Introduction
Tumor necrosis factor-related apoptosis-inducing
ligand (TRAIL/Apo2L) is a member of the tumor necrosis factor (TNF)
superfamily and known to induce apoptosis of cancer cells without
significant toxicity toward normal cells (1–3). The
TRAIL induces apoptosis through an extrinsic pathway by binding to
the death receptors (DR) 4 and DR5. Activation of DR4 or DR5
recruits Fas-associated death domain (FADD) protein and
procaspase-8, thereby forming a death-inducing signaling complex
(DISC), which leads to activation of the caspase cascade (1,4).
Caspase activation can be suppressed by inhibitors of apoptosis
protein (IAP) family members, as well as by anti-apoptotic B-cell
lymphoma 2 (Bcl-2) family proteins (5). Application of TRAIL to cancer
treatment is currently under intensive clinical evaluation.
Especially, human recombinant TRAIL or high-affinity agonist
monoclonal antibodies against DRs are now in phase I/II clinical
trials (6–9). However, recent studies have shown
that many types of cancer cells are resistant to the apoptotic
effects of TRAIL including colorectal cancer (CRC) cells (10). TRAIL resistance in CRC cells can
occur at several steps in signaling cascade such as deficient
receptor redistribution to the membrane, mutation of caspase-8,
cellular fas-associated death domain-like
interleukin-1-β-converting enzyme-inhibitory protein (cFLIP)
expression, Bax deficiency, or through X-linked inhibitor of
apoptosis protein (XIAP) expression have been reported (11–15).
Therefore, overcoming TRAIL resistance is a major challenge for the
development of effective TRAIL-based therapeutic strategies.
Parthenolide (PT), a natural product, has been used
for the treatment of fever and inflammatory disease. It is well
known to inhibit interleukin-1 (IL-1) and tumor necrosis
factor-α-mediated nuclear factor-κB (NF-κB) activation, which is
responsible for its inflammatory activity (16–18).
For over two decades, it is known that anticancer property of PT is
through induction of apoptotic cell death in a number of human
cancer cells (19–23). Apoptotic effect of PT is associated
with inhibition of NF-κB and the activator of transcription 3
(STAT3), enhanced oxidative stress, and mitochondria-mediated
apoptosis (19,20,24–26).
In our previous studies, we found that PT can be a potential
chemopreventive and therapeutic agent for CRC treatment by inducing
apoptosis through mitochondrial dysfunction and inhibition of
angiogenesis (20,27).
In recent years, various investigations of combined
therapy using PT were reported; PT sensitizes cancer cells to the
non-steroidal anti-inflammatory drugs (NSAIDs), anticancer drug
toxicity and radiation (25,28–33).
Previously we have shown that combination of PT and 5-FU can
overcome 5-FU resistance in human CRC cells and that
intra-peritoneal injection of PT and 5-FU significantly inhibits
tumor growth in the xenograft model (34). However, the effect and functional
role of PT on TRAIL-induced apoptosis in CRC cells has not been
reported.
In this study, we evaluated effects of combination
therapy with PT and TRAIL on TRAIL-sensitive and -insensitive human
CRC cells to gain insight into a potential treatment for CRC,
especially TRAIL-resistant CRC. We also investigated the molecular
mechanism of enhancing sensitivity of TRAIL by PT.
Materials and methods
Chemicals and reagents
PT and z-VAD-FMK were from Calbiochem (San Diego,
CA, USA). TRAIL was purchased from Pepprotech (Rocky Hill, NJ,
USA). Parthenolide was dissolved in dimethylsulfoxide (DMSO; Sigma,
St. Louis, MO, USA) to a concentration of 100 μM and stored in the
dark at −20°C. Annexin-V-FITC and propidium iodide (PI) were
purchased from Invitrogen (Eugene, OR, USA). Hoechst 33258 was from
Sigma. Levels of DR4 and DR5 protein were analyzed using a
respective specific antibody from ProSci Inc. (San Diego, CA, USA).
Other antibodies were from Santa Cruz Biotechnology (Santa Cruz,
CA, USA).
