Introduction
Deregulation of cell cycle progression is a
universal characteristic of cancer, and the majority of human
cancers have abnormalities in one or more components associated
with CDK activity (1). Therefore,
synthetic inhibitors of CDK activity present as an appropriate
strategy in the development of new cancer therapeutic methods.
Based on this hypothesis, we synthesized
2-[1,1′-biphenyl]-4-yl-N-[5-(1,1-dioxo-1λ6-isothiazolidin-2-yl)-1H-indazol-3-yl]acetamide
(BAI), one of the 3,5-diaminoindazole derivatives, as a novel CDK
inhibitor (2). Our previous
studies (3,4) showed that BAI potentially inhibited
proliferation at nanomolar concentrations in AMC-HN4, AMC-HN6,
A549, Caki, and HCT116 cells.
Ras proteins regulate diverse cellular pathways that
are important in the growth and spread of malignancies, including
cell proliferation, cell cycle regulation, cell survival,
angiogenesis, cell migration (5).
It has been reported that carcinogenesis is correlated with
activation of Ras in various human cancers (6,7).
Because farnesylation of Ras plays an important role in the
conversion of Ras to its biologically active form, the inhibition
of farnesyltransferase has been studied as a specific molecular
targeting therapy for the treatment of various cancers (8–10).
Recent studies revealed that several classes of farnesyltransferase
inhibitors (FTIs) have anti-proliferative effect on human cancers
(11–14). Furthermore, the anticancer effect
of FTI can be enhanced by co-treatment with various
chemotherapeutic drugs resulting in a synergistic apoptotic
response in various tumor cells (15–18).
Especially, it has been revealed that the synergistic effect of a
cdk inhibitor enhanced FTI-induced apoptosis (18). Novel strategies in developing FTIs
have led to a new component in which LB42708, pyrrole-based orally
active FTI, has a potent apoptotic effect (14,19).
In the present study, we aimed to elucidate the
phenomenon by which the combinational therapy with BAI and LB42708
inhibited growth of human cancer cells, as well as to uncover the
molecular biological basis of mechanism for their apoptosis
induction in cancer cells.
Materials and methods
Cell lines and culture
The human non-small cell lung cancer (HNSCLC) A549
was obtained from the American Type Culture Collection (ATCC,
Rockville, MD, USA) and grown in RPM-1640 medium supplemented with
10% heated-inactivated fetal bovine serum (FBS), 2 mM L-glutamine,
100 μg/μl streptomycin and 100 μg/μl penicillin. Human renal clear
cell carcinoma Caki was obtained from the ATCC and grown in
Dulbecco’s modified Eagle’s medium (DMEM), containing 10%
heat-inactivated FBS, 20 mM HEPES buffer and 100 μg/μl streptomycin
and 100 μg/μl penicillin.
Drugs and materials
2-[1,1′-biphenyl]-4-yl-N-[5-(1,1-dioxo-1λ6-isothiazolidin-2-yl)-1H-indazol-3-yl]acetamide
(BAI) was kindly supplied by Dr J.H. Lee (Keimyung University,
Daegu, Korea). LB42708 was purchased from TOCRIS. Anti-cIAP-1,
anti-cIAP-2, anti-Bcl-2, anti-Mcl-1, anti-AIF, anti-HSC70, and
anti-β-actin antibodies were purchased from Santa Cruz
Biotechnology (Santa Cruz, CA, USA). Anti-PARP and anti-ERK
antibodies were purchased from Cell Signaling Technology (Danvers,
MA, USA). Anti-OxPhos Complex II subunit (QPs2) antibody was
purchase from Molecular Probes (Eugene, OR, USA). Anti-cellular
FLICE-like inhibitory protein (c-FLIP) antibody was purchased from
Alexis (San Diego, CA, USA). Anti-XIAP and anti-cytochrome c
antibodies were purchased from BD Biosciences Pharmingen (San
Diego, CA, USA). Antibodies against the following proteins were
purchased from the indicated suppliers: pro-caspase-3 from Santa
Cruz Biotechnology. z-VAD-fmk was purchased from Biomol (Plymouth
Meeting, PA, USA).
Western blot analysis
Cellular lysates were prepared by suspending
0.3×106 cells in 80 μl of lysis buffer (137 mM NaCl, 15
mM EGTA, 0.1 mM sodium orthovanadate, 15 mM MgCl2, 0.1%
Triton X-100, 25 mM MOPS, 100 μM phenylmethylsulfonyl fluoride and
20 μM leupeptin, adjusted to pH 7.2). The cells were disrupted by
vortexing and extracted at 4°C for 30 min. The proteins were
electrotransferred to Immobilon-P membranes (Millipore Corp.,
Bedford, MA, USA). Detection of specific proteins was carried out
with an ECL Western blotting kit according to the manufacturer’s
instructions.
Construction of the expression vectors
for c-FLIP (L), c-FLIP (s) and Mcl-1 (L)
The human cDNAs encoding c-FLIP (L) and c-FLIP (s)
were PCR amplified from plasmids [pCA-FLAG-hFLIP (L) and
pCA-FLAG-hFLIP (s); kindly provided by Dr S.I. Park, Korea Centers
for Disease Control and Prevention, Seoul, Korea] containing these
sequences with the specific primers. c-FLIP (L) and c-FLIP (s) cDNA
fragment were digested with KpnI and XhoI and
subcloned into the pcDNA 3.1 (+) vector (Invitrogen), and the
resulting constructs were confirmed by nucleotide sequencing. The
human cDNA for Mcl-1 (L) was PCR amplified using the following
specific primers: Mcl-1 (sense) 5′-GCGACTGGCAAAGCTTGGC CTCAA-3′ and
(anti-sense) 5′-CAACTCTAGAAACTGGT TTTGGTG-3′. The human Mcl-1 (L)
cDNA fragments were subcloned into the pcDNA 3.1 (+) vector
(Invitrogen, Calsbad, CA, USA).
Establishment of the stable cell lines
overexpressing CrmA, Bcl-2, Mcl-1 (L), c-FLIP (L), or c-FLIP
(s)
Caki cells were transfected with the following: a
mammalian expression vector containing CrmA cDNA, a pMAX vector
containing the human Bcl-2 gene (provided by Dr Rakesh
Srivastava, NIH/NIA), a vector containing Mcl-1 (L), and a vector
encoding Flag-tagged c-FLIP (L) or c-FLIP (s). Stable cell lines
overexpressing CrmA, Bcl-2, Mcl-1 (L), c-FLIP (L), or c-FLIP (s)
were selected with fresh medium containing 500 μg/ml G418
(Calbiochem, Madison, WI, USA) for 4 weeks. Overexpression of CrmA,
Bcl-2, Mcl-1 (L), c-FLIP (L), or c-FLIP (s) was analyzed by western
blotting using anti-CrmA (BD Pharmingen), anti-Bcl-2 (Santa Cruz),
anti-Mcl-1 (Santa Cruz), anti-Flag (Sigma, St. Louis, MO, USA), or
anti-c-FLIP (Alexis) antibody, respectively.
Flow cytometric analysis
Approximately 0.3×106 cells were
suspended in 100 μl PBS, and 200 μl of 95% ethanol was added while
vortexing. The cells were incubated at 4°C for 1 h, washed with
PBS, and resuspended in 250 μl of 1.12% sodium citrate buffer (pH
8.4) together with 12.5 μg RNase. Incubation was continued at 37°C
for 30 min. The cellular DNA was then stained by applying 250 μl
propidium iodide (50 μg/ml) for 30 min at room temperature. The
stained cells were analyzed by a FACScan flow cytometer for
relative DNA content based on red fluorescence.
DEVDase activity assay
To evaluate caspase-3 activity, cell lysates were
prepared after their respective treatment with various drugs.
Assays were performed in 96-well microtiter plates by incubating 20
μg cell lysates in 100 μl reaction buffer [1% NP-40, 20 mM Tris-HCl
(pH 7.5), 137 mM NaCl and 10% glycerol)] containing the caspase-3
substrate (DEVD-pNA) at 5 μM. Lysates were incubated at 37°C for 2
h. Thereafter, the absorbance at 405 nM was measured with a
spectrophotometer.
