which contributes to
continued prostate cancer cell growth despite castrate level of
androgens. Synthesized in the early 1990s, and approved for use in
CRPC patients in 2011, abiraterone treatment promotes a global
blockade of androgen production through irreversible inhibition of
17 α-hydroxylase/C17,20 lyase (CYP17), a key enzyme involved in
androgen synthesis. CYP17 is expressed in testicular, adrenal, and
prostatic tumor tissues and inhibition of CYP17 results in a
profound decrease of circulating androgens. Currently, abiraterone
is used in mCRPC patients both prior to and post chemotherapy with
or without mild symptoms to delay symptomatic disease progression
(23,24). Side effects following abiraterone
treatment include hypertension, decreased serum potassium, edema
and increased adrenocorticotropic hormone (ACTH) release. Other
frequent adverse events include fatigue, fluid retention,
hypokalemia, hypertension, cardiac disorders and liver enzyme
increases (24,25).
Combination therapy with other androgen signaling
pathway inhibitors, albeit with different mechanisms of action, for
example enzalutamide and/or other therapeutic agents, are currently
ongoing or in the planning phase (26).
Enzalutamide
Enzalutamide affects multiple steps in the AR
signaling pathway including: i) competitive inhibition of androgen
binding to the AR; ii) inhibition of nuclear translocation of the
AR into the nucleus; iii) reduction of AR association with DNA; and
iv) in vitro suppression of growth and induction of
apoptosis in cell lines with AR gene amplifications (27).
Enzalutamid is well tolerated, most frequent
side-effects reported were fatigue (33.6%), cardiac disorder
(6.1%), myocardial infarction (0.3%), abnormalities like AST/ALT
increase and bilirubin increase (1%). Seizures were rare and
occurred in 0.6% of patients (28).
Enzalutamide is used in the post-chemotherapy
setting and was found to improve progression-free survival (PFS),
quality of life (QOL) and overall survival (OS) (29). Results of the PREVAIL phase III
trial showed a significantly improved PFS and OS also in
chemotherapy-naive patients (6).
ARN-509
ARN-509 is a novel AR receptor antagonist that,
unlike bicalutamide, does not show any agonist functions in the
context of CRPC with AR overexpression. ARN-509 inhibits AR nuclear
translocation and transcriptional activity and a phase I trial
showed a favorable safety and side effect profile (26,30,31).
4. Phosphatidylinositol-3-kinase (PI3K)
signaling
One of the key pathways essential to cell
proliferation, survival, and metabolism is the phosphatidylinositol
3-kinase (PI3K) pathway (32). The
PI3K pathway is the second most frequently deregulated signaling
pathway in prostate cancer, behind only the AR signaling pathway
(7). The PI3K pathway is normally
activated following growth factor stimulation of receptor tyrosine
kinases ultimately resulting in the conversion of membrane
phosphatidylinositol-bis-phosphate (PI(3,4)P2; PIP2) to
phosphatidylinositol-tri-phosphate (PI(3,4,5)P3; PIP3) by PI3K.
This conversion of PIP2 to PIP3 by PI3K can be reversed by the
tumor suppressor phosphatase and tensin homolog deleted on
chromosome ten (PTEN) phosphatase, which functions to negatively
regulate PI3K pathway activation. The formation of PIP3 mediates
the activation of AKT, which is the central effector of many
downstream signaling pathways regulating protein synthesis, cell
cycle, cell death, cell growth and cell survival (32).
Another important target of the PI3K signaling
pathway is the serine/threonine kinase mTOR (mammalian target of
rapamycin). mTOR occurs in two complexes, the TORC1 complex (mTOR
bound to Raptor) and the mTORC2 complex (mTOR bound to Rictor).
Both mTORC1 and mTORC2 are substrates of AKT: when activated,
mTORC1 regulates protein translation, while mTORC2 can
phosphorylate AKT and provide positive feedback to this branch of
the signaling network (33).
Approximately 34–40% of primary and 74–100% of
metastatic prostate cancers harbor alterations of components of the
PI3K pathway, generally promoting aberrant pathway activation
(7). These genetic aberrations
frequently involve loss-of-function mutations, deletions, or
epigenetic silencing of PTEN, a negative regulator of PI3K, and/or
activating mutations in PIK3CA, the catalytic p110α kinase subunit
(7,34–36).