Cell culture and treatment
Human CRC cell lines, HT-29 and HCT 116 cells
(American Type Culture Collection, Rockville, MD, USA) were
employed as TRAIL-resistant and TRAIL-sensitive CRC cells,
respectively. The cells were cultured in RPMI-1640 medium
supplemented with 10% FBS, 100 U penicillin and 100 U streptomycin.
For treatment of cells with PT or TRAIL or PT plus TRAIL, cells
were sub-cultured in RPMI-1640 medium without FBS for 12 h. PT and
TRAIL in the stock were diluted with FBS-free medium to achieve
designated concentrations. Same concentration of DMSO was applied
to the cells as a control.
Cell viability assay
Human CRC cells were plated at a density of
1.0×104 cells per well in 96-well plates. Cells were
treated with PT and/or TRAIL for 24 h, the medium was removed, and
200 μl of fresh medium plus 20 μl of 3-(4,
5-dimethylthiazol-2yl)-2, 5-diphenyltetrazolium bromide (MTT, 2.5
mg dissolved in 50 μl of dimethylsulfoxide, Sigma) were added to
each well. After incubation for 4 h at 37°C, the culture medium
containing MTT was withdrawn and 200 μl of dimethylsulfoxide (DMSO)
was added, followed by shaking until the crystals were dissolved.
Viable cells were detected by measuring absorbance at 570 nm using
a microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Annexin-V-fluorescein staining
After being incubated with single or dual agent for
24 h, the cells were trypsinized, collected, washed with ice-cold
PBS, suspended in a 500 μl Annexin V binding buffer containing 5 μl
of Annexin V-FITC, and incubated for 15 min at room temperature in
the dark. The fluorescence was measured using a BD LSR flow
cytometer and processed with CellQuest software for analysis.
Cell cycle and sub-G1 analysis
Cell cycle and sub-G1 distribution were determined
by staining of DNA with propidium iodide (PI; Sigma-Aldrich) (Ex/Em
= 488 nm/617 nm). PI is a fluorescent biomolecule that can be used
to stain DNA. In brief, 1×106 cells were incubated with
single or dual agents for 24 h. Cells were then washed with
phosphate-buffered saline (PBS) and fixed in 70% ethanol overnight.
Cells were washed again with PBS and then incubated with PI (10
μg/ml) with simultaneous treatment of RNase at 37°C for 1 h. The
percentage of cells in different phases of the cell cycle or having
sub-G1 DNA content was measured with a BD LSR flow cytometer and
analyzed using CellQuest software.
Hoechst 33258 staining
Apoptotic feature of cancer cells was assessed by
determining DNA condensation using Hoechst 33258. The cells were
treated with single or dual agents for 24 h and then stained with
Hoechst 33258 (1 μg/ml) at 37°C for 10 min. Nuclear morphology was
examined under a Confocal Laser Scanning Microscope (Carl Zeiss,
Germany) to identify cells undergoing apoptosis.
Quantification of death receptor
expression on cell surface
In order to quantify the cell surface expression of
death receptors, DR4 and DR5, cells were harvested by
trypsinization, washed in PBS and incubated for 30 min a 4°C with
phycoerythrin (PE)-conjugated monoclonal anti-human DR4 and DR5
antibody (eBioscences, San Diego, CA, USA). Non-immune mouse IgG
was used as the negative control. The fluorescence was measured
using a BD LSR flow cytometer and processed with CellQuest software
for analysis.
Cell extraction and western blotting
After being incubated with single or dual agent for
24 h, the cells were collected, washed twice with PBS, and then
lysed for 30 min on ice in a lysis buffer (50 mM Tris-HCl pH 8.0,
150 mM EDTA, 1% Triton X-100, 0.5% SDS and protease inhibitor
cocktail). The protein concentration in cell lysates was measured
by using Protein Quantification kit from Bio-Rad. Total 50 μg
proteins were loaded onto an SDS-PAGE gel. After transferring and
blocking, the membrane was probed with various antibodies
(anti-DR4, anti-DR5, anti-Bcl2, anti-Bax, anti-cytochrome C,
anti-p53, anti-caspase-3, anti-caspase-8, anti-caspase-9 and
anti-actin antibody). The binding of antibody to antigen was
detected by using enhanced ECL prime (Amersham, UK), captured, and
analyzed by the Las-3000 luminescent Image Analyzer (Fuji Film,
Tokyo, Japan).
