Introduction
The anthracycline antibiotic doxorubicin (DOX) is
one of the most effective chemotherapeutic agents in the treatment
of numerous solid tumors and hematological malignancies (1,2).
However, its clinical use has been compromised due to its
dose-dependent cardiotoxicity, which appears to be progressive and
permanent (1,3,4).
Since the first heart failure was reported in children treated with
DOX in 1967 (2), the
anthracyclines have been classified as the most common cardiotoxic
chemotherapeutic agents. Total dose of DOX within a patient’s
lifetime is restricted not to exceed 450–500 mg/m2
because cardiotoxicity is related to the cumulative dose (5). Thus, patients who have already
received the maximum cumulative dose of DOX cannot receive further
DOX therapy even if they may benefit from DOX treatment.
The precise molecular mechanisms underlying
anthracycline-induced cardiotoxicity are not fully understood
because the cause of cardiotoxicity is complex and multifactorial.
The most common hypothesis is that the formation of reactive oxygen
species (ROS) such as superoxide anion (•O2−)
and hydrogen peroxide (H2O2) cause oxidative
damage to the cellular components and membranes in heart tissue and
reduction of energy in cardiomyocytes, which ultimately lead to
cardiomyopathy and congestive heart failure (6–8). In
addition to the formation of ROS, more recent studies suggest that
cardiotoxicity is associated with DNA damage via DOX interaction
with nuclear DNA of cardiac cells, which interferes with DNA
replication and transcription, potentially triggering myocyte
apoptosis (9–11).
Since DOX-induced cardiotoxicity is a major limiting
factor in the use of DOX, new strategies to prevent or reverse the
cardiotoxic side-effects of DOX have been explored (12–14).
Several antioxidants or iron chelators including N-acetylcysteine,
ascorbic acid, and dexrazoxane have been shown to alleviate
anthracycline-induced cardiotoxicity by reducing the oxidative
stress (15–17). Besides antioxidants, lipid-lowering
reagents such as probucol and statins also exhibit favorable
cardioprotective effects against anthracycline-induced
cardiotoxicity (18–20).
Recently, deep sea water (DSW) has gained much
scientific interest for therapeutic intervention due to its
enrichment in nutrients and minerals. DSW is obtained from a clean
area at a depth of >200 m and is rich in minerals such as
calcium (Ca), magnesium (Mg), potassium (K), sodium (Na), zinc
(Zn), etc. (21). Now, it is well
recognized that DSW has health benefits in lowering of blood
cholesterol and preventing obesity and atherosclerosis (22,23).
Moreover, our previous studies showed the inhibitory effects of DSW
on the metastatic potential of human breast cancer cell lines
(24) and on the
carcinogen-induced expression of cyclooxygenase-2 (COX-2),
transforming growth factor-β (TGF-β), and urokinase plasminogen
activator (uPA) in HT-29 colorectal cancer cells (25). Although the precise mechanisms
mediating these biological activities of DSW have not been
clarified yet, it is presumed that its activities may be derived
from the combined ionic action of several minerals. In particular,
Mg and Ca may play important roles in mediating the biological
activities of DSW because they are the main mineral ions present in
DSW. Since it is well known that Mg plays important roles in
regulating cardiac muscle function and maintaining adequate
electrophysiology (26,27), the present study was directed to
evaluate the protective effect of DSW against DOX-induced
cardiotoxicity in H9c2 cardiomyocytes.
Materials and methods
Preparation of DSW
DSW was supplied by the Marine Deep Ocean Water
Application Research Center in the Korea Institute of Ocean Science
and Technology (Goseong, Gyeongsangnam-do, Korea). DSW was taken
from the sea in Goseong at a depth of 500 m and subjected to a
process of filtrations, reverse osmosis, and concentration by
electrolysis to achieve desalinated water and 4,000 hardness DSW.
Mg and Ca within DSW were present in the ratio of 3:1 and the
hardness of DSW was determined from the concentration of Ca and Mg
ions. The following equation was used to calculate the hardness of
DSW in this study: Hardness of DSW (mg/l) = Mg (mg/l) × 4.1 + Ca
(mg/l) × 2.5. DSW of hardness 1,500 was prepared by diluting
hardness 4,000 DSW with desalinated DSW (hardness 0) and dissolved
Dulbecco’s modified Eagle’s medium (DMEM) powder with 1%
antibiotic-antimycotic solution. Further serial dilutions were
performed to achieve hardness 200–800 DSW media from 1,500 hardness
DSW with desalinated media (hardness 0).
Cell culture
H9c2 rat cardiomyocytes were purchased from the
Korean Cell Line Bank (Seoul, Korea). Cells were cultured in DMEM
(WelGENE, Daegu, Korea) supplemented with 10% fetal bovine serum
(Invitrogen Life Technologies, Carlsbad, CA, USA) and 1%
antibiotic-antimycotic solution (WelGENE). MCF-7 and MDA-MB-231
human breast cancer cell lines were purchased from the Korean Cell
Line Bank. MCF-7 cells were cultured in DMEM supplemented with 10%
fetal bovine serum and 10 μg/ml insulin, while MDA-MB-231 cells
were cultured in DMEM supplemented with 10% fetal bovine serum
without insulin.