Cell viability assay
The anti-proliferative effect of the BAI or FTI on
Caki cells was investigated using a live cell movie analyzer, JuLI™
Br (NanoEnTek Inc., Seoul, Korea). Briefly, the cells were plated
in 6-well culture plates at a density of 0.3×106
cells/well in medium and allowed to attach for 10 h. The cells
treated with BAI in the presence or absence of FTI for 24 h. During
this study, JuLi Br recorded images of Caki cells at 5 minute
intervals, and confluences were also measured.
RNA isolation and quantitative real-time
PCR
Total cellular RNA was extracted from tissues using
the TRIzol reagent (Molecular Research Center, Inc., Cincinnati,
OH, USA). RNA was quantified using Nanodrop 1000 (Thermo
Scientific, Wilmington, DE, USA). Each cDNA was synthesized form 2
μg of total RNA using M-MLV reverse transcriptase (Promega,
Madison, WI, USA) according to the manufacturer’s protocol. By
using the specific primer pairs described in Table I and SYBR Green Premix (Toyobo,
Japan). Quantitative real-time PCR (qPCR) was performed on the
LightCycler® 480 real-time PCR system (Roche
Diagnostics, Mannheim, Germany). β-actin was used as a housekeeping
gene for normalization, and a no template sample was used as a
negative control.
 | Table IPrimer sequences of miRNA machinery
components used in quantitative PCR. |
Table I
Primer sequences of miRNA machinery
components used in quantitative PCR.
| Components | Position | Sequences |
|---|
| XIAP | Forward |
5′-ACCGTGCGGTGCTTTAGTT-3′ |
| Reverse |
5′-TGCGTGGCACTATTTTCAAGATA-3′ |
| Mcl-1 (L) | Forward |
5′-GTGCCTTTGTGGCTAAACACT-3′ |
| Reverse |
5′-AGTCCCGTTTTGTCCTTACGA-3′ |
| Bcl-2 | Forward |
5′-GCCTTCTTTGAGTTCGGTGG-3′ |
| Reverse |
5′-ATCTCCCGGTTGACGCTCT-3′ |
| c-FLIP (L) | Forward |
5′-GAGGCTCCCAGAGTGTGTATGG-3′ |
| Reverse |
5′-TGGCCCTCTGACACCACATAG-3′ |
| c-FLIP (s) | Forward |
5′-AATGTTCTCCAAGCAGCAATCC-3′ |
| Reverse |
5′-CCAAGAATTTTCAGATCAGGACAAT-3′ |
| β-actin | Forward |
5′-CAGCCATGTACGTTGCTATCCAGG-3′ |
| Reverse |
5′-AGGTCCAGACGCAGGATGGCATG-3′ |
Small interfering RNA
The XIAP small-interfering RNA (siRNA) duplexes were
obtained from Cell Signaling Technology. Control siRNA duplexes
used in this study were purchased from Invitrogen and had the
following sequences: green fluorescent protein (GFP), AAG ACC CGC
GCC GAG GUG AAG. Cells were transfected with siRNA oligonucleotides
using Lipofectamine RNAiMAX (Invitrogen) according to the
manufacturer’s recommendations.
Determination of the mitochondrial
membrane potential by rhodamine 123
Rhodamine 123 (Molecular Probes) uptake by
mitochondria is directly proportional to its membrane potential.
Caki cells subjected to 4 and 10 h after treatment were incubated
with rhodamine 123 (5 μM) for 30 min in the dark at 37°C. The cells
were harvested and suspended in PBS. The mitochondrial membrane
potential was subsequently analyzed using a flow cytometer (BD
Bioscience).
Analysis of mitochondrial cytochrome c
release
Caki cells (0.3×106) were harvested,
washed once with ice-cold PBS and gently lysed for 2 min in 80 μl
ice-cold lysis buffer (250 mM sucrose, 1 mM EDTA, 20 mM Tris-HCl pH
7.2, 1 mM DTT, 10 mM KCl, 1.5 mM MgCl2, 5 μg/ml
pepstatin, 10 μg/ml leupeptin, 2 μg/ml aprotinin). Lysates were
centrifuged at 12,000 g at 4°C for 10 min to obtain the
supernatants (cytosolic extracts free of mitochondria) and the
pellets (fraction that contains mitochondria). The resulting
cytosolic fractions were used for western blot analysis with an
anti-cytochrome c antibody.
Densitometry
The intensities of corresponding bands were
quantified using the ImageJ program (National Institutes of Health,
MD, USA) according to the manufacturer’s instructions.
Statistical analysis
All data are presented as mean ± SD. Significant
differences between the groups were determined using the unpaired
Student’s t-test. A value of *P<0.005 was accepted as
indication of statistical significance. All the data shown in the
figures were obtained from at least two independent experiments
with a similar pattern.
Results
Co-treatment of BAI and LB42708 induces
apoptosis
In order to investigate the effect of co-treatment
with BAI and FTI on Caki cells, Caki cells were treated with BAI
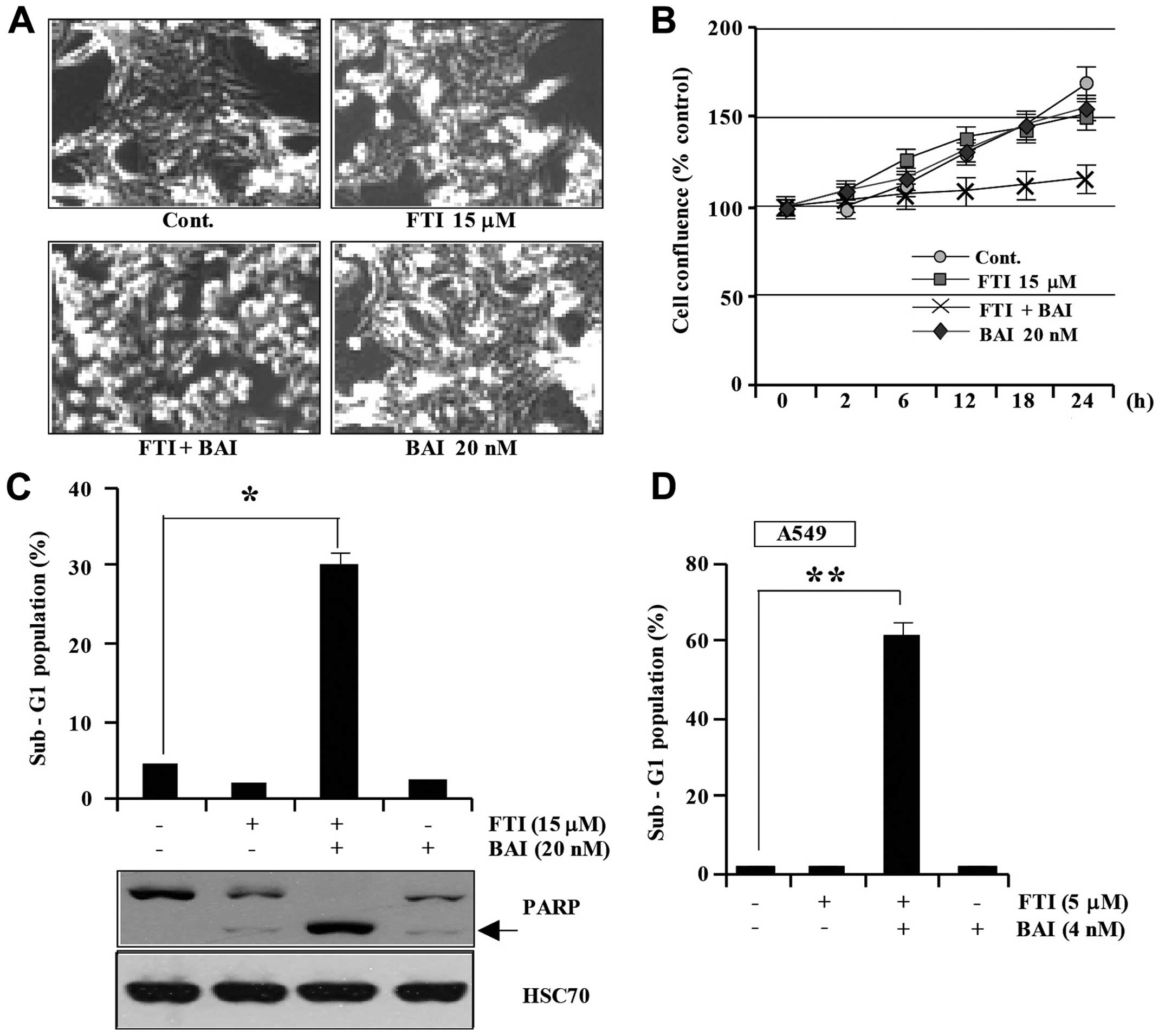
alone, LB42708 alone, or BAI plus LB42708. As shown in Fig. 1A, the combined treatment of Caki
cells with BAI and LB42708 induced morphological features of
apoptosis including cell shrinkage, rounding, and detachment of the
cell from the plate and marked inhibited proliferation (Fig. 1A). Additionally, co-treatment with
BAI and LB42708 inhibited cell viability, while BAI treatment alone
or LB42708 treatment alone did not reduce cell viability (Fig. 1B). Furthermore, we analyzed the
occurrence of apoptosis in Caki cells using flow cytometric
analysis to detect hypodiploid cell populations. As shown in
Fig. 1C, co-treatment of Caki
cells with BAI and LB42708 resulted in a significantly increased
accumulation of sub-G1 phase cells, whereas treatment with BAI
alone or LB42708 alone rarely increased accumulation of sub-G1
phase cells. Exposure to co-treatment of BAI and LB42708 led to
increased cleaved form of PARP (Fig.