Importantly, it has recently been shown that
inhibition of the PI3K pathway in PTEN-deficient prostate cancer
activates AR signaling by relieving feedback inhibition of the
receptor tyrosine kinases HER2 and HER3. Conversely, inhibition of
AR signaling was shown to activate AKT signaling through a reduced
expression of the AKT phosphatase PHLPP (11). Reciprocal feedback regulation of
the PI3K and AR pathways provides a compelling explanation for the
poor efficacy of single-pathway inhibition therapy, for instance
inhibition of the AR pathway alone, in PTEN-null cancers and the
substantially improved antitumoral efficacy of combined PI3K/AR
pathway inhibition. This pathway crosstalk may also be partially
responsible for induction of castration resistance, which further
underscores the importance of developing PI3K pathway targeting
agents for the treatment of prostate cancer patients.
5. Pharmacological inhibition of PI3K
signaling
The PI3K pathway is target-rich and a number of
efforts have been made to exploit this fact for anticancer therapy.
The PI3Ks are grouped into three classes of enzymes (I-III),
leading to the development of both pan- and isoform-specific PI3K
inhibitors as well as dual PI3K/TORC1/2 inhibitors (37). Several substances are currently
under investigation as single agents or in combination with
abiraterone (7–9).
The pan-PI3K inhibitor BKM120 (buparlisib) is
currently under investigation in men with CRPC as single agent and
in combination with abiraterone in two clinical trials
(NCT01385293, NCT01634061). Treatment-related adverse events in
BKM120-treated patients were most commonly fatigue, nausea, rash,
hyperglycemia, diarrhea, anorexia and mood alterations (37–39).
The latter underlines the need for close observation for
psychiatric symptoms in patients treated with PI3K inhibitors that
are able to cross the blood-brain barrier. Metabolic adverse events
were most often reversible and not acutely toxic (40).
Isoform-specific PI3K inhbitors may be particularly
relevant in patients in which PIK3CA is mutated (up to 16%) but
redundancy between different isoform needs to be considered
(7). Dual PI3K/mTOR inhibitor such
as BEZ235 or GDC-0980 lead to a profound inhibition of PI3K
signaling and and have so far been well-tolerated. BEZ235 or
GDC-0980 in combination with abiraterone is currently under
investigation in a phase I/II clinical trials in men with mCRPC
(NCT01717898). AKT inhibitors such as perifosine have so far not
led to significant clinical responses (37,41).
Again, combination therapies between antihormonal substances and
PI3K inhibitors appear to be the most promising avenues for future
drug development and results from ongoing phase II trials will be
instrumental to corroborate this notion. Inhibitors of TORC1 such
as rapamycin and its analogs everolimus or temsirolimus did not
show significant antitumoral effects when used as single agents in
men with CRPC (37,41).
6. Ras/Raf/MEK/ERK signaling
The Ras/Raf/MEK/ERK signaling pathway regulates
fundamental cellular processes, including proliferation,
differentiation, and cell survival and is frequently activated in
several cancer types including prostate cancer. Increased
activation of this pathway correlates with a poor prognosis and
tumor invasiveness (42–44). Ras/Raf/MEK/ERK signaling is
stimulated by growth factor receptor activation of the small GTPase
Ras, which in turn activates the protein kinase Raf leading to the
activation of the dual specificity MAPK kinase (MEK1/MEK2), and
finally phosphorylation of extracellular signal-regulated kinases
(ERK1/ERK2) (45). The PI3K and
Ras/Raf/MEK/ERK pathways have been shown to interact extensively
and frequent co-activation of these two pathways has been observed
in prostate cancer (46). Both
pathways share common activation signals, such as receptor tyrosine
kinase mediated activation, and also appear to provide compensatory
signaling when one or the other is inhibited (47). Approximately 43% of primary and 90%
of metastatic prostate tumors were found to harbor genetic
alterations of the Ras/Raf/MEK/ERK pathway (7).