Cell treatment
After H9c2 cells were pre-treated with DSW of
various hardness for 24 h, 0.25 μM DOX (Sigma, St. Louis, MO, USA)
was added to the cells. Cells were further incubated for 24 h and
harvested for RNA isolation or preparation of protein lysates. For
inhibition of the PI3K/Akt-signaling pathway, cells were treated
with 10 μM LY294002 (LC Laboratories, Woburn, MA, USA) with 0.25 μM
DOX and cultured for 24 h before cell harvest.
Cell viability assay
Cells were seeded in 96-well plates and incubated at
37°C for 24 h. The cells were treated with conditioned media
containing DSW of various hardness (200, 400, 800, 1,500) for 24 h
prior to adding 0.25 μM DOX. Cells were further incubated for 24 or
48 h and their cell viabilities were measured by MTT assay (Sigma).
Absorbance at 570 nm was measured in a Multi-Detection Microplate
Reader (Molecular Devices, Sunnyvale, CA, USA).
Measurement of intracellular ROS
production
After cells were treated as indicated above, cells
were trypsinized and incubated with 20 μM 2′,7′-dichlorofluorescin
diacetate (DCF-DA) (Sigma) for 1 h at 37°C in the dark. After
incubation, cells were immediately washed and resuspended in PBS.
Intracellular ROS production was detected on a FACSCalibur (BD
Biosciences, San Jose, CA, USA) by the fluorescent intensity of DCF
measured at 525 nm.
Quantitative real-time PCR
The mRNA expression of multi-drug resistance protein
1 (MDR1) was determined by quantitative real-time PCR. Cells were
grown and treated in 6-well plates as indicated above. Total RNA
was extracted with easy-BLUE™ Total RNA Extraction kit (Intron
Biotechnology, Inc., Gyeonggi, Korea) and cDNA was synthesized with
reverse transcriptase (Takara Bio, Inc., Shiga, Japan). The
real-time PCR reactions were performed using QuantiMix SYBR-Green
kit (Philekorea, Daejeon, Korea) in Eco Real-Time PCR (Illumina,
San Diego, CA, USA). mRNA expression level of MDR1 was calculated
after normalizing with glyceraldehyde-3-phosphate dehydrogenase
(GAPDH). The utilized primer sequences were as follows: MDR1
forward, 5′-GATGGAATTGATAATGTGGACA-3′ and MDR1 reverse,
5′-GTACGTCGTCATCCAGAGC-3′; GAPDH forward,
5′-AACTTTGGCATCGTGGAAGG-3′ and GAPDH reverse,
5′-TACATTGGGGGTAGGAACAC-3′.
Western blot analysis
Cells were grown and treated in 6-well plates as
indicated above. Cells were lysed with RIPA buffer (50 mM NaCl, 1%
Triton X-100, 1% Na deoxycholate, 0.1% SDS, 50 mM Tris-HCl pH 7.5
and 2 mM EDTA). Phosphatase and protease inhibitor cocktail
(GenDEPOT, Barker, TX, USA) were added immediately before use.
Lysates were cleared of debris at 13,000 rpm for 10 min, and
protein concentrations were determined using bicinchoninic acid
reagent (Sigma). Proteins were separated by SDS-PAGE (8–15% gels)
and transferred onto polyvinylidene difluoride (PVDF) membranes at
100 V for 45 min. Membranes were blocked in 5% milk in TBS-Tween
(50 mM Tris-HCl, 150 mM NaCl, 0.1% Tween-20) for 1 h at room
temperature. The following primary antibodies were incubated with
blots overnight at 4°C: Anti-rabbit phospho-H2A histone family
member X (H2AX), phospho-p38, total-p38, phospho-protein kinase B
(Akt), total-Akt, phospho-extracellular signal-regulated kinase 1/2
(ERK1/2), total-ERK1/2, B-cell lymphoma-extra large (Bcl-xL),
cleaved cysteine-aspartic acid protease-3 (caspase-3), and
poly(ADP-ribose) polymerase (PARP) (Cell Signaling Technology,
Inc., Beverly, MA, USA). HRP-conjugated secondary anti-rabbit
antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA)
diluted 1:5,000 was incubated with blots for 1 h at room
temperature. Blots were developed using Luminescent Image Analyzer
LAS-4000 (Fujifilm, Tokyo, Japan).
Statistical analysis
The Student’s t-test was used for statistical
analysis of the data. P<0.05 was considered significant.