1C). To generalize these phenomena, we investigated whether BAI
enhances LB42708-induced apoptosis in a synergistic fashion in
another human cancer cell type. The combined treatment strongly
induced apoptosis in A549 cells (Fig.
1D). These results suggest that BAI can sensitize various
malignant cancer cells to LB42708-induced apoptosis.
BAI plus LB42708-induced apoptosis is not
associated with ER stress or ROS generation
Endoplasmic reticulum (ER) stress-mediated apoptosis
is a well known mechanism of cell death (20). So, to evaluate whether BAI plus
LB42708-induced apoptosis is involved in ER stress, cyclosporine A
(CsA) was used as a potent inhibitor of ER stress-induced
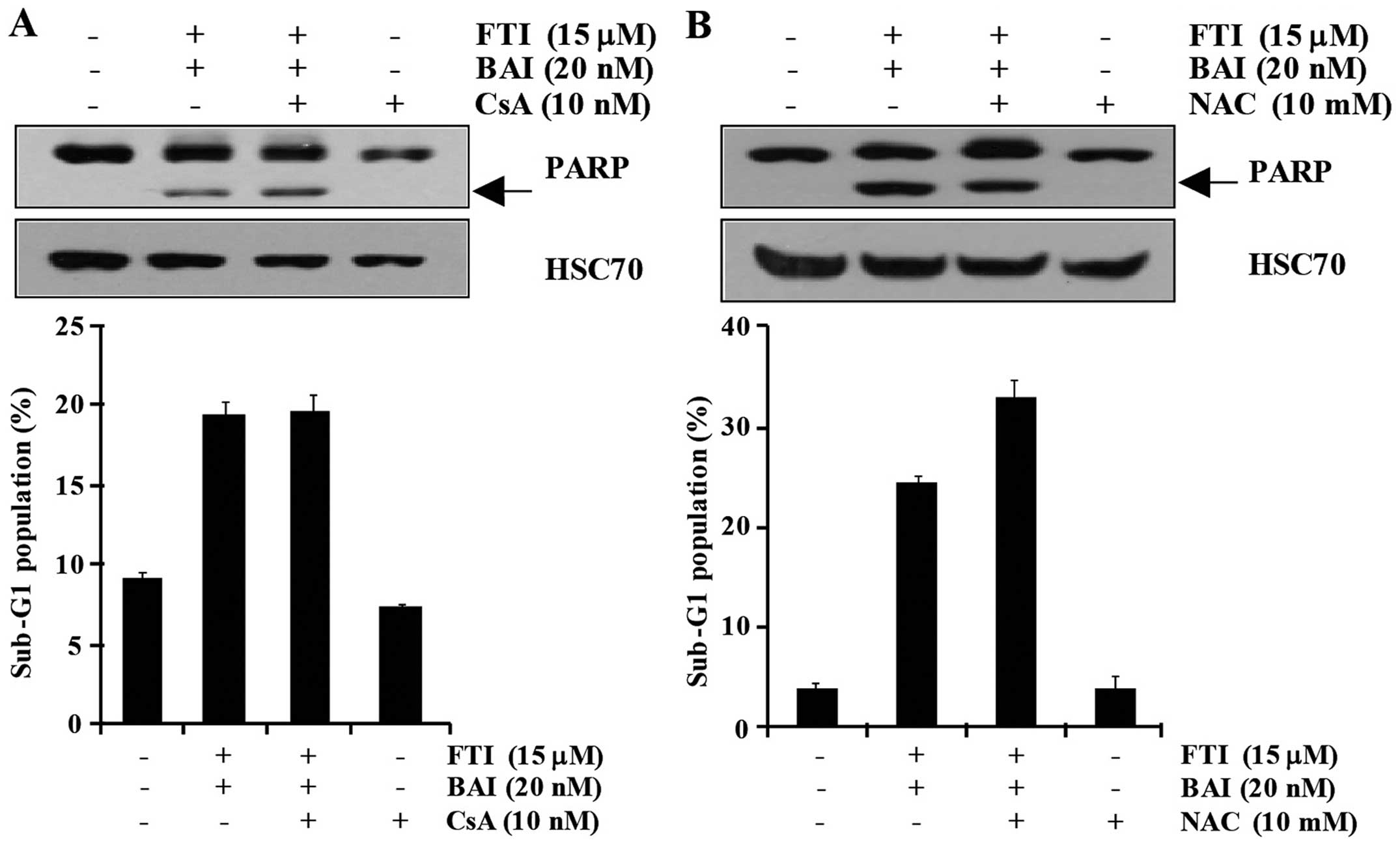
apoptosis. As shown in Fig. 2A,
the apoptosis induced by the combined treatment with BAI and
LB42708 similarly occurred in CsA-pretreated cells. Moreover, PARP
cleavage was not diminished by pretreatment of CsA (Fig. 2A). Reactive oxygen species (ROS),
natural byproducts of the normal metabolism of oxygen, play an
important role in apoptosis under both physiologic and pathologic
conditions (21). Therefore, we
examined whether ROS generation is involved in BAI plus
LB42708-induced apoptosis. As shown in Fig. 2B, pretreatment with
N-acetylcysteine (NAC) only slightly inhibited BAI plus
LB42708-induced apoptosis and PARP cleavage. Therefore, these
results clearly indicate that the combination of BAI and
LB42708-induced apoptosis is not associated with ER stress and ROS
generation.