Statistical analysis
The data are presented as the mean ± standard error
(SE) of at least three independent experiments done in duplicate.
Representative blots are shown. The data were entered into the
Microsoft Excel 5.0, and SPSS software was used to perform the
two-tailed t-tests or the analysis of the variance, where
appropriate. P-value <0.05 was considered significant.
Results
TRAIL sensitivity of human colorectal
cancer cells and effect of PT on TRAIL-induced cell death
A panel of 4 human colorectal cancer cell lines
(HT-29, SW480, SW620 and HCT116) were screened for TRAIL
sensitivity by determining viability at various concentrations (0,
5, 10, 25, 50 or 100 ng/ml) for 24 h using the MTT assay. Of the 4
cell lines, 3 showed inhibition of viability in a dose-dependent
manner to TRAIL treatment (Fig.
1A). Especially, HCT116 sensitively responded to TRAIL, showing
~70% of growth inhibition at 100 ng/ml concentration. On the
contrary, treatment with TRAIL induced marginal cell death in HT-29
cells. These results indicate that HT-29 cells are TRAIL-resistant
and HCT116 cells are TRAIL-sensitive.
To determine the synergistic effect of PT on
TRAIL-induced cell death, HT-29 and HCT116 cells were incubated in
the absence or presence of PT (10 μM) and TRAIL (with 5, 25 and 40
ng/ml) for 24 h. Co-treatment of PT with TRAIL significantly
increased death of not only HCT116 cells but also HT-29 cells,
suggesting that PT may sensitize HT-29 cells to TRAIL (Fig. 1B).
Effect of PT on TRAIL-induced
apoptosis
To ascertain the above observations, Annexin V
analysis was performed using FACScan. We found that treatment of
HT-29 cells with PT and TRAIL alone induced 15.29 and 8.6%
apoptosis in HT-29 cells, respectively, and 14.77 and 22.01% in
HCT116 cells, respectively. In agreement with cell growth
inhibition, treatment with TRAIL plus PT dramatically increased the
Annexin V-positive cells (41.86%) by 5-fold than treatment with
TRAIL, indicating that PT promotes TRAIL-induced apoptosis in HT-29
cells. Similar combination effect on apoptotic cell death was found
in HCT116 cells (Fig. 2A).
We also evaluated cell cycle modifications induced
by PT and TRAIL on TRAIL-resistant and -sensitive cells. After 24-h
incubation with PT or PT plus TRAIL, cells were analyzed by PI
staining using flow cytometric analysis (FCM). Treatment with PT
and/or TRAIL resulted in the presence of a sub-G1 population,
suggestive of apoptotic cell death. Similarly, FCM revealed that PT
significantly enhanced TRAIL-induced apoptosis >3-fold in HT-29
cells (8.07 versus 27.77%). In addition, higher sub-G1 arrested
cells were detected.
HCT116 cells treated with PT plus TRAIL were
arrested at higher level than single treatment in sub-G1 stage of
the cell cycle. Peaks accounting for 11.34 and 8.07% of HT-29 cells
and 5.6 and 18.05% of HCT116 cells of the overall cell population
were detectable in treated with PT or TRAIL, respectively. In
addition, a peak accounting for 27.77% was observed in treated with
combination of HT-29 and HCT116 cells, indicating that combination
treatment dramatically promotes apoptosis (Fig. 2B).
To understand the mechanism of cell death induced by
combination treatment, apoptotic nuclear morphology was evaluated
after Hoechst 33258 staining. After treatment with single agent,
HT-29 cells were regular in morphology and grew fully in patches
and were confluent (Fig. 2C).
However, treatment with PT and TRAIL together, HT-29 cells
exhibited apoptotic characteristics, such as cell shrinkage,
nuclear condensation and fragmentation. In HCT116 cells, treatment
with TRAIL or PT plus TRAIL exhibited apoptotic nuclear
morphologies while treatment with PT alone showed regular nuclear
morphology.
Effect of PT on the expression of death
receptor in CRC cells
In order to ascertain the mechanism by which PT
sensitizes CRC cells to TRAIL, we investigated the level of
expression of death receptors on the cell surface. Levels of DR4
and DR5 expression were analyzed by flow cytometry using PE
conjugated anti-human DR4 and DR5 antibody (Fig. 3A). Cell surface expression of DR4
(green line) and DR5 (pink line) was found in both of cell lines.