Results
DSW protects H9c2 rat cardiomyocytes from
DOX-induced cell death without interfering with anticancer effects
of DOX
To evaluate whether DSW itself has harmful effects
on normal cardiomyocyte cells, we first measured viabilities of
H9c2 rat cardiomyocytes after treatment with DSW of different
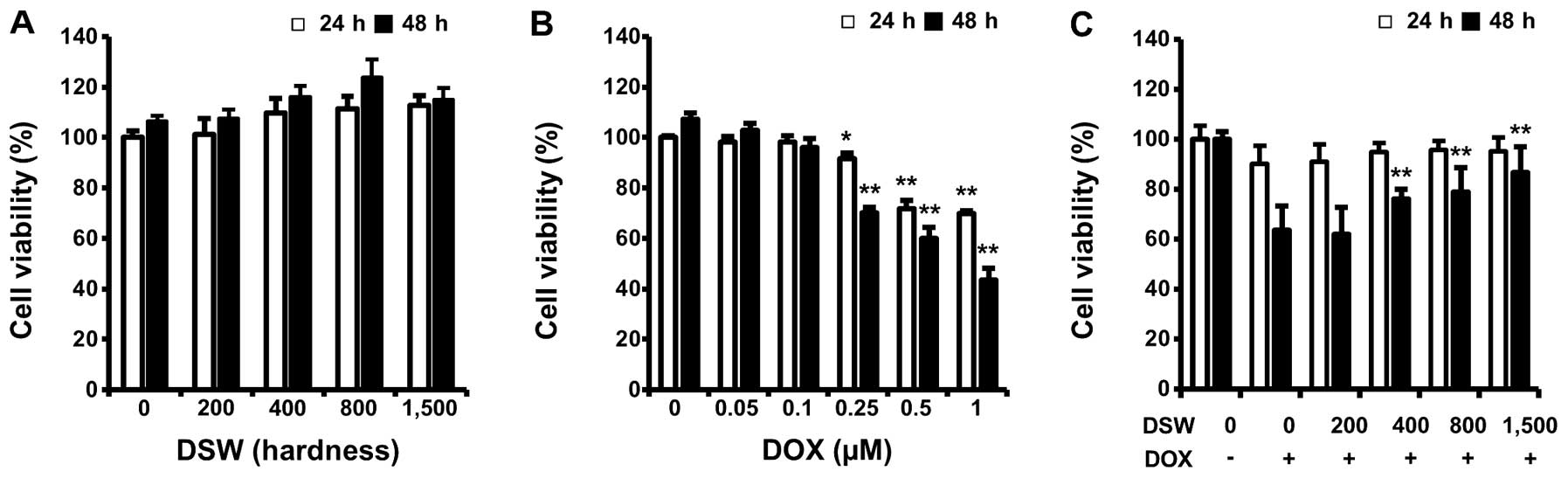
hardness for 24 or 48 h. As shown in Fig. 1A, cell proliferation was not
affected by treatment with DSW even when cells were treated with
DSW of 1,500 hardness for 48 h (Fig.
1A). However, treatment with DOX significantly reduced the
viability of cardiomyocytes dose-dependently (Fig. 1B). To investigate whether DSW is
able to protect cardiomyocytes from DOX-induced cell death, we
treated cells with conditioned media containing DSW of various
hardness for 24 h prior to adding 0.25 μM DOX and further incubated
cells for another 24 or 48 h before measuring cell viability by MTT
assay. DSW clearly protected H9c2 cardiomyocytes from DOX-induced
cell death, showing increased cell viability from 60 to >90% in
the presence of DSW of 1,500 hardness at 48 h (Fig. 1C).
Since DSW exhibited cardioprotection by inhibiting
DOX-induced cell death, we tested possible interference of DSW with
antitumor effects of DOX in the MCF-7 and MDA-MB-231 human breast
cancer cells. Interestingly, mildly enhanced antitumor effects of
DOX were observed in both MCF-7 and MDA-MB-231 cells in a
dose-dependent manner, exhibiting ~10% decrease of cell viability
at DSW of 1,500 hardness (Fig. 2A and
B). This result manifested that the antitumor effects of DOX
were not impaired by DSW. Taken together, our data suggested that
DSW provides cardioprotection in cardiomyocytes exposed to DOX,
without interfering with the antitumor activity of DOX in human
breast cancer cells.
Protective effect of DSW is associated
with DNA damage responses rather than ROS generation or expression
of MDR1
To elucidate the molecular mechanisms involved in
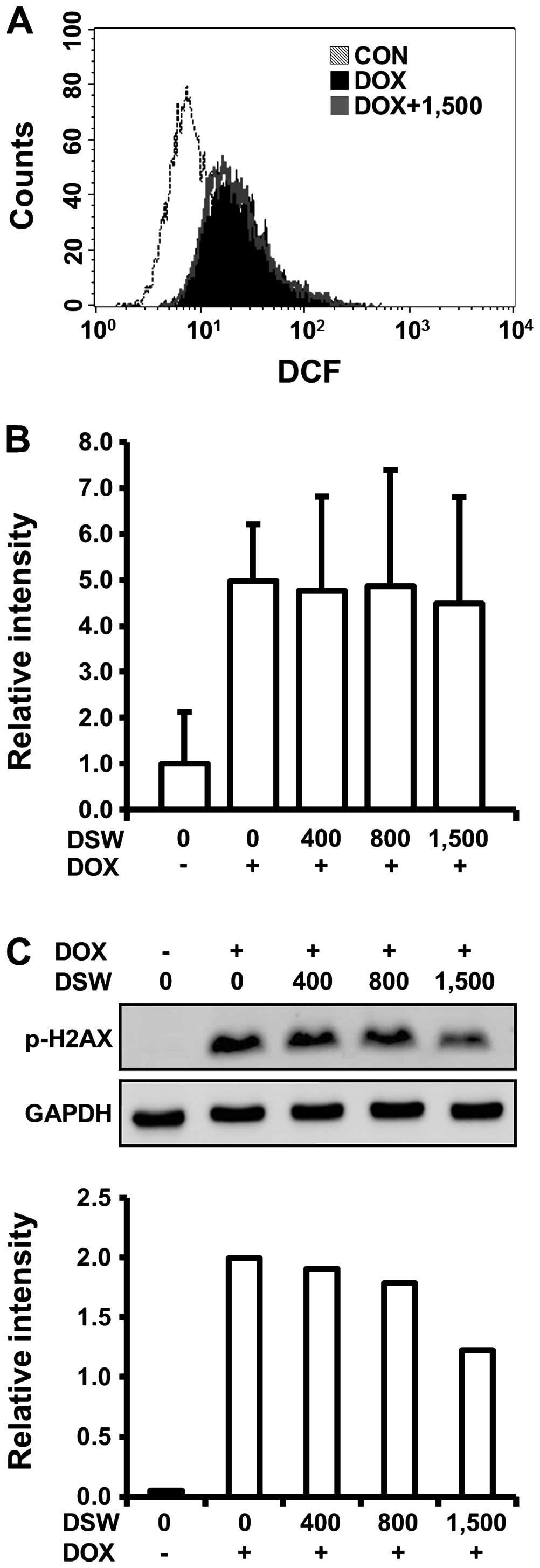
the cardioprotective effects of DSW, we first monitored the
generation of ROS in H9c2 cells treated with DOX and DSW since a
number of recent reviews described the involvement of ROS in the
mechanism of DOX-induced cardiotoxicity. In flow cytometry analysis
using DCF-DA reagent, which can be converted to fluorescent DCF in
a reaction with intracellular ROS, we observed that DOX caused a
right-shift of the fluorescence intensity of DCF signal compared to