BAI plus LB42708-induced apoptosis is
mediated by DEVDase-dependent pathway
To address the significance of caspase activation in
the combination of BAI and LB42708- induced apoptosis, we used a
general and potent inhibitor of caspases, z-VAD-fmk
(benzyloxycarbonyl-Val-Ala-Aspfluoromethyl ketone). As shown in
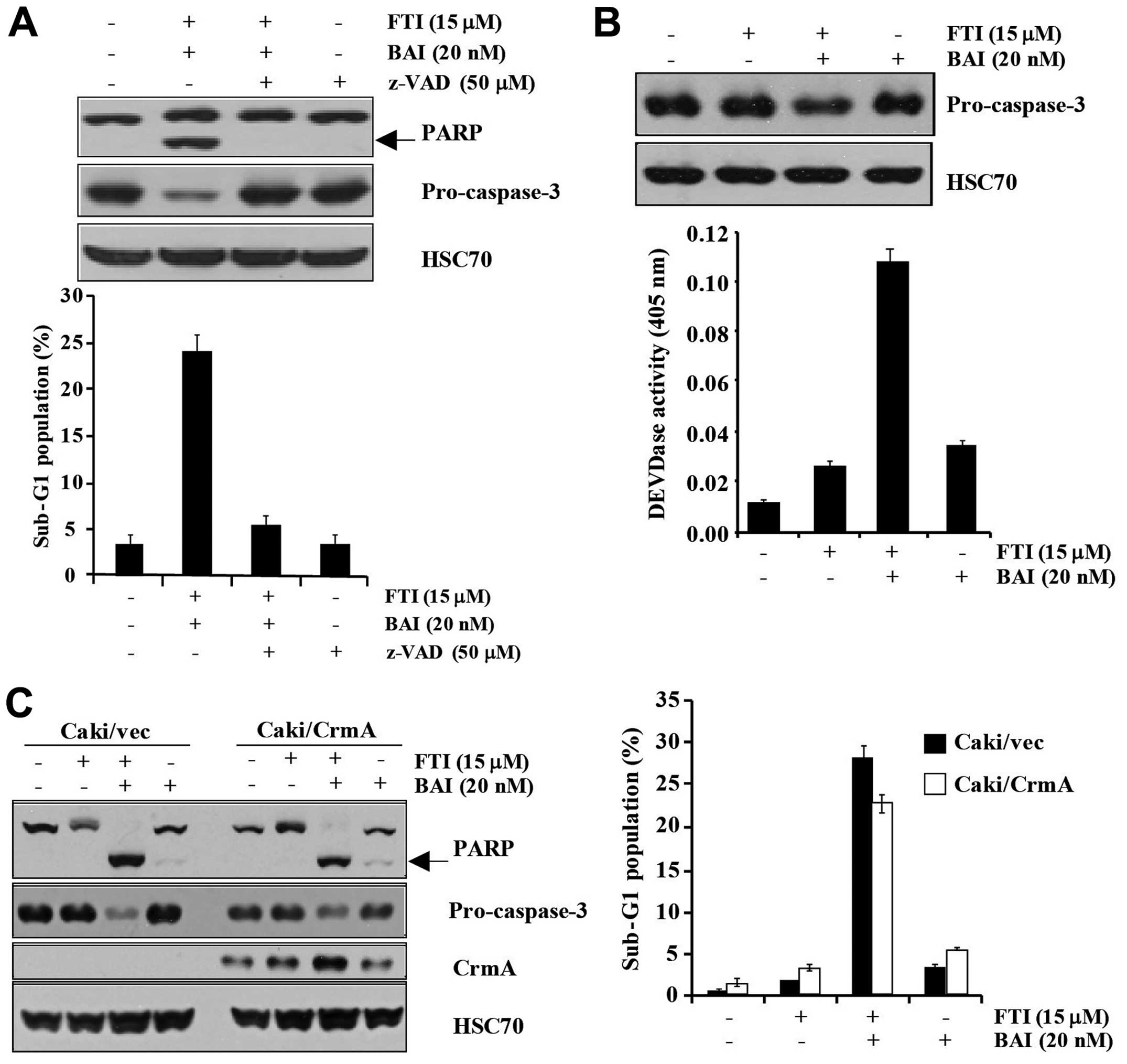
Fig. 3A, co-treatment of BAI and
LB42708-induced apoptotic population was markedly inhibited by
pretreatment with z-VAD-fmk. Additionally, the degradation of
pro-caspase-3 and PARP was completely blocked by pretreatment of
z-VAD-fmk (Fig. 3A). Not only did
the BAI plus LB42708 induce increased DEVDase activity, but it also
induced the degradation of pro-caspase-3 (Fig. 3B). These results suggest that BAI
plus LB42708-induced apoptosis is mediated by the
caspase-3-dependent pathway. To investigate which specific caspase
is associated in BAI plus LB42708-induced apoptosis, CrmA
overexpressing Caki cells were used. Overexpression of CrmA could
not attenuate the apoptosis induced by BAI plus LB42708 (Fig. 3C). Collectively, our data suggested
that BAI plus LB42708-induced apoptosis is involved in activation
of caspase-3 and -7, with only slight activation of caspase-1, -4,
-5, -8, -9 and -10.
Modulation of mitochondrial transmembrane
potential in BAI plus LB42708-induced apoptosis
The induction of cell death is generally associated
with, and probably mediated, by perturbations of the mitochondrial
function, a manifestation of which is the dissipation of the
transmembrane potential (ΔΨm). Hence, we wanted to
analyze ΔΨm during apoptosis induction in BAI plus
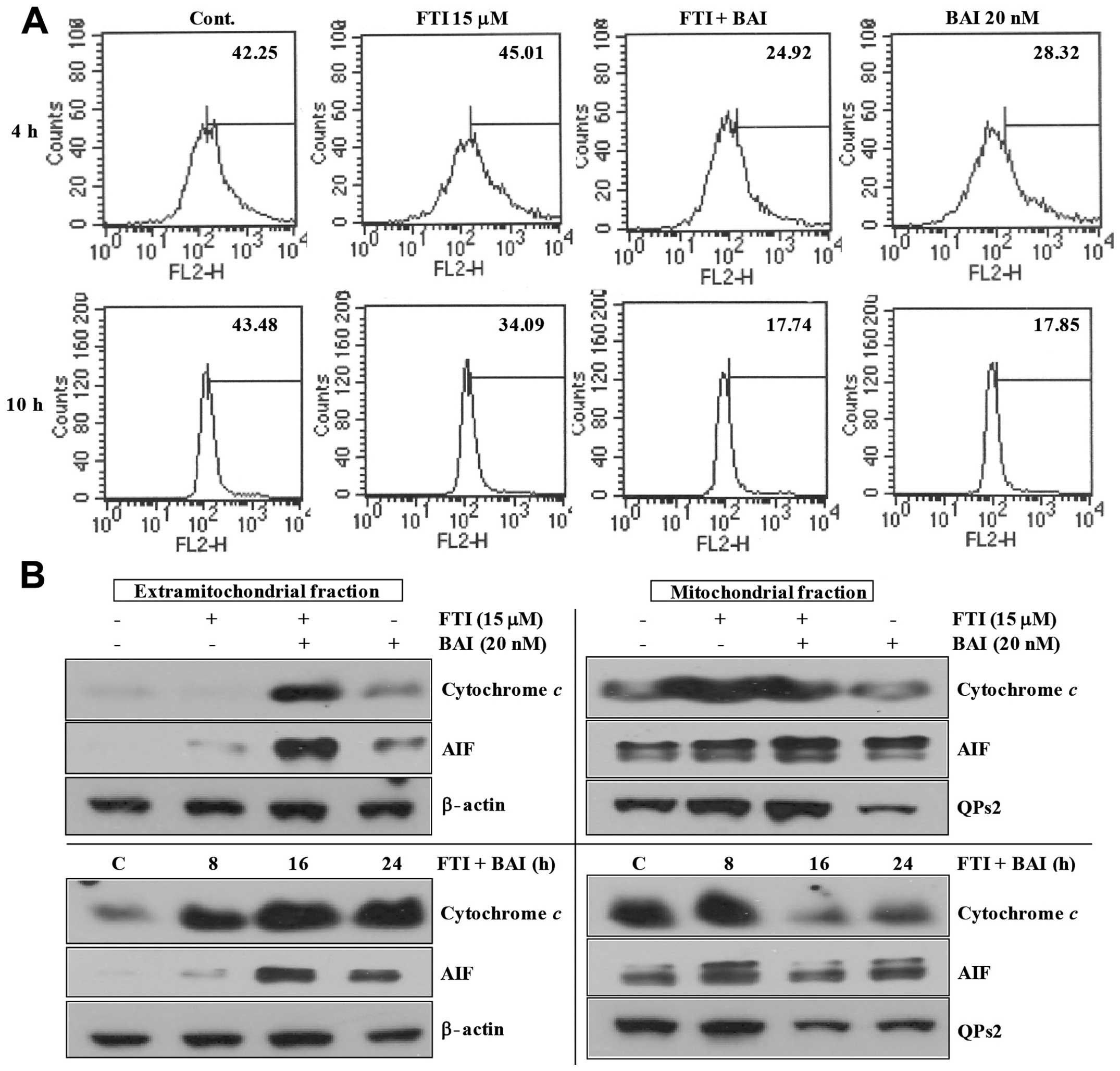
LB42708-treated Caki cells. As shown in Fig. 4A, co-treatment of BAI and LB42708
markedly decreased transmembrane potential (ΔΨm). It has
been reported that mitochondria play an important role in apoptosis
by releasing apoptogenic effectors such as cytochrome c and
apoptosis-inducing factor (AIF) (22). As shown in Fig. 4B, co-treatment of BAI and LB42708
remarkably induced time-dependent release of cytochrome c
and AIF into the cytoplasm. Taken together, these results indicate
that mitochondria may have an important role in BAI plus
LB42708-induced apoptosis.