Although members of the Ras family are rarely
mutated in general as well as in prostate cancer, the expression of
important growth factor receptors promoting Ras activation, such as
EGFR, FGFR and PDGFR, is frequently upregulated in prostate cancers
(48). Further, ERK1/ERK2
activation is associated with increasing Gleason score and tumor
stage (49). This is consistent
with a functional role for the Ras/Raf/MEK/ERK signaling pathway as
prostate cancer progresses to a more advanced, androgen-independent
stage.
An activated Ras/Raf/MEK/ERK pathway could therefore
provide a selective advantage to tumor cells under
androgen-deprivation pressure This notion is corroborated by the
finding that the Ras/Raf/MEK/ERK pathway can sustain the
transcription of androgen-responsive genes, and therefore prostate
cancer cell proliferation, even when the AR is inhibited (49,50).
7. Pharmacological inhibition of the
Ras/Raf/MEK/ERK pathway
Efforts to target Ras directly have not been
successful in the clinic to date, but recent clinical trials with
Raf and MEK inhibitors have suggested that targeting these
downstream Ras effectors could be promising.
Single agent therapy with MEK inhibitor alone showed
a compensatory upregulation of the PI3K pathway as well as several
others including NF-κB and hedgehog as an expression of cellular
pro-survival mechanisms (51).
Dual inhibition of both MEK and PI3K/mTOR signaling increases
apoptosis in cell lines and dual inhibition of AKT/mTOR and ERK has
been demonstrated to lead to effective growth inhibition in mouse
models of prostate cancer (46,52).
Furthermore, ERK inhibition was found to enhance docetaxel-induced
cytotoxicity in androgen-independent prostate cancer cells
(63).
Phase III trials have been performed with
GSK-1120212 (trametinib), which is an FDA-approved MEK1/2
inhibitor, for metastatic melanoma showing a favorable safety
profile. Main adverse events were acneiform skin alterations,
diarrhea, peripheral edema, hypertension and transient mild cardiac
dysfunction. Most toxicities did not require drug discontinuation
(54–57). Trials are going to be initiated in
patients with different tumor entities (58). Other MEK inhibitors such as and
MEK162 (binimetinib) and PD-0325901 are likewise in phase I/II
clinical trials.
8. Retinoblastoma protein (pRB) pathway
The retinoblastoma gene (RB1) is a tumor suppressor
gene with somatic alterations in multiple cancers whose protein
product (pRB) inhibits cell cycle progression (59). In prostate cancer, pRB inactivation
through genomic deletion of RB1 has been reported in approximately
20–60% of tumors and is associated with transition to CRPC and poor
clinical outcome (60,61). pRB activity is predominately
controlled by cell cycle regulated cyclin-dependent kinase (CDK)
activity (58). When CDK activity
is inhibited, pRB remains in a hypophosphorylated and active state
repressing E2F-mediated gene transcription thereby inhibiting cell
cycle progression (58).
Inactivation of pRB prevents cells from restraining cellular
proliferation and leads to an aberrant expression of E2F-responsive
genes (62). Overexpression of
cyclins also contributes to the deregulation of pRB function in
prostate cancer (7,63,64).
Remarkably, CRPC tissue has been found to show an altered
repertoire of AR binding sites that were enriched for E2F motifs,
which can lead to cyclin/CDK hyperactivity, pRB inactivation and
uncontrolled proliferation (61).
Although it was demonstrated that pRB-deficient
tumors respond poorly to hormone therapy, increasing evidence
suggests that tumors with reduced pRB expression exhibit a more
beneficial initial response to chemotherapy (65). Thus, RB1/pRB status could be a
predictive marker of response to chemotherapy, with a potential to
influence clinical decision-making, and a potential biomarker of
transition to castration resistance. In addition, treatment with
anti-androgens can lead to more aggressive tumors with
neuroendocrine differentiation which is often associated with a
loss of pRB protein expression (66).
9. Pharmacological inhibition of pRB
signaling
Numerous studies have explored CDK inhibitors to
target tumor cells with inactivated pRB and E2F-induced cyclin/CDK
hyperactivity. The pan-CDK (CDK1, CDK2, CDK4/6) inhibitor
flavoperidol was found to enhance apoptosis in androgen-independent
PC-3 prostate cancer cells (67).