Interestingly, the level of DR5 expression was significantly higher
than the DR4 level. Moreover, the level of DR5 expression on HCT116
cells was much higher than the level on HT-29 cells. Supporting the
data, western blotting results were well correlated with flow
cytometry analysis (Fig. 3B).
In addition, effects of PT on DR4 and DR5 expression
in HT-29 and HCT116 cells were analyzed by western blotting.
Treatment with PT (10 μM) markedly changed the level of DR5 protein
in both cell lines in a time-dependent manner (Fig. 3C). The PT treatment increased the
level of DR5 protein up to 6 h in HT-29 cells and 3 h in HCT116
cells but the effect disappeared after that. On the contrary, the
level of DR4 protein did not change in either cell line after
treatment with PT.
We also investigated whether treatment with PT
increases surface expression of the TRAIL receptors. In these
experiments, cells treated with PT (10 μM) for 3 h were incubated
for 30 min with anti-human DR4 or DR5 antibody conjugated with PE
and analyzed by flow cytometry. The PT treatment did not induce
surface expression of DR4, while the cell surface expression of DR5
were increased in both cell lines (Fig. 3D). Together, these observations
suggest that TRAIL sensitivity is influenced by the level of DR5
expression.
PT enhances TRAIL-induced apoptosis via
activation of caspase cascade
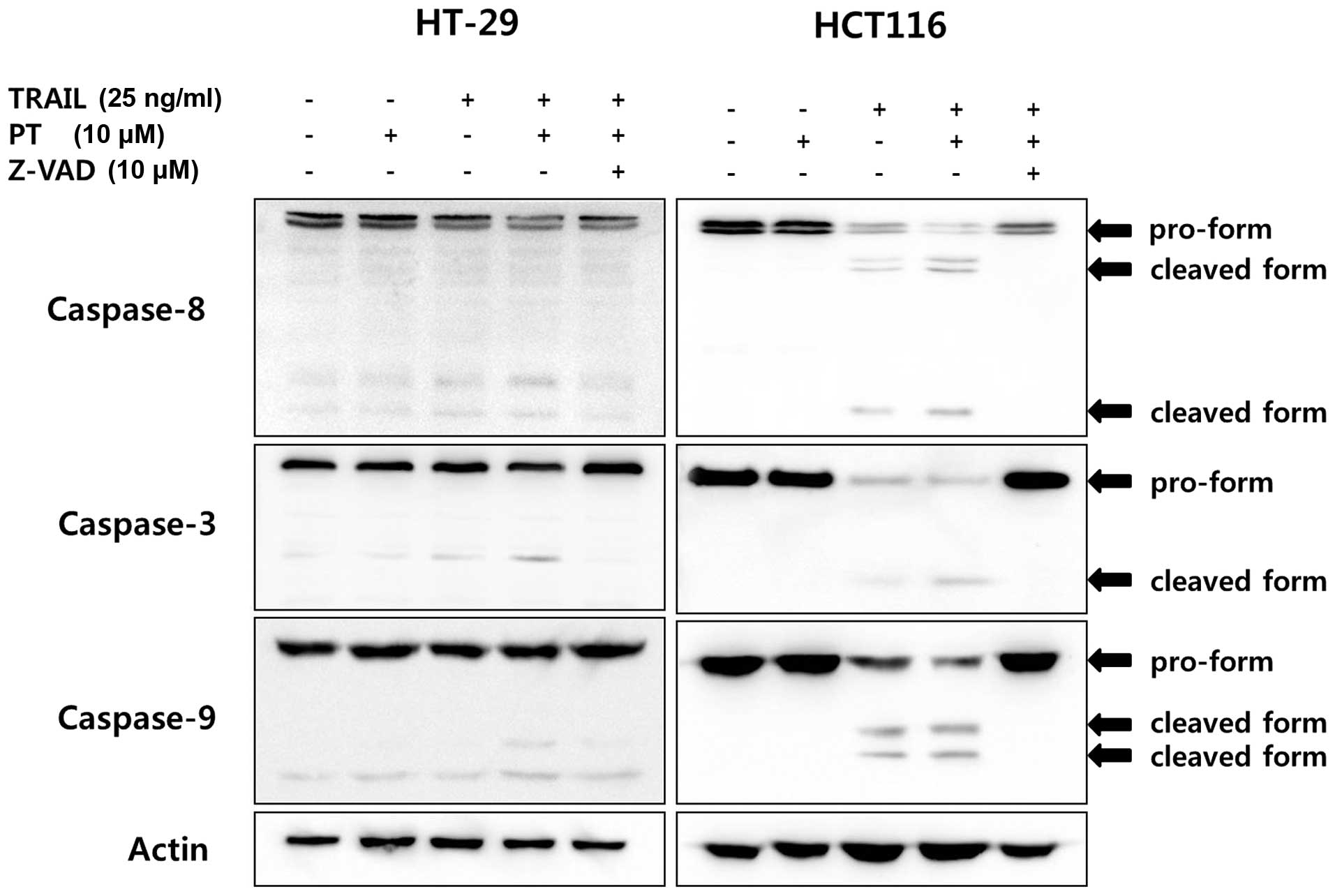
Many anticancer agents are capable of initiating
caspase activation and inducing apoptotic cell death (35). The effects of PT, TRAIL or PT plus
TRAIL on caspase activation in TRAIL-resistant and -sensitive cells
were examined. Western blot analysis of HT-29 cells treated with PT
plus TRAIL revealed that levels of cleaved caspase-3, -8 and -9
were significantly increased compared the levels in the cells
treated PT or TRAIL alone (Fig.
4). Although, in HCT116 cells, TRAIL alone had effects on the
activation of the caspase (-3, -8 and -9) cleavage, the combination
treatment significantly increased the activation of caspases.
Western blot analysis of HCT116 cells treated with
TRAIL or PT plus TRAL showed that levels of cleaved caspase-3, -8
and -9 were significantly increased compared to the levels of
control and PT treated cells. In addition, the decrease in the
levels of caspase-3, -8 and -9 in cells treated with PT plus TRAIL