untreated cells, confirming the generation of ROS by DOX. However,
this DOX-induced ROS generation was not diminished by pre-treatment
with DSW of 1,500 hardness, suggesting that the cardioprotective
effects of DSW are independent of ROS generation (Fig. 3A). We also tested whether DSW
enhanced the mRNA expression of drug transporter MDR1, which plays
an important role in the protection of cardiac tissue by inhibiting
accumulation of DOX within tissues (28). Although the expression of MDR1 was
induced by stimulation of the cells with DOX, its further induction
with DSW treatment was not observed, suggesting that DSW has no
effect on inhibiting accumulation of DOX within H9c2 cells
(Fig. 3B).
Several studies suggested that induction of DNA
damage is an early event in DOX-induced lethal cardiomyocyte injury
(10). Since H2AX is rapidly
phosphorylated in response to DNA damage and its phosphorylation is
frequently used as a marker for DNA damage (29), we analyzed H2AX phosphorylation
(γ-H2AX) by western blot analysis to test whether the
cardioprotective effects of DSW are associated with the response of
DNA damage in H9c2 cells. As expected, DOX exposure significantly
induced the phosphorylation of H2AX. However, pre-treatment with
DSW attenuated this response in a dose-dependent manner, exhibiting
~50% decreased phosphorylation of H2AX at DSW of 1,500 hardness
(Fig. 3C).
The inhibitory effect of DSW on
DOX-induced DNA damage subsequently attenuates apoptotic
signaling
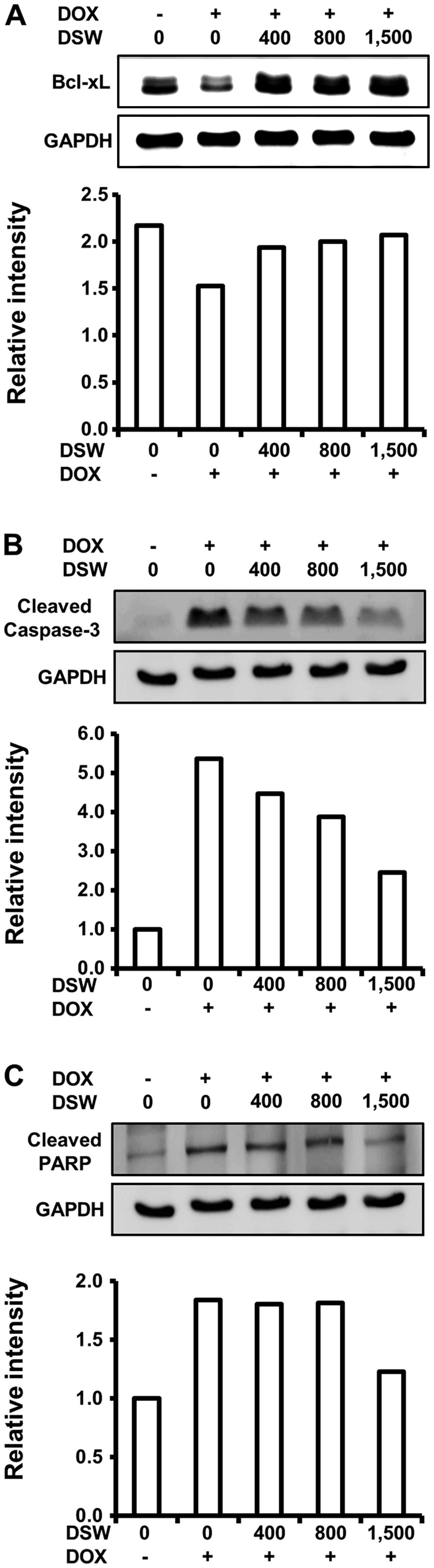
As DSW suppressed DOX-induced DNA damage, which
triggers the cell death program, we assessed the effects of DSW on
apoptosis signaling. We first analyzed the expression of Bcl-xL,
which is an anti-apoptotic protein that inhibits the release of
mitochondrial cytochrome c into the cytosol. Western blot
analysis demonstrated that DOX downregulated the expression of
Bcl-xL, which was effectively restored to control level by DSW of
1,500 hardness (Fig. 4A). We
further studied the inhibitory effect of DSW on DOX-triggered
apoptosis by evaluating the activation of caspase-3 and PARP
fragmentation. Treatment of cardiomyocytes with DOX resulted in a
remarkable increase in the levels of cleaved caspase-3, as well as
fragmentation of its substrate, PARP. However, DSW effectively
suppressed the activation of caspase-3 (Fig. 4B), leading to subsequent inhibition
of PARP fragmentation (Fig. 4C).