An increase in apoptosis and a decrease in angiogenesis was
detected when combined with docetaxel in a mouse model of prostate
cancer (68,69). In addition, it has been shown that
CDK1 can phosphorylate the AR at serine 5115 which was associated
with unfavorable patient outcome. The CDK inhibitor roscovitine was
found to reduce AR phosphorylation, which could be exploited
clinically (70). A selective
CDK4/6 inhibitor, PD-0332991 (palbociclib), showed a growth
suppressive effect in prostate cancer xenografts. Phase I studies
in colorectal cancer and multiple myeloma are ongoing (71). The CDK4/6 inhibitor LEE011 is
currently under investigation in several phase I and II trials for
advanced solid tumor, breast cancer, lymphoma and melanoma
(72). Possible side effects known
from clinical trials with roscovitine are nausea, vomiting,
transient elevations in serum creatinine and liver enzymes.
10. Conclusions and outlook
Due to extensive crosstalk between signaling
pathways such as the AR and PI3K pathways, PI3K and Ras/Raf/MEK/ERK
pathway and pRB and AR signaling, single-agent therapy is likely to
result in an activation of mechanisms that thwart the antitumoral
response, lead to drug resistance and ultimately treatment failure.
Future drug and clinical trial development therefore needs to
reflect the extensive feedback mechanisms that exist between the
four most frequently altered signaling nodes in prostate cancer.
However, there are a number of questions and concerns that need to
be addressed. First and foremost, it will be important to determine
the safety, tolerability and the actual clinical effectiveness of
treatment regimen that encompass two or more pathway-targeting
drugs. Given that unexpected and paradoxical survival pathway
activation in response to targeted agents has been reported in
other malignancies (73–76), it is possible that, depending on
the genetic background and intratumoral heterogeneity, combination
therapies that have been found to be effective in vitro and
in preclinical models may not show the same efficacy when used in
cancer patients.
Second, toxicity profiles for combination targeted
therapies need to be carefully analyzed and weighed against the
clinical benefit. Whether and to what extent a dose reduction of
individual compounds is permissable without impeding the
oncological effectiveness as it has been shown for ‘classical’
chemotherapeutic agents remains to be determined. Nonetheless, it
is worth remembering that combination drug therapies were a major
breakthrough in the chemotherapeutic treatment of several
malignancies including Hodgkin’s lymphoma and several others. So
far, toxicity profiles of newer generation pathway inhibitors
appear to be manageable but unexpected pharmacological interactions
and resistance mechanisms always need to be considered.
Third, regulatory limitations are a major concern.
Effective drugs may not be manufactured by the same pharmaceutical
company and may lack approval in certain geographic regions. How to
deal with this problem and whether multi-pathway targeting drugs
are an attainable solution remains to be determined.
Nonetheless, from a purely biological point of view,
novel treatment strategies for prostate cancer need to reflect the
highly disorganized and profoundly interconnected signaling
landscape that has emerged from genome-wide analyses.
Acknowledgements
Work in the authors’ laboratory is supported by the
Medical Faculty Heidelberg.
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar
|
|
2
|
Prostate Cancer Trialists’ Collaborative
Group. Maximum androgen blockade in advanced prostate cancer: an
overview of the randomised trials. Lancet. 355:1491–1498. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Berthold DR, Pond GR, Soban F, de Wit R,
Eisenberger M and Tannock IF: Docetaxel plus prednisone or
mitoxantrone plus prednisone for advanced prostate cancer: updated
survival in the TAX 327 study. J Clin Oncol. 26:242–245. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tannock IF, de Wit R, Berry WR, et al:
Docetaxel plus prednisone or mitoxantrone plus prednisone for
advanced prostate cancer. N Engl J Med. 351:1502–1512. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ryan CJ, Smith MR, de Bono JS, et al:
Abiraterone in metastatic prostate cancer without previous
chemotherapy. N Engl J Med. 368:138–148. 2013. View Article : Google Scholar
|
|
6
|
Beer TM, Armstrong AJ, Sternberg CN, et
al: Enzalutamide in men with chemotherapy-naive metastatic prostate
cancer (mCRPC): Results of phase III PREVAIL study. J Clin Oncol.