was significantly blocked by pretreatment of a general caspase
inhibitor, Z-VAD-FMK (Fig. 4).
PT enhances TRAIL-induced expression of
proteins involved in apotosis
To evaluate the mechanisms responsible for apoptosis
by combination of TRAIL and PT, we examined the expression level of
several pro-apoptosis and anti-apoptosis proteins in
TRAIL-resistant and -sensitive cells, HT-29 and HCT116 cells,
respectively. Western blot analysis showed that the level of Bcl-2
in both cell lines was significantly decreased by treatment with PT
plus TRAIL compared to the level in the cells treated with PT or
TRAIL alone. In contrast, the expression of Bax was significantly
increased by treatment with PT plus TRAIL compared to the level in
the cells treated with PT or TRAIL alone (Fig. 4, first and second panel).
One of the consequences following the changes of
Bcl-2 family members is the dissipation of mitochondrial potential
and release of the mitochondrial proapoptotic protein, cytochrome
C. Treatment with PT plus TRAIL significantly increased the release
of cytochrome C of both of cell lines compared to that of control
and single drug treated cells (Fig.
4, third panel).
In addition, the p53 gene, which is inactivated in a
majority of human cancers, has been recognized as a hallmark of
apoptosis (36). The level of p53
was significantly increased by treatment with PT plus TRAIL
compared to the level in cells treated with PT or TRAIL alone
(Fig. 4, fourth panel). These
results indicated that the apoptosis induced by the combination of
TRAIL and PT may be associated with the mitochondrial pathway.
Discussion
In CRC, chemotherapy is currently used to reduce
tumor recurrence and prolong survival. However, due to drug
resistance, investigations of new chemotherapy strategies are
required. Compared to other cancer cells, CRC cells show increased
resistance to chemotherapeutic agents (37). The molecular mechanisms of drug
resistance in colon cancer cells are still unknown.
Drug combination therapies play a prominent role in
overcoming of drug resistance in cancer treatment. Exploration of
the molecular mechanisms underlying the synergistic effects
achieved by a drug combination would fuel efforts to rationally
develop combination therapeutics that could significantly improve
patient outcomes in cancer.