Modulation of IAP family, Bcl-2 family,
and c-FLIPs in BAI plus LB42708-induced apoptosis
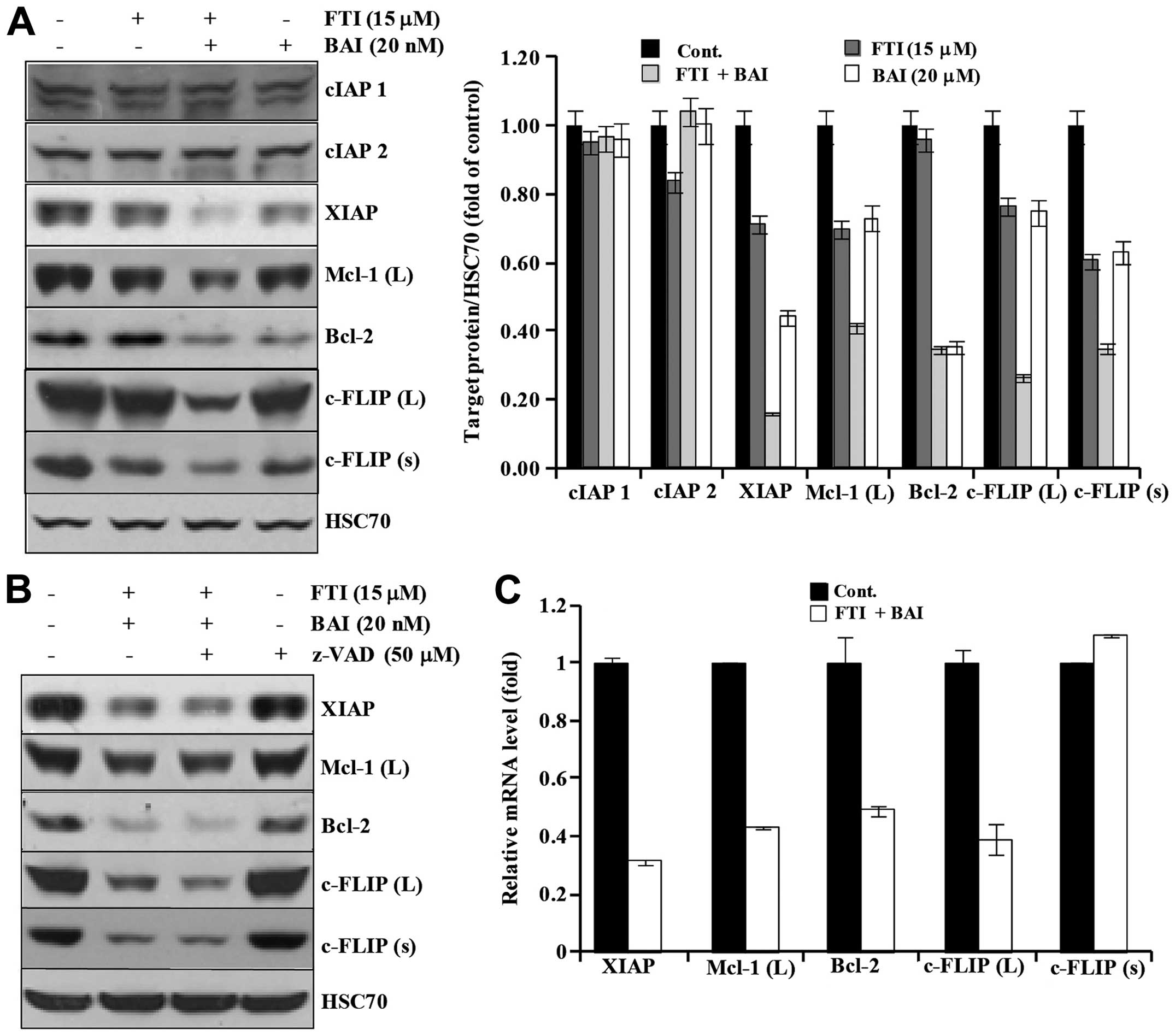
Next, we investigated whether co-treatment of BAI
and LB42708 could modulate the expression of IAP family, Bcl-2
family, and c-FLIPs proteins. As shown in Fig. 5A, BAI plus LB42708 did not alter
the expression levels of IAP family proteins such as cIAP1 and
cIAP2, but XIAP. Moreover, the expression of Mcl-1 (L), Bcl-2,
c-FLIP (L), and c-FLIP (s) were markedly decreased in the BAI plus
LB42708-treated cells (Fig. 5A).
To further examine whether the downregulation of apoptosis-related
proteins by co-treatment of BAI and LB42708 is associated with
activation of caspases, a pan-caspase inhibitor, z-VAD-fmk was
used. As shown in Fig. 5B,
pretreatment with z-VAD-fmk had no effect on reduction of those
proteins by BAI plus LB42708, implying that the downregulation of
those proteins induced by BAI plus LB42708 is not associated with
the caspase activity. This led us to promptly identify the effect
of co-treatment with BAI and LB42708 on transcriptional regulation
of XIAP, Mcl-1 (L), c-FLIP (L), c-FLIP (s) and Bcl-2 in Caki cells.
As shown in Fig. 5C, results of
real-time PCR analysis revealed that XIAP, Mcl-1 (L), c-FLIP (L)
and Bcl-2 transcripts were remarkably reduced in BAI plus
LB42708-treated Caki cells. However, c-FLIP (s) mRNA expression
level was not altered in BAI plus LB42708-treated Caki cells. These
results suggested that co-treatment with BAI and LB42708
downregulates XIAP, Mcl-1 (L), c-FLIP (L), and Bcl-2 at their
transcriptional level and c-FLIP (s) at post-transcriptional level
in Caki cells.
The downregulations of Bcl-2 and c-FLIP
(L) are associated with BAI plus-LB42708-induced apoptosis
To confirm the functional role played by
downregulated apoptosis regulatory proteins in BAI plus
LB42708-induced apoptosis, we employed Caki renal carcinoma cells
engineered for overexpression of Mcl-1 (L), c-FLIP (L), c-FLIP (s),
and Bcl-2 (Fig. 6A–D). For the
control model, vector-transfected control cells (Caki/vector) were
engineered (Fig. 6A–D). As shown
in Fig. 6A and B, overexpression
of Mcl-1 (L) and c-FLIP (s) could not attenuate the apoptosis
induced by co-treatment with BAI and LB42708. Collectively, these
results indicate that downregulations of Mcl-1 (L) and c-FLIP (s)
is not associated with the BAI plus LB42708-induced apoptosis in
Caki cells. Next, to examine the functional significance of BAI
plus LB42708- induced XIAP downregulation, we employed the siRNA
duplex against XIAP mRNA. Caki cells were transfected with XIAP
siRNA and co-treated with or without BAI and LB42708. Immunoblot
analysis demonstrated that transfection of siRNA against XIAP
resulted in a suppression of XIAP expression in Caki cells as
compared to cells transfected with control GFP siRNA (Fig. 6C). Under these conditions, the BAI
plus LB42708-induced accumulation of the sub-G1 phase and PLC-γ1
cleavage were similar in cells transfected with XIAP siRNA as
compared to control siRNA-transfected cells (Fig. 6C). Thus, these results suggest that
downregulation of XIAP protein is not associated with the BAI plus
LB42708- induced apoptosis in Caki cells. It has been reported that
Bcl-2 is an anti-apoptotic member of the family and its aberrant
expression has been linked to a variety of different cancers, and
cancer cell lines (23). Moreover,
c-FLIP has been found to act as a survival factor and to be
overexpressed in several types of cancers (24). To investigate whether the decreased
expression levels of Bcl-2 and c-FLIP (L) are important to induce
apoptosis in BAI plus LB42708-co-treated Caki cells, we established
Bcl-2 and c-FLIP (L) overexpressing cells. As shown in Fig. 6D, Caki/c-FLIP (L) cells treated
with BAI plus LB42708 partially inhibited apoptosis in comparison
with Cali/vector cells. Overexpression of Bcl-2 significantly
blocked the apoptosis induced by co-treatment with BAI and LB42708.
Additionally, PARP cleavage was attenuated by Bcl-2 overexpression
(Fig. 6E). Therefore, these
results indicate that downregulations of mainly Bcl-2 and partially
c-FLIP (L) induced by BAI plus LB42708 play a critical role in the
synergistic effect in growth inhibition of Caki cell line.