32(Suppl 4): abs. LBA1ˆ. 2014.PubMed/NCBI
|
|
7
|
Taylor BS, Schultz N, Hieronymus H, et al:
Integrative genomic profiling of human prostate cancer. Cancer
Cell. 18:11–22. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mulholland DJ, Kobayashi N, Ruscetti M, et
al: Pten loss and RAS/MAPK activation cooperate to promote EMT and
metastasis initiated from prostate cancer stem/progenitor cells.
Cancer Res. 72:1878–1889. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Carver BS, Chapinski C, Wongvipat J, et
al: Reciprocal feedback regulation of PI3K and androgen receptor
signaling in PTEN-deficient prostate cancer. Cancer Cell.
19:575–586. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bennett NC, Gardiner RA, Hooper JD,
Johnson DW and Gobe GC: Molecular cell biology of androgen receptor
signalling. Int J Biochem Cell Biol. 42:813–827. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ryan CJ and Tindall DJ: Androgen receptor
rediscovered: the new biology and targeting the androgen receptor
therapeutically. J Clin Oncol. 29:3651–3658. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Koivisto P, Kononen J, Palmberg C, et al:
Androgen receptor gene amplification: a possible molecular
mechanism for androgen deprivation therapy failure in prostate
cancer. Cancer Res. 57:314–319. 1997.PubMed/NCBI
|
|
13
|
Edwards J, Krishna NS, Grigor KM and
Bartlett JM: Androgen receptor gene amplification and protein
expression in hormone refractory prostate cancer. Br J Cancer.
89:552–556. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dehm SM, Schmidt LJ, Heemers HV, Vessella
RL and Tindall DJ: Splicing of a novel androgen receptor exon
generates a constitutively active androgen receptor that mediates
prostate cancer therapy resistance. Cancer Res. 68:5469–5477. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sun S, Sprenger CC, Vessella RL, et al:
Castration resistance in human prostate cancer is conferred by a
frequently occurring androgen receptor splice variant. J Clin
Invest. 120:2715–2730. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen S, Xu Y, Yuan X, Bubley GJ and Balk
SP: Androgen receptor phosphorylation and stabilization in prostate
cancer by cyclin-dependent kinase 1. Proc Natl Acad Sci USA.
103:15969–15974. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fujimoto N, Mizokami A, Harada S and
Matsumoto T: Different expression of androgen receptor coactivators
in human prostate. Urology. 58:289–294. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Taplin ME, Rajeshkumar B, Halabi S, et al:
Androgen receptor mutations in androgen-independent prostate
cancer: Cancer and Leukemia Group B Study 9663. J Clin Oncol.
21:2673–2678. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sack JS, Kish KF, Wang C, et al:
Crystallographic structures of the ligand-binding domains of the
androgen receptor and its T877A mutant complexed with the natural
agonist dihydrotes-tosterone. Proc Natl Acad Sci USA. 98:4904–4909.
2001. View Article : Google Scholar
|
|
20
|
Stein MN, Patel N, Bershadskiy A, Sokoloff
A and Singer EA: Androgen synthesis inhibitors in the treatment of
castration-resistant prostate cancer. Asian J Androl. 16:387–400.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Abreu-Martin MT, Chari A, Palladino AA,
Craft NA and Sawyers CL: Mitogen-activated protein kinase kinase
kinase 1 activates androgen receptor-dependent transcription and
apoptosis in prostate cancer. Mol Cell Biol. 19:5143–5154.