Recent studies have demonstrated that PT has
anticancer activity and induces apoptosis through wide range of
intracellular signals in cancer cells (22,38).
We have also found that PT induces apoptosis through mitochondrial
dysfunction in human CRC cells and inhibits tumor growth and
angiogenesis in a CRC xenograft model (20). PT is considered to be a promising
candidate as a new type of chemotherapeutic agent for cancer
treatment. It would be interesting to investigate whether treatment
with PT alone or in combination with an anticancer drug can
overcome drug resistance. In this study, we found that combination
treatment with PT and TRAIL resulted in the increase of apoptosis
via caspase activation and that PT enhanced TRAIL sensitivity by
upregulation of DR5 in TRAIL-resistant and -sensitive cells. These
observations indicate that combination of TRAIL and PT may overcome
TRAIL resistance in CRC and be an effective therapeutic strategy
for patients with CRC.
Interactions between TRAIL and PT have been examined
in a limited number of preclinical studies. A study in breast
cancer cells has demonstrated that PT reverses resistance of breast
cancer cells to TRAIL through c-Jun N-terminal kinase (JNK)
activation (39). PT also
sensitizes hepatocellular carcinoma cells to TRAIL through
inhibition of STAT3 (28).
However, the effect of PT on TRAIL-induced apoptosis remains to be
understood. Therefore, in this study, we focused on the mechanism
of apoptosis induced by PT, evaluating regulation of TRAIL
receptors and mitochondrial apoptotic pathway using TRAIL-resistant
and -sensitive human CRC cells.
Our results showed that TRAIL inhibited growth of
HCT116 cells (a TRAIL-sensitive cell line) in a dose-dependent
manner; however, the inhibition did not occur in TRAIL-resistant
HT-29 cells. These results are very similar to the findings
reported on the TRAIL sensitivity of human CRC cells (40–42).
Interestingly, our results also indicated that combination of PT
and TRAIL reduced cell growth and increased apoptotic cell death of
not only TRAIL-sensitive cells but also TRAIL-resistant cells.
TRAIL mediates apoptotic cell death through
enhancing expression of the death receptors DR4 and DR5, which are
expressed on the surface of cancer cells (43,44).
The binding of TRAIL to DR4 and DR5 leads to activation of
caspases, which in turn cleaves and activates executioner caspases
that mediate apoptosis (45,46).
Several studies have provided evidence that DR upregulation is a
promising strategy for sensitizing TRAIL resistance in cancer cells
(47–51). Garcinol, a polyisoprenylated
benzophenone derivative, can potentiate TRAIL-induced apoptotic
cell death of human CRC cell through upregulation of DR4 and DR5
(47). Quercetin, a ubiquitous
bioactive plant flavonoid, enhances TRAIL-induced apoptotic cell
death in prostate cancer cells via expression of DR5 (48,49).
Snake venom toxin from Vipera lebetina turanica sensitizes
cancer cells to TRAIL through upregulation of death receptors and
downregulation of survival proteins (51). In the present study, our data
indicated that PT markedly increased the expression of DR5 protein
at early stage, while PT did not affect the expression of DR4. In
particular, the effect of PT on the expression of DRs at various
time-points has not been evaluated. Moreover, analysis by flow
cytometry permitted to establish that PT upregulated cell surface
expression of DR5 and that PT was ineffective on DR4 expression.
These findings suggest that upregulation of surface expression of
DR5 is the prominent event by which PT sensitizes human CRC cells
to TRAIL-induced apoptosis.
Many anticancer agents are capable of initiating
activation of caspase cascade and inducing apoptotic cell death
(52). Caspase-3 and -9, terminal
factor of apoptosis, exist as an inactive precursor in cytoplasm,
which is activated during apoptosis and takes part in apoptosis
induced by multiple factors. Moreover, caspase-8 activation is the
initial step in the TRAIL-mediated caspase activation cascade, and
lack of caspase-8 expression has been reported to cause TRAIL
resistance (53). To understand
the mechanism by which PT and TRAIL induce apoptosis, we examined
its effect on the activation and cleavage of these caspases. The
results showed that the levels of cleaved form of caspase-3, -8 and
-9 were increased by combination treatment with PT and TRAIL
especially in TRAIL-resistant cells, decreasing the levels of
pro-forms. The cleavage of caspases was prevented by pretreatment
with the pancaspase inhibitor Z-VAD-FMK. Our findings suggest that
PT and TRAIL-induced apoptosis is mediated by enhancing the
apoptotic sensitivity to TRAIL via caspase-dependent pathway.