These data suggest that DSW can counteract DOX-triggered apoptosis
in H9c2 cells.
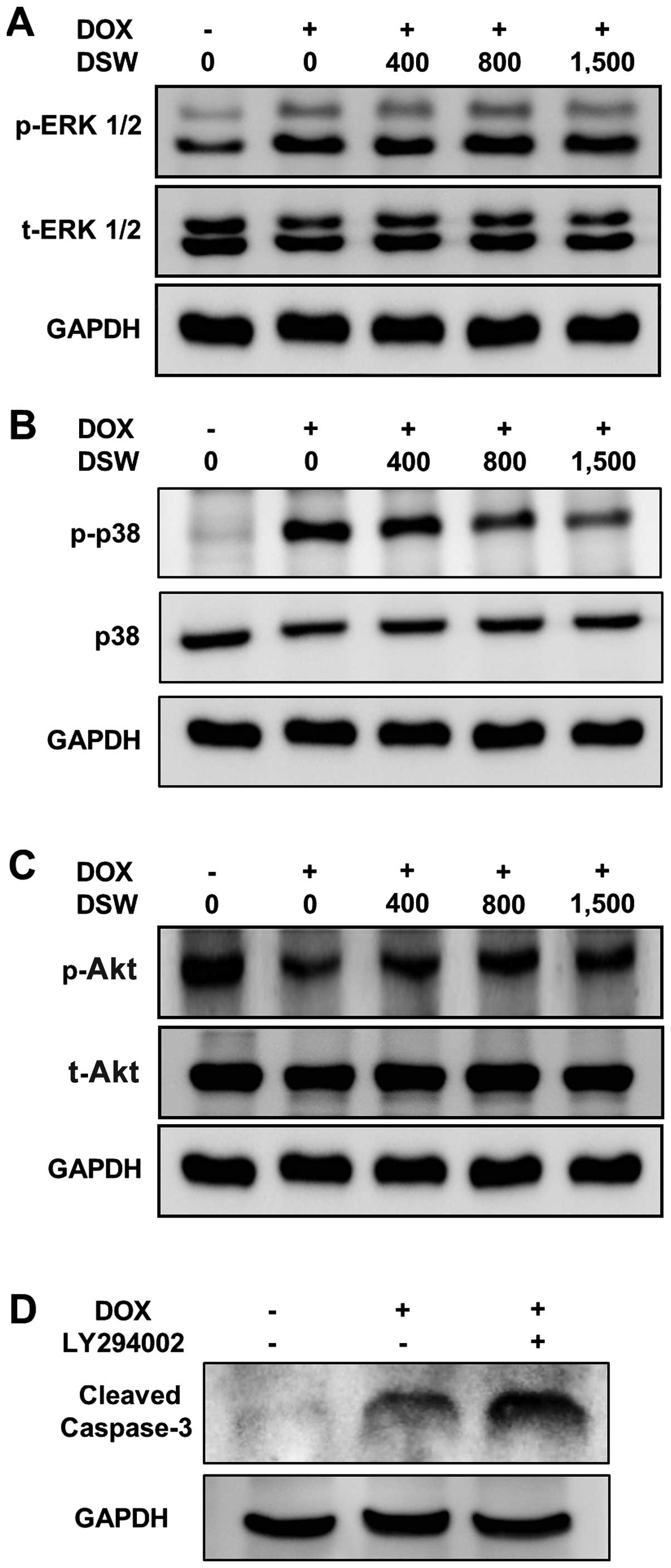
DSW rescues the Akt-signaling pathway to
protect cells from DOX-induced cell death
Several studies have shown that the PI3K/Akt- and
MAP kinase-signaling pathways are involved in DOX-induced apoptosis
(11,30,31).
Increased MEK-ERK1/2 activity is responsible for the DOX-induced
apoptosis (11), while increased
PI3K/Akt activity is protective against DOX-induced cardiomyocyte
apoptosis (31). Thus, to get more
detailed insights into the mechanism underlying the protective
effects of DSW against DOX-induced apoptosis, we further analyzed
the effects of DSW on PI3K/Akt- and MAP kinase-signaling pathways.
Although DOX significantly induced phosphorylation of ERK1/2, no
inhibitory effect of DSW on the protein level of phosphorylated
ERK1/2 was observed (Fig. 5A).