1999.
|
|
22
|
Bakin RE, Gioeli D, Sikes RA, Bissonette
EA and Weber MJ: Constitutive activation of the
Ras/mitogen-activated protein kinase signaling pathway promotes
androgen hypersensitivity in LNCaP prostate cancer cells. Cancer
Res. 63:1981–1989. 2003.
|
|
23
|
Araujo JC, Trudel GC, Saad F, et al:
Docetaxel and dasatinib or placebo in men with metastatic
castration-resistant prostate cancer (READY): a randomised,
double-blind phase 3 trial. Lancet Oncol. 14:1307–1316. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cea R: Interim analysis results of
COU-AA-302, a randomized, phase 3 study of abiraterone acetate (AA)
in chemotherapie-naive patients with metastatic
castration-resistant prostate cancer (mCRPC). J Clin Oncol. (Suppl
30): abs LBA4518. 2012.
|
|
25
|
De Bono JS, Logothetis CJ, Molina A, et
al: Abiraterone and increased survival in metastatic prostate
cancer. N Engl J Med. 364:1995–2005. 2011.
|
|
26
|
Sridhar SS, Freedland SJ, Gleave ME, et
al: Castration-resistant prostate cancer: from new pathophysiology
to new treatment. Eur Urol. 65:289–299. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tran C, Ouk S, Clegg NJ, et al:
Development of a second-generation antiandrogen for treatment of
advanced prostate cancer. Science. 324:787–790. 2009. View Article : Google Scholar
|
|
28
|
De Bono JS: Primary, secondary, and
quality-of-life endpoint results from the phase III AFFIRM study of
MDV3100, an androgen receptor signaling inhibitor. J Clin Oncol.
(Suppl 30): abs 4519. 2012.
|
|
29
|
Scher HI, Fizazi K, Saad F, et al:
Increased survival with enzalutamide in prostate cancer after
chemotherapy. N Engl J Med. 367:1187–1197. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Clegg NJ, Wongvipat J, Joseph JD, et al:
ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer
Res. 72:1494–1503. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rathkopf DE, Morris MJ, Fox JJ, et al:
Phase I study of ARN-509, a novel antiandrogen, in the treatment of
castration-resistant prostate cancer. J Clin Oncol.
31:3525–3530
|
|
32
|
Wong KK, Engelman JA and Cantley LC:
Targeting the PI3K signaling pathway in cancer. Curr Opin Genet
Dev. 20:87–90. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
LoPiccolo J, Blumenthal GM, Bernstein WB
and Dennis PA: Targeting the PI3K/Akt/mTOR pathway: effective
combinations and clinical considerations. Drug Resist Updat.
11:32–50. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vlietstra RJ, van Alewijk DC, Hermans KG,
van Steenbrugge GJ and Trapman J: Frequent inactivation of PTEN in
prostate cancer cell lines and xenografts. Cancer Res.
58:2720–2723. 1998.PubMed/NCBI
|
|
35
|
Verhagen PC, van Duijn PW, Hermans KG, et
al: The PTEN gene in locally progressive prostate cancer is
preferentially inactivated by bi-allelic gene deletion. J Pathol.
208:699–707. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Konishi N, Nakamura M, Kishi M, Nishimine
M, Ishida E and Shimada K: Heterogeneous methylation and deletion
patterns of the INK4a/ARF locus within prostate carcinomas. Am J
Pathol. 160:1207–1214. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bitting RL and Armstrong AJ: Targeting the
PI3K/Akt/mTOR pathway in castration-resistant prostate cancer.
Endocr Relat Cancer. 20:R83–R99. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ando Y, Inada-Inoue M, Mitsuma A, et al:
Phase I dose-escalation study of buparlisib (BKM120), an oral
pan-class I PI3K inhibitor, in Japanese patients with advanced
solid tumors. Cancer Sci. 105:347–353
|
|
39
|
Bendell JC, Rodon J, Burris HA, et al:
Phase I, dose-escalation study of BKM120, an oral pan-class I PI3K
inhibitor, in patients with advanced solid tumors. J Clin Oncol.
30:282–290. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Busaidy NL, Farooki A, Dowlati A, et al:
Management of metabolic effects associated with anticancer agents
targeting the PI3K-Akt-mTOR pathway. J Clin Oncol. 30:2919–2928.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Armstrong GT, Kawashima T, Leisenring W,
et al: Aging and risk of severe, disabling, life-threatening, and
fatal events in the childhood cancer survivor study. J Clin Oncol.
32:1218–1227. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
McCubrey JA, Steelman LS, Chappell WH, et
al: Roles of the Raf/MEK/ERK pathway in cell growth, malignant
transformation and drug resistance. Biochim Biophys Acta.