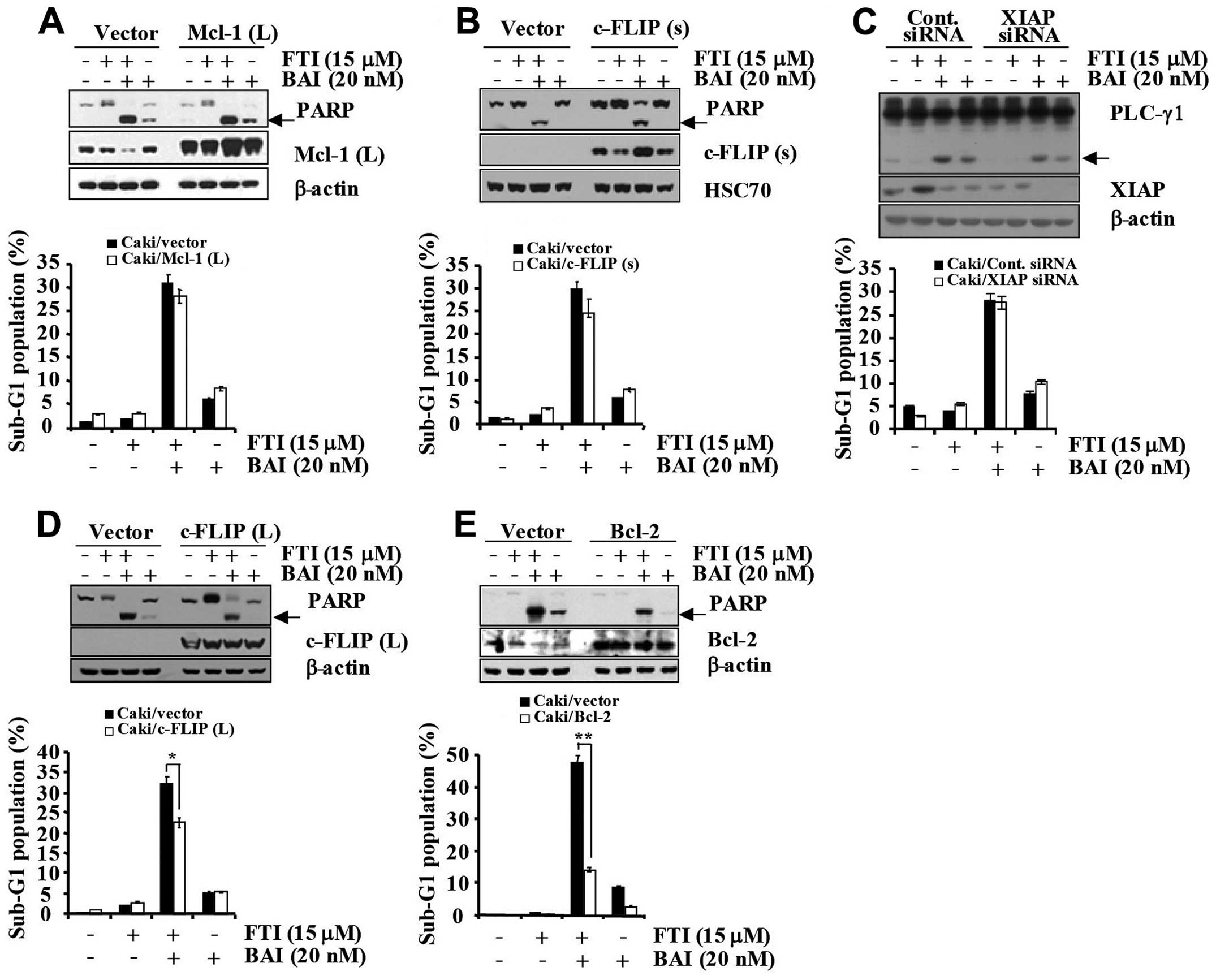 | Figure 6BAI plus LB42708 induces apoptosis
mainly via Bcl-2 and partially by c-FLIP (L) dependent pathway in
Caki cells. (A) Caki/vector and Caki/Mcl-1 (L) cells were treated
with BAI in the presence or absence of LB42708 for 24 h. Whole cell
lysates obtained from Caki cells stably transfected with a Mcl-1
(L) expression vector or the empty vector were subjected to
SDS-PAGE, transferred to membranes, and immunoblotted using the
respective antibody. Cleaved form of PARP is indicated by an arrow.
β-actin was used as a protein loading control (A, upper panel). The
sub-G1 fraction was measured by flow cytometry (A, lower panel) as
an indicator of the level of apoptosis. (B) Caki/vector and
Caki/c-FLIP (s) cells were treated with BAI in the presence or
absence of LB42708 for 24 h. Whole cell lysates obtained from Caki
cells stably transfected with a c-FLIP (s) expression vector or the
empty vector were subjected to SDS-PAGE, transferred to membranes,
and immunoblotted using the respective antibody. Cleaved form of
PARP is indicated by an arrow. HSC70 was used as a protein loading
control (B, upper panel). The sub-G1 fraction was measured by flow
cytometry (B, lower panel) as an indicator of the level of
apoptosis. (C) Caki cells were transfected with XIAP siRNA or GFP
siRNA. Thirty hours after transfection, cells were treated with BAI
in the presence or absence of LB42708 for 24 h. Whole cell lysates
obtained from the Caki/Cont siRNA or Caki/XIAP siRNA cells were
subjected to SDS-PAGE, transferred to membranes, and immunoblotted
using the respective antibody. Cleaved form of PLC-γ1 is indicated
by an arrow. β-actin was used as a protein loading control (C,
upper panel). The sub-G1 fraction was measured by flow cytometry
(C, lower panel) as an indicator of the level of apoptosis. (D)
Caki/vector and Caki/c-FLIP (L) cells were treated with BAI in the
presence or absence of LB42708 for 24 h. Whole cell lysates
obtained from Caki cells stably transfected with a c-FLIP (L)
expression vector or the empty vector were subjected to SDS-PAGE,
transferred to membranes, and immunoblotted using the respective
antibody. Cleaved form of PARP is indicated by arrow. β-actin was
used as a protein loading control (D, upper panel). The sub-G1
fraction was measured by flow cytometry (D, lower panel) as an
indicator of the level of apoptosis. (E) Caki/vector and Caki/Bcl-2
cells were treated with BAI in the presence or absence of LB42708
for 24 h. Whole cell lysates obtained from Caki cells stably
transfected with a Bcl-2 expression vector or the empty vector were
subjected to SDS-PAGE, transferred to membranes, and immunoblotted
using respective antibody. Cleaved form of PARP is indicated by an
arrow. β-actin was used as a protein loading control (E, upper
panel). The sub-G1 fraction was measured by flow cytometry (E,
lower panel) as an indicator of the level of apoptosis. (D and E)
The values represent the mean ± SD from three independent samples.
*P<0.005 compared to control. **P<0.005
compared to control. Data are mean values from three independent
experiments and bars represent the standard deviations. |
Discussion
In the present study, we have investigated the
synergistic effect of BAI plus a FTI, LB42708 in human renal cell
carcinoma Caki cell line. We showed that the combined treatment of
BAI and LB42708 induces significant apoptosis in Caki cells. In
this process, the activation of caspases, the release of cytochrome
c from mitochondria and downregulation of the
apoptosis-related proteins were noted. Importantly, we provided
evidence that the combination of BAI and LB42708 induced apoptosis
through the downregulation of Bcl-2 and c-FLIP (L).
Mutations of ras occur in approximately 30%
of all human cancers, leading to increased invasion and metastasis,
and decreased apoptosis since mutant Ras proteins continuously
activate the downstream effector pathways such as cell
proliferation (25). Since
farnesylation is required for Ras activation, FTI has been
identified to suppress farnesylation of Ras protein, which play a
major role in the proliferation and differentiation of transformed
cells (26) and, thus FTI has been
investigated as a promising cancer therapeutic drug (11–13).
There is accumulating evidence demonstrating the additive or
synergistic effects of FTIs when used in combination with other
drugs (18,27,28)
or especially the Cdk inhibitors, olomoucine and roscovitine
(18,29). Thus, we investigated whether
co-treatment of the novel cdk inhibitor BAI and the novel FTI
LB42708 could have synergistic effect on induction of apoptosis in
human cancer cells.