Similar induction was observed in the phosphorylation of p38 in
DOX-treated cardiomyocytes. However, treatment with DSW
significantly decreased the phosphorylation of p38 (Fig. 5B), suggesting that DSW protects
cells from DOX-induced cell death by suppressing the activation of
p38. In contrast to the roles of MAP kinase-signaling pathways, the
PI3K/Akt-signaling pathway is a major cell survival signal in
cardiomyocytes (31–33). Our data also showed that DOX
significantly decreased the phosphorylation of Akt. However, DSW
treatment rescued the activation of Akt from DOX-mediated Akt
suppression (Fig. 5C). To confirm
the protective role of PI3K/Akt-signaling pathway against
DOX-induced apoptosis, we blocked this signal with LY294002.
Blocking PI3K/Akt-signaling pathway with LY294002 in DOX-treated
H9c2 cells significantly increased the cleavage of caspase-3
(Fig. 5D), resulting in decreased
cell viability (data not shown). These data suggest that the
PI3K/Akt-signaling pathway is protective against the progression of
apoptosis and DSW rescues cardiomyocytes from DOX-induced cell
death via the restoration of Akt activation.
Discussion
In the present study, we demonstrated that DSW
provides a cardioprotective effect against DOX-induced
cardiotoxicity in rat H9c2 cardiac muscle cells without interfering
with the antitumor activities of DOX. This protective effect of DSW
appears to be mediated through the inhibition of DNA damage rather
than suppression of ROS, resulting in subsequent inhibition of
DOX-induced apoptotic signaling. Moreover, DSW rescues the
Akt-signaling pathway to protect cells from DOX-induced cell
death.
Since oxidative stress is generally accepted as the
major mechanism by which DOX causes toxicity to the heart, numerous
antioxidants have been investigated as cardioprotective agents to
prevent or reverse the cardiotoxic side-effects of DOX. However,
administration of antioxidants with DOX has failed to show
favorable outcomes in clinical studies, implying the involvement of
additional mechanisms in the cardiotoxic action of DOX. More recent
studies suggest that DNA damage plays an important role in
mediating DOX-induced cardiomyocyte death through a pathway
involving p53 and the mitochondria (10). A cardioprotective effect through
direct inhibition of DOX-induced DNA damage was demonstrated by
Huelsenbeck et al showing that lovastatin, which is a widely
used lipid-lowering drug, effectively protects against DOX-induced
cardiac damage by inhibiting DNA damage in a Rac1-dependent manner
without suppressing ROS generation in both in vitro and
in vivo studies (18).
Since DSW also exhibits a cardioprotective effect through the
inhibition of DNA damage rather than ROS generation, inhibiting
DOX-induced DNA damage may provide alternative cardioprotective
strategies besides antioxidant therapy in DOX treatment.
It is not clear which component of DSW is
responsible for the protective effects against DOX-induced
cardiotoxicity. However, it is assumed that the combined ionic
action of several minerals such as Ca, Mg, K, and Na may play
important roles in mediating diverse biological effects of DSW
including its cardioprotective effect. Indeed, it is now well known
that these essential metal ions are crucial to maintain cellular
functions and their deficiency is considered to be a potential
health hazard. In particular, Mg and Ca may be the primary minerals
responsible for the protective effect of DSW against DOX-induced
cardiotoxicity due to their profound existence within DSW. Mg is an
essential intracellular ion necessary for normal cellular function
(26). Its crucial role in
regulating cardiac function is well established in studies of
several cardiovascular diseases, including hypertension, stroke and
atherosclerosis (34,35). Moreover, it has been shown that
chronic dietary Mg deficiency results in cardiac apoptosis in the
rat heart (27), whereas Mg
treatment contributes to the improvement of the prognosis of heart
failure patients (36). Ca
deficiency is also known to be associated with increased apoptosis
in multiple cell lines. Although it has been suggested that an
excessive level of intracellular ionized Ca may be responsible for
triggering apoptotic machinery within cells, emerging evidence
emphasizes that loss of Ca is a greater determinant in apoptotic
cell death than high level of intracellular ionized Ca (37,38).
The requirement of both Mg and Ca in preventing apoptotic cell
death has been proven by Feng et al (39). They demonstrated that deficiency of
Mg and Ca induce apoptosis in Chinese hamster ovary cells but the
restoration of Mg and Ca protects cells from apoptotic cell death.
Furthermore, a clinical study has shown that supplementation of Mg
and Ca in drinking water decreased the mortality of breast cancer
patients (40). In this study, the
cardioprotective effect of DSW was escalated corresponding to the
hardness of DSW, which reflects the concentration of Ca and Mg.
Compared to the effect of desalinated DSW (hardness 0), the
cardioprotective effect was maximized in cardiomyocytes treated
with 1,500 hardness DSW, in which Ca concentration is ~100 mg/l and
the amount of Mg is ~300 mg/l. Thus, the synergistic action of Mg
and Ca within DSW may be mainly responsible for its
cardioprotective effects against DOX-induced cell death. However,
the possible involvement of the other trace elements in DSW also
needs to be considered. Our data showed that inhibition of
DOX-induced DNA damage is one of the mechanisms underlying the
cardioprotective effects of DSW. To elucidate a more detailed
mechanism of DSW-mediated cardioprotection against DOX-induced
cardiotoxicity, further investigations will be needed. So far,
diverse beneficial effects of DSW have been revealed and applied in
many fields such as food processing and cosmetics (21). However, to our knowledge, this is
the first study to show the cardioprotective effects of DSW against
DOX-induced cardiotoxicity, suggesting that DSW has some promise as
a novel protective supplement to extend the use of DOX therapy in
patients who may benefit from further DOX treatment.