1773:1263–1284. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
McCubrey JA, Steelman LS, Abrams SL, et
al: Roles of the Raf/MEK/ERK and PI3K/PTEN/AKT pathways in
malignant transformation and drug resistance. Adv Enzyme Regul.
46:249–279. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ramsay AK, McCracken SR, Soofi M, et al:
ERK5 signalling in prostate cancer promotes an invasive phenotype.
Br J Cancer. 104:664–672. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kim EK and Choi EJ: Pathological roles of
MAPK signaling pathways in human diseases. Biochim Biophys Acta.
1802:396–405. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kinkade CW, Castillo-Martin M, Puzio-Kuter
A, et al: Targeting AKT/mTOR and ERK MAPK signaling inhibits
hormone-refractory prostate cancer in a preclinical mouse model. J
Clin Invest. 118:3051–3064. 2008.PubMed/NCBI
|
|
47
|
Shimizu T, Tolcher AW, Papadopoulos KP, et
al: The clinical effect of the dual-targeting strategy involving
PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced
cancer. Clin Cancer Res. 18:2316–2325. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Drake JM, Graham NA, Stoyanova T, et al:
Oncogene-specific activation of tyrosine kinase networks during
prostate cancer progression. Proc Natl Acad Sci USA. 109:1643–1648.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Carey AM, Pramanik R, Nicholson LJ, et al:
Ras-MEK-ERK signaling cascade regulates androgen receptor
element-inducible gene transcription and DNA synthesis in prostate
cancer cells. Int J Cancer. 121:520–527. 2007. View Article : Google Scholar
|
|
50
|
Weber MJ and Gioeli D: Ras signaling in
prostate cancer progression. J Cell Biochem. 91:13–25. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Gioeli D, Wunderlich W, Sebolt-Leopold J,
et al: Compensatory pathways induced by MEK inhibition are
effective drug targets for combination therapy against
castration-resistant prostate cancer. Mol Cancer Ther.
10:1581–1590. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Steelman LS, Abrams SL, Shelton JG, et al:
Dominant roles of the Raf/MEK/ERK pathway in cell cycle
progression, prevention of apoptosis and sensitivity to
chemotherapeutic drugs. Cell Cycle. 9:1629–1638. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zelivianski S, Spellman M, Kellerman M, et
al: ERK inhibitor PD98059 enhances docetaxel-induced apoptosis of
androgen-independent human prostate cancer cells. Int J Cancer.
107:478–485. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Menzies AM and Long GV: Dabrafenib and
Trametinib, alone and in combination for BRAF-mutant metastatic
melanoma. Clin Cancer Res. 20:2035–2043. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Schadendorf D, Amonkar MM, Milhem M, et
al: Functional and symptom impact of trametinib versus chemotherapy
in BRAF V600E advanced or metastatic melanoma: quality-of-life
analyses of the METRIC study. Ann Oncol. 25:700–706
|
|
56
|
Anforth R, Liu M, Nguyen B, et al:
Acneiform eruptions: A common cutaneous toxicity of the MEK
inhibitor trametinib. Australas J Dermatol. View Article : Google Scholar : 2013.[Epub ahead
of print].
|
|
57
|
Kim KB, Kefford R, Pavlick AC, et al:
Phase II study of the MEK1/MEK2 inhibitor Trametinib in patients
with metastatic BRAF-mutant cutaneous melanoma previously treated
with or without a BRAF inhibitor. J Clin Oncol. 31:482–489. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Giacinti C and Giordano A: RB and cell
cycle progression. Oncogene. 25:5220–5227. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Sellers WR and Kaelin WG Jr: Role of the
retinoblastoma protein in the pathogenesis of human cancer. J Clin
Oncol. 15:3301–3312. 1997.PubMed/NCBI
|
|
60
|
Grasso CS, Wu YM, Robinson DR, et al: The
mutational landscape of lethal castration-resistant prostate
cancer. Nature. 487:239–243. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Sharma A, Yeow WS, Ertel A, et al: The
retinoblastoma tumor suppressor controls androgen signaling and
human prostate cancer progression. J Clin Invest. 120:4478–4492.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Dyson N: The regulation of E2F by
pRB-family proteins. Genes Dev. 12:2245–2262. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Mashal RD, Lester S, Corless C, et al:
Expression of cell cycle-regulated proteins in prostate cancer.