In the initial experiments, we have demonstrated
that co-treatment with BAI and LB42708 shows strong growth
inhibitory effects on Caki and A549 cells (Fig. 1). Importantly, further biochemical
analyses for apoptotic marker such as accumulation of sub-G1 phase
cells illustrated that co-treatment with BAI and LB42708 strongly
induces apoptosis in Caki cells (Fig.
1C). Recently, it is shown that reduction of cancer cell
viability induced by manumycin A, an inhibitor of
farnesyltransferase is correlated with ER stress (30). Furthermore, it has been revealed
that a Cdk inhibitor, flavopiridol induced ER stress (31). Therefore, we checked whether BAI
plus LB42708 can induce ER stress-mediated apoptosis in Caki cells.
At the present study, however, CsA pretreatment did not inhibit BAI
plus LB42708-induced apoptosis in Caki cells (Fig. 2A). These results imply that BAI
plus LB42708 do not induce ER stress-mediated apoptosis in Caki
cells. Reactive oxygen species (ROS) is a very important mediator
of apoptosis in various cancer cells (32–34).
Several reports have demonstrated that a FTI induces apoptosis via
induction of reactive oxygen species (ROS), which mediated DNA
damage (35,36). Thus, we evaluated whether BAI and
LB42708-mediated apoptosis is associated with ROS generation. In
our study, BAI plus LB4270-induced apoptosis was not associated
with the ROS generation (Fig.
2B).
Next, we elucidated which specific caspase is
involved in BAI plus LB42708-induced apoptosis. z-VAD-fmk can
irreversibly inhibit various caspases, such as caspase-1, -3
(37,38), -7 (39), -8 (40), and -9 (41). Additionally, CrmA has been revealed
to be a potent selective inhibitor of caspase-1, -4 and -5 (group I
caspases) and most group III caspases (caspase-8, -9, and -10)
(42,43). As shown in Fig. 3, z-VADfmk almost completely
inhibited BAI plus LB42708-induced apoptosis, however,
CrmA-overexpressing cells did not attenuate the proportion of
apoptosis induced by BAI plus LB42708. Therefore, these results
suggest that the activation of caspase-3 or -7 is mostly involved
in BAI plus LB42708- induced apoptosis.
It has been reported that a cdk inhibitor,
roscovitine synergized with FTI to release cytochrome c from
mitochondria and enhanced FTI-induced apoptosis (18). In this study, we verified whether
BAI plus LB42708 can induce cytochrome c release from
mitochondria. As shown in Fig. 4,
co-treatment of BAI and LB42708 induced decreased transmembrane
potential and increased cytochrome c release from
mitochondria.
Evidence suggests that the members of IAP and/or
Bcl-2 family are involved in apoptosis of cancer cells. XIAP, a
member of the IAP family, plays an important role in cellular
survival by modulating death-signaling pathways at the
post-mitochondrial level (44). It
has been reported that Mcl-1 is a member of the Bcl-2 family and
has an anti-proliferative effect (45). Additionally, c-FLIP is known to be
an inhibitory protein of death receptor-mediated apoptosis via
inhibition of caspase-8 activation as well as mitochondria-mediated
apoptosis induced by chemotherapeutic agents in cancer cells
(46,47). However, downregulation of Mcl-1
(L), c-FLIP (s), and XIAP proteins is likely to be unrelated to the
BAI plus LB42708-induced apoptosis and/or growth inhibition because
overexpression or knockdown of the proteins in the presence of BAI
plus LB42708 failed to rescue or enhance BAI plus LB42708-induced
apoptosis (Fig. 6). However, in
contrast to c-FLIP (s), c-FLIP (L) overexpression partially blocked
apoptosis induced by BAI plus LB42708 (Fig. 6D). Evidence recently indicated that
Bcl-2 is a pro-survival member of the family and its aberrant
expression has been implicated in cancer (48). In the present study, Bcl-2 played
an important role in BAI plus LB42708-induced apoptosis of Caki
cells.
Next, we investigated whether which specific
sensitizing mechanism of BAI on LB42708-mediated apoptosis is
involved. While BAI treatment alone had no effect on apoptosis
(Fig. 1B and C), BAI alone
treatment markedly downregulated expression levels of XIAP, Mcl-1
(L), c-FLIP (L), c-FLIP (s), and Bcl-2 proteins (Fig. 5A). However, we found that BAI alone
treatment hardly reduced the expression of XIAP, c-FLIP (L), c-FLIP
(s), and Bcl-2 mRNA in comparison with the decreased levels of each
protein, except Mcl-1 (L) (data not shown). Among these proteins,
overexpression of c-FLIP (L) partially inhibited BAI plus
LB42708-induced apoptosis (Fig.
6D) and overexpression of Bcl-2 significantly attenuated the
apoptosis induced by co-treatment with BAI and LB42708 in Caki
cells (Fig. 6E). Although, further
assessment is needed to confirm the regulating mechanism of BAI on
the apoptosis-related protein, BAI probably sensitized
LB42708-mediated apoptosis mainly through downregulation of Bcl-2
and partially via c-FLIP (L) at post-transcriptional level in Caki
cells.
Collectively, an important finding in this study is
that the novel Cdk inhibitor BAI synergistically with the FTI
LB42708 induce apoptosis of human cancer cell lines such as Caki
and A549. Therefore, the combination of BAI and LB42708 exerting
synergistic effects on growth of cancer suggest that it could be
applied as a new cancer therapeutic strategy.
Acknowledgements
This study was supported by Basic Science Research
Program through the National Research Foundation of Korea (NRF)
funded by the Ministry of Education, Science and Technology
(2012R1A1A2004758).
References
|
1
|
Malumbres M and Barbacid M: To cycle or
not to cycle: a critical decision in cancer. Nat Rev Cancer.
1:222–231. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lee J, Choi H, Kim KH, et al: Synthesis
and biological evaluation of 3,5-diaminoindazoles as
cyclin-dependent kinase inhibitors. Bioorg Med Chem Lett.
18:2292–2295. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shin HC, Song DW, Baek WK, et al:
Anticancer activity and differentially expressed genes in head and
neck cancer cells treated with a novel cyclin-dependent kinase
inhibitor. Chemotherapy. 55:353–362. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kim S, Lee J, Jang BC, Kwon TK and Park
JW: BAI, a novel cyclin-dependent kinase inhibitor induces
apoptosis in A549 cells through activation of caspases and
inactivation of Akt. J Cell Biochem. 114:282–293. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bharate SB, Singh B and Vishwakarma RA:
Modulation of K-Ras signaling by natural products. Curr Med Chem.
19:2273–2291. 2012.PubMed/NCBI
|
|
6
|
Almoguera C, Shibata D, Forrester K,
Martin J, Arnheim N and Perucho M: Most human carcinomas of the
exocrine pancreas contain mutant c-K-ras genes. Cell. 53:549–554.
1988. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nelson MA, Wymer J and Clements N Jr:
Detection of K-ras gene mutations in non-neoplastic lung tissue and
lung cancers. Cancer Lett. 103:115–121. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gibbs JB, Oliff A and Kohl NE:
Farnesyltransferase inhibitors: Ras research yields a potential
cancer therapeutic. Cell. 77:175–178. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tamanoi F: Inhibitors of Ras
farnesyltransferases. Trends Biochem Sci. 18:349–353. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Travis J: Novel anticancer agents move
closer to reality. Science. 260:1877–1878. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Carloni V, Vizzutti F and Pantaleo P:
Farnesyltransferase inhibitor, ABT-100, is a potent liver cancer
chemopreventive agent. Clin Cancer Res. 11:4266–4274. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lin NH, Wang L, Cohen J, et al: Synthesis
and biological evaluation of
4-[(3-methyl-3H-imidazol-4-yl)-(2-phenylethynyl-benzyloxy)-
methyl]-benzonitrile as novel farnesyltransferase inhibitor. Bioorg
Med Chem Lett. 13:3821–3825. 2003.
|
|
13
|
Lin NH, Wang L, Wang X, et al: Synthesis
and biological evaluation of
1-benzyl-5-(3-biphenyl-2-yl-propyl)-1H-imidazole as novel
farnesyltransferase inhibitor. Bioorg Med Chem Lett. 14:5057–5062.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim HS, Kim JW, Gang J, et al: The
farnesyltransferase inhibitor, LB42708, inhibits growth and induces
apoptosis irreversibly in H-ras and K-ras-transformed rat
intestinal epithelial cells. Toxicol Appl Pharmacol. 215:317–329.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Balasis ME, Forinash KD, Chen YA, et al:
Combination of farnesyltransferase and Akt inhibitors is
synergistic in breast cancer cells and causes significant breast
tumor regression in ErbB2 transgenic mice. Clin Cancer Res.