Acknowledgements
This study was supported by the project entitled
‘Development of Technology for support of DSW industry (PJT200014)’
from the Ministry of Land, Transport and Maritime Affairs,
Korea.
Abbreviations:
|
DOX
|
doxorubicin
|
|
DSW
|
deep sea water
|
|
ROS
|
reactive oxygen species
|
|
DCF-DA
|
2′,7′-dichlorofluorescin diacetate
|
|
MDR1
|
multi-drug resistance protein 1
|
|
H2AX
|
H2A histone family member X
|
|
Bcl-xL
|
B-cell lymphoma-extra large
|
|
caspase-3
|
cysteine-aspartic acid protease-3
|
|
PARP
|
poly(ADPribose) polymerase
|
|
Akt
|
protein kinase B
|
|
ERK1/2
|
extracellular signal-regulated kinase
1/2
|
|
GAPDH
|
glyceraldehyde-3-phosphate
dehydrogenase
|
References
|
1
|
Minotti G, Menna P, Salvatorelli E, Cairo
G and Gianni L: Anthracyclines: molecular advances and
pharmacologic developments in antitumor activity and
cardiotoxicity. Pharmacol Rev. 56:185–229. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tan C, Tasaka H, Yu KP, Murphy ML and
Karnofsky DA: Daunomycin, an antitumor antibiotic, in the treatment
of neoplastic disease. Clinical evaluation with special reference
to childhood leukemia. Cancer. 20:333–353. 1967. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gharib MI and Burnett AK:
Chemotherapy-induced cardiotoxicity: current practice and prospects
of prophylaxis. Eur J Heart Fail. 4:235–242. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Singal PK, Li T, Kumar D, Danelisen I and
Iliskovic N: Adriamycin-induced heart failure: mechanism and
modulation. Mol Cell Biochem. 207:77–86. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Von Hoff DD, Layard MW, Basa P, et al:
Risk factors for doxorubicin-induced congestive heart failure. Ann
Intern Med. 91:710–717. 1979.PubMed/NCBI
|
|
6
|
Rajagopalan S, Politi PM, Sinha BK and
Myers CE: Adriamycin-induced free radical formation in the perfused
rat heart: implications for cardiotoxicity. Cancer Res.
48:4766–4769. 1988.PubMed/NCBI
|
|
7
|
Simůnek T, Stérba M, Popelová O, Adamcová
M, Hrdina R and Gersl V: Anthracycline-induced cardiotoxicity:
overview of studies examining the roles of oxidative stress and
free cellular iron. Pharmacol Rep. 61:154–171. 2009.PubMed/NCBI
|
|
8
|
Vásquez-Vivar J, Martasek P, Hogg N,
Masters BS, Pritchard KA Jr and Kalyanaraman B: Endothelial nitric
oxide synthase-dependent superoxide generation from adriamycin.
Biochemistry. 36:11293–11297. 1997.PubMed/NCBI
|
|
9
|
Goto S, Ihara Y, Urata Y, et al:
Doxorubicin-induced DNA intercalation and scavenging by nuclear
glutathione S-transferase pi. FASEB J. 15:2702–2714. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
L’Ecuyer T, Sanjeev S, Thomas R, et al:
DNA damage is an early event in doxorubicin-induced cardiac myocyte
death. Am J Physiol Heart Circ Physiol. 291:H1273–H1280.
2006.PubMed/NCBI
|
|
11
|
Liu J, Mao W, Ding B and Liang CS:
ERKs/p53 signal transduction pathway is involved in
doxorubicin-induced apoptosis in H9c2 cells and cardiomyocytes. Am
J Physiol Heart Circ Physiol. 295:H1956–H1965. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chularojmontri L, Gerdprasert O and
Wattanapitayakul SK: Pummelo protects doxorubicin-induced cardiac
cell death by reducing oxidative stress, modifying glutathione
transferase expression, and preventing cellular senescence. Evid
Based Complement Alternat Med. 2013:2548352013.
|
|
13
|
Das J, Ghosh J, Manna P and Sil PC:
Taurine suppresses doxorubicin-triggered oxidative stress and
cardiac apoptosis in rat via up-regulation of PI3-K/Akt and
inhibition of p53, p38-JNK. Biochem Pharmacol. 81:891–909. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ghosh J, Das J, Manna P and Sil PC: The
protective role of arjunolic acid against doxorubicin induced
intracellular ROS dependent JNK-p38 and p53-mediated cardiac
apoptosis. Biomaterials. 32:4857–4866. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Doroshow JH, Locker GY, Ifrim I and Myers
CE: Prevention of doxorubicin cardiac toxicity in the mouse by
N-acetylcysteine. J Clin Invest. 68:1053–1064. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shimpo K, Nagatsu T, Yamada K, et al:
Ascorbic acid and adriamycin toxicity. Am J Clin Nutr. 54(Suppl 6):
1298S–1301S. 1991.PubMed/NCBI
|
|
17
|
Swain SM and Vici P: The current and
future role of dexrazoxane as a cardioprotectant in anthracycline
treatment: expert panel review. J Cancer Res Clin Oncol. 130:1–7.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huelsenbeck J, Henninger C, Schad A,
Lackner KJ, Kaina B and Fritz G: Inhibition of Rac1 signaling by
lovastatin protects against anthracycline-induced cardiac toxicity.