Cancer Res. 56:4159–4163. 1996.PubMed/NCBI
|
|
64
|
Drobnjak M, Osman I, Scher HI, Fazzari M
and Cordon-Cardo C: Overexpression of cyclin D1 is associated with
metastatic prostate cancer to bone. Clin Cancer Res. 6:1891–1895.
2000.PubMed/NCBI
|
|
65
|
Aparicio A, Den RB and Knudsen KE: Time to
stratify? The retinoblastoma protein in castrate-resistant prostate
cancer. Nat Rev Urol. 8:562–568. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Tan HL, Sood A, Rahimi HA, et al: Rb loss
is characteristic of prostatic small cell neuroendocrine carcinoma.
Clin Cancer Res. 20:890–903. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Nathwani SM, Cloonan SM, Stronach M, et
al: Novel microtubule-targeting agents, pyrrolo-1,5-benzoxazepines,
induce cell cycle arrest and apoptosis in prostate cancer cells.
Oncol Rep. 24:1499–1507. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Reiner T, de las Pozas A and Perez-Stable
C: Sequential combinations of flavopiridol and docetaxel inhibit
prostate tumors, induce apoptosis, and decrease angiogenesis in the
Ggamma/T-15 transgenic mouse model of prostate cancer. Prostate.
66:1487–1497. 2006. View Article : Google Scholar
|
|
69
|
Gomez LA, de Las Pozas A and Perez-Stable
C: Sequential combination of flavopiridol and docetaxel reduces the
levels of X-linked inhibitor of apoptosis and AKT proteins and
stimulates apoptosis in human LNCaP prostate cancer cells. Mol
Cancer Ther. 5:1216–1226. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Willder JM, Heng SJ, McCall P, Adams CE,
Tannahill C, Fyffe G, Seywright M, Horgan PG, Leung HY, Underwood
MA and Edwards J: Androgen receptor phosphorylation at serine 515
by Cdk1 predicts biochemical relapse in prostate cancer patients.
Br J Cancer. 15:139–148. 2013.PubMed/NCBI
|
|
71
|
Gilbert MR, Dignam J, Pugh S, et al: Reply
to m. C Chamberlain J Clin Oncol. 32:1634–1635. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Vici P, Capomolla E, Foggi P, et al: High
activity of salvage treatment with biweekly paclitaxel-gemcitabine
combination in heavily pretreated breast cancer patients. J Exp
Clin Cancer Res. 25:39–44. 2006.PubMed/NCBI
|
|
73
|
Wang X, Hawk N, Yue P, et al: Overcoming
mTOR inhibition-induced paradoxical activation of survival
signaling pathways enhances mTOR inhibitors’ anticancer efficacy.
Cancer Biol Ther. 7:1952–1958. 2008.PubMed/NCBI
|
|
74
|
Chu S, Holtz M, Gupta M and Bhatia R:
BCR/ABL kinase inhibition by imatinib mesylate enhances MAP kinase
activity in chronic myelogenous leukemia CD34+ cells.
Blood. 103:3167–3174. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Yu C, Krystal G, Varticovksi L, et al:
Pharmacologic mitogen-activated protein/extracellular
signal-regulated kinase kinase/ mitogen-activated protein kinase
inhibitors interact synergistically with STI571 to induce apoptosis
in Bcr/Abl-expressing human leukemia cells. Cancer Res. 62:188–199.
2002.
|
|
76
|
Packer LM, Rana S, Hayward R, et al:
Nilotinib and MEK inhibitors induce synthetic lethality through
paradoxical activation of RAF in drug-resistant chronic myeloid
leukemia. Cancer Cell. 20:715–727. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Dickson MA and Schwartz GK: Development of
cell-cycle inhibitors for cancer therapy. Curr Oncol. 16:36–43.
2009.PubMed/NCBI
|