Previous studies have reported that the Bcl-2 family
members play an important role in PT action in CRC. The
mitochondrial pathway is regulated by the Bcl-2 family, which is
divided into two groups, the anti-apoptotic members (Bcl-2 and
Bcl-xl) and pro-apoptotic members (Bax, BAD, BAK and Bid) (54,55).
Level of Bcl-2 is often enhanced in tumors, with a possible
substitution of Bcl-2 by Bcl-xL in the most aggressive tumors
(56,57). Mutation in Bax has been reported in
some colon cancers, the majority of which have a defect in DNA
mismatch repair which is readily detected by mutations in
repetitive sequences (58).
Therefore, the regulation of Bcl-2 and Bax expression has an
important role in chemotherapy, and it could be a measure of
chemotherapeutic effect. In this study, the results showed that
expression level of Bcl-2 in HCT116 and HT-29 cells treated with PT
plus TRAIL was decreased while the level of Bax was increased.
These results demonstrate that resistance of TRAIL CRC cells
(HCT116 cells) is overcome by combining with PT and that the
combination treatment-induced apoptosis is under the control of the
mitochondrial pathway.
The tumor suppressor gene p53 is involved in G1
growth arrest by inducing the cyclin-dependent kinase inhibitor p21
and also in apoptosis through transactivation of the pro-apoptotic
Bax gene in response to DNA-damage (58,59).
The p53 gene, which is inactivated in a majority of human cancers,
has been proposed as an accurate indicator of response of CRC to
anticancer drug (36). In the
present study, that results showed that the level of p53 was
enhanced by combination treatment with PT and TRAIL in
TRAILresistant and -sensitive cells.
In conclusion, we investigated effects of
combination of PT and TRAIL on cell growth and apoptotic cell death
using TRAIL-resistant and -sensitive human CRC cells. Treatment
with PT dramatically increased the surface expression of DR5
protein in both cell types. Moreover, combination of PT and TRAIL
upregulated expression of proteins involved in the mitochondrial
apoptotic pathway and increased caspase activation. Taken together,
these results suggest that PT sensitizes CRC cells resistant to
TRAIL, therefore, we believe that combined treatment with PT and
TRAIL could represent a new therapeutic strategy for CRC
treatment.
Acknowledgements
This study was supported by Fund of Chonbuk National
University Hospital Research Institute of Clinical Medicine. The
authors thank Professor Mie-Jae Im (Department of Internal
Medicine, Research Center for Pulmonary Disorders, Chonbuk National
University Medical School) for kindly proofreading and
contribution.
References
|
1
|
Johnstone RW, Frew AJ and Smyth MJ: The
TRAIL apoptotic pathway in cancer onset, progression and therapy.
Nat Rev Cancer. 8:782–798. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Koschny R, Walczak H and Ganten TM: The
promise of TRAIL - potential and risks of a novel anticancer
therapy. J Mol Med (Berl). 85:923–935. 2007. View Article : Google Scholar
|
|
3
|
Walczak H, Miller RE, Ariail K, et al:
Tumoricidal activity of tumor necrosis factor-related
apoptosis-inducing ligand in vivo. Nat Med. 5:157–163. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gonzalvez F and Ashkenazi A: New insights
into apoptosis signaling by Apo2L/TRAIL. Oncogene. 29:4752–4765.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shi Y: Mechanisms of caspase activation
and inhibition during apoptosis. Mol Cell. 9:459–470. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ashkenazi A, Holland P and Eckhardt SG:
Ligand-based targeting of apoptosis in cancer: the potential of
recombinant human apoptosis ligand 2/Tumor necrosis factor-related
apoptosis-inducing ligand (rhApo2L/TRAIL). J Clin Oncol.
26:3621–3630. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pan Y, Xu R, Peach M, et al: Evaluation of
pharmacodynamic biomarkers in