17:2852–2862. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Krzykowska-Petitjean K, Malecki J, Bentke
A, Ostrowska B and Laidler P: Tipifarnib and tanespimycin show
synergic proapoptotic activity in U937 cells. J Cancer Res Clin
Oncol. 138:537–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sackova V, Kulikova L, Kello M, Uhrinova I
and Fedorocko P: Enhanced antiproliferative and apoptotic response
of HT-29 adenocarcinoma cells to combination of photoactivated
hypericin and farnesyltransferase inhibitor manumycin A. Int J Mol
Sci. 12:8388–8405. 2011. View Article : Google Scholar
|
|
18
|
Edamatsu H, Gau CL, Nemoto T, Guo L and
Tamanoi F: Cdk inhibitors, roscovitine and olomoucine, synergize
with farnesyltransferase inhibitor (FTI) to induce efficient
apoptosis of human cancer cell lines. Oncogene. 19:3059–3068. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee H, Lee J, Lee S, et al: A novel class
of highly potent, selective, and non-peptidic inhibitor of Ras
farnesyltransferase (FTase). Bioorg Med Chem Lett. 11:3069–3072.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hetz C: The unfolded protein response:
controlling cell fate decisions under ER stress and beyond. Nat Rev
Mol Cell Biol. 13:89–102. 2012.PubMed/NCBI
|
|
21
|
Simon HU, Haj-Yehia A and Levi-Schaffer F:
Role of reactive oxygen species (ROS) in apoptosis induction.
Apoptosis. 5:415–418. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Le Bras M, Rouy I and Brenner C: The
modulation of interorganelle cross-talk to control apoptosis. Med
Chem. 2:1–12. 2006.PubMed/NCBI
|
|
23
|
Bodur C and Basaga H: Bcl-2 inhibitors:
emerging drugs in cancer therapy. Curr Med Chem. 19:1804–1820.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ili CG, Brebi P, Tapia O, et al: Cellular
FLICE-like inhibitory protein long form (c-FLIPL) overexpression is
related to cervical cancer progression. Int J Gynecol Pathol.
32:316–322. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Khosravi-Far R and Der CJ: The Ras signal
transduction pathway. Cancer Metastasis Rev. 13:67–89. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cohen LH, Pieterman E, van Leeuwen RE, et
al: Inhibitors of prenylation of Ras and other G-proteins and their
application as therapeutics. Biochem Pharmacol. 60:1061–1068. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Moasser MM, Sepp-Lorenzino L, Kohl NE, et
al: Farnesyl transferase inhibitors cause enhanced mitotic
sensitivity to taxol and epothilones. Proc Natl Acad Sci USA.
95:1369–1374. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Smalley KSM and Eisen TG: Farnesyl
transferase inhibitor SCH66336 is cytostatic, pro-apoptotic and
enhances chemosensitivity to cisplatin in melanoma cells. Int J
Cancer. 105:165–175. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wesierska-Gadek J, Maurer M and Schmid G:
Inhibition of farnesyl protein transferase sensitizes human MCF-7
breast cancer cells to roscovitine-mediated cell cycle arrest. J
Cell Biochem. 102:736–747. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Singha PK, Pandeswara S, Venkatachalam MA
and Saikumar P: Manumycin A inhibits triple-negative breast cancer
growth through LC3-mediated cytoplasmic vacuolation death. Cell
Death Dis. 4:e4572013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mahoney E, Lucas DM, Gupta SV, et al: ER
stress and autophagy: new discoveries in the mechanism of action
and drug resistance of the cyclin-dependent kinase inhibitor
flavopiridol. Blood. 120:1262–1273. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kuo PL, Chen CY and Hsu YL:
Isoobtusilactone A induces cell cycle arrest and apoptosis through
reactive oxygen species/apoptosis signal-regulating kinase 1
signaling pathway in human breast cancer cells. Cancer Res.
67:7406–7420. 2007. View Article : Google Scholar
|
|
33
|
Yodkeeree S, Sung B, Limtrakul P and
Aggarwal BB: Zerumbone enhances TRAIL-induced apoptosis through the
induction of death receptors in human colon cancer cells: Evidence
for an essential role of reactive oxygen species. Cancer Res.
69:6581–6589. 2009. View Article : Google Scholar
|
|
34
|
Min KJ, Kim HS, Park EJ and Kwon TK:
Melatonin enhances thapsigargin-induced apoptosis through reactive
oxygen species-mediated upregulation of CCAAT-enhancer-binding
protein homologous protein in human renal cancer cells. J Pineal
Res. 53:99–106. 2012. View Article : Google Scholar
|
|
35
|
She M, Yang H, Sun L and Yeung SC: Redox
control of manumycin A-induced apoptosis in anaplastic thyroid
cancer cells: involvement of the xenobiotic apoptotic pathway.
Cancer Biol Ther. 5:275–280. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pan J, She M, Xu ZX, Sun L and Yeung SC:
Farnesyltransferase inhibitors induce DNA damage via reactive
oxygen species in human cancer cells. Cancer Res. 65:3671–3681.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Slee EA, Zhu H, Chow SC, MacFarlane M,
Nicholson DW and Cohen GM: Benzyloxycarbonyl-Val-Ala-Asp (OMe)
fluoromethylketone (Z-VAD.FMK) inhibits apoptosis by blocking the
processing of CPP32. Biochem J. 315:21–24. 1996.PubMed/NCBI
|
|
38
|
Yang B, El Nahas AM, Fisher M, et al:
Inhibitors directed towards caspase-1 and -3 are less effective
than pan caspase inhibition in preventing renal proximal tubular
cell apoptosis. Nephron Exp Nephrol. 96:e39–e51. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yang PM, Tseng HH, Peng CW, Chen WS and
Chiu SJ: Dietary flavonoid fisetin targets caspase-3-deficient
human breast cancer MCF-7 cells by induction of
caspase-7-associated apoptosis and inhibition of autophagy. Int J
Oncol. 40:469–478. 2012.PubMed/NCBI
|
|
40
|
Sawai H: Differential effects of caspase
inhibitors on TNF-induced necroptosis. Biochem Biophys Res Commun.
432:451–455. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Uchiyama R, Kawamura I, Fujimura T, et al:
Involvement of caspase-9 in the inhibition of necrosis of RAW 264
cells infected with Mycobacterium tuberculosis. Infect Immun.
75:2894–2902. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhou Q, Snipas S, Orth K, Muzio M, Dixit
VM and Salvesen GS: Target protease specificity of the viral serpin
CrmA. Analysis of five caspases. J Biol Chem. 272:7797–7800. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Garcia-Calvo M, Peterson EP, Leiting B,
Ruel R, Nicholson DW and Thornberry NA: Inhibition of human
caspases by peptide-based and macromolecular inhibitors. J Biol
Chem. 273:32608–32613. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Schimmer AD, Dalili S, Batey RA and Riedl
SJ: Targeting XIAP for the treatment of malignancy. Cell Death
Differ. 13:179–188. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zinkel S, Gross A and Yang E: BCL2 family
in DNA damage and cell cycle control. Cell Death Differ.
13:1351–1359. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Jonsson G, Paulie S and Grandien A: High
level of cFLIP correlates with resistance to death receptor-induced
apoptosis in bladder carcinoma cells. Anticancer Res. 23:1213–1218.
2003.PubMed/NCBI
|
|
47
|
Longley DB, Wilson TR, McEwan M, et al:
c-FLIP inhibits chemotherapy-induced colorectal cancer cell death.
Oncogene. 25:838–848. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Thomas S, Quinn BA, Das SK, et al:
Targeting the Bcl-2 family for cancer therapy. Expert Opin Ther
Targets. 17:61–75. 2013. View Article : Google Scholar
|