Cell Death Dis. 2:e1902011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Siveski-Iliskovic N, Hill M, Chow DA and
Singal PK: Probucol protects against adriamycin cardiomyopathy
without interfering with its antitumor effect. Circulation.
91:10–15. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Siveski-Iliskovic N, Kaul N and Singal PK:
Probucol promotes endogenous antioxidants and provides protection
against adriamycin-induced cardiomyopathy in rats. Circulation.
89:2829–2835. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nakasone T and Akeda S: The application of
deep sea water in Japan. UJNR Technical Report. 28:69–75. 1999.
|
|
22
|
Hwang HS, Kim SH, Yoo YG, et al:
Inhibitory effect of deep-sea water on differentiation of 3T3-L1
adipocytes. Mar Biotechnol (NY). 11:161–168. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Radhakrishnan G, Yamamoto M, Maeda H, et
al: Intake of dissolved organic matter from deep sea water inhibits
atherosclerosis progression. Biochem Biophys Res Commun. 387:25–30.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim S, Chun SY, Lee DH, Lee KS and Nam KS:
Mineral-enriched deep-sea water inhibits the metastatic potential
of human breast cancer cell lines. Int J Oncol. 43:1691–1700.
2013.PubMed/NCBI
|
|
25
|
Lee KS, Shin JS, Kwon YS, Moon DS and Nam
KS: Suppression of cancer progression and metastasis in HT-29 human
colorectal adenocarcinoma by deep sea water. Biotechnol Bioproc
Eng. 18:194–200. 2013. View Article : Google Scholar
|
|
26
|
Hartwig A: Role of magnesium in genomic
stability. Mutat Res. 475:113–121. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tejero-Taldo MI, Chmielinska JJ and
Weglicki WB: Chronic dietary Mg2+ deficiency induces
cardiac apoptosis in the rat heart. Magnes Res. 20:208–212.
2007.
|
|
28
|
van Asperen J, van Tellingen O, Tijssen F,
Schinkel AH and Beijnen JH: Increased accumulation of doxorubicin
and doxorubicinol in cardiac tissue of mice lacking mdr1a
P-glycoprotein. Br J Cancer. 79:108–113. 1999.PubMed/NCBI
|
|
29
|
Olive PL: Detection of DNA damage in
individual cells by analysis of histone H2AX phosphorylation.
Methods Cell Biol. 75:355–373. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Brantley-Finley C, Lyle CS, Du L, et al:
The JNK, ERK and p53 pathways play distinct roles in apoptosis
mediated by the antitumor agents vinblastine, doxorubicin, and
etoposide. Biochem Pharmacol. 66:459–469. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Negoro S, Oh H, Tone E, et al:
Glycoprotein 130 regulates cardiac myocyte survival in
doxorubicin-induced apoptosis through phosphatidylinositol
3-kinase/Akt phosphorylation and Bcl-xL/caspase-3 interaction.
Circulation. 103:555–561. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ahmed NN, Grimes HL, Bellacosa A, Chan TO
and Tsichlis PN: Transduction of interleukin-2 antiapoptotic and
proliferative signals via Akt protein kinase. Proc Natl Acad Sci
USA. 94:3627–3632. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang Y: Mitogen-activated protein kinases
in heart development and diseases. Circulation. 116:1413–1423.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Saris NE, Mervaala E, Karppanen H, Khawaja
JA and Lewenstam A: Magnesium. An update on physiological, clinical
and analytical aspects. Clin Chim Acta. 294:1–26. 2000.PubMed/NCBI
|
|
35
|
Song Y, Manson JE, Cook NR, Albert CM,
Buring JE and Liu S: Dietary magnesium intake and risk of
cardiovascular disease among women. Am J Cardiol. 96:1135–1141.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Almoznino-Sarafian D, Sarafian G, Berman
S, et al: Magnesium administration may improve heart rate
variability in patients with heart failure. Nutr Metab Cardiovasc
Dis. 19:641–645. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kluck RM, McDougall CA, Harmon BV and
Halliday JW: Calcium chelators induce apoptosis - evidence that
raised intracellular ionised calcium is not essential for
apoptosis. Biochim Biophys Acta. 1223:247–254. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Turner CP, Connell J, Blackstone K and
Ringler SL: Loss of calcium and increased apoptosis within the same
neuron. Brain Res. 1128:50–60. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Feng H, Guo L, Gao H and Li XA: Deficiency
of calcium and magnesium induces apoptosis via scavenger receptor
BI. Life Sci. 88:606–612. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yang CY, Chiu HF, Cheng MF, Hsu TY and Wu
TN: Calcium and magnesium in drinking water and the risk of death
from breast cancer. J Toxicol Environ Health A. 60:231–241. 2000.
View Article : Google Scholar : PubMed/NCBI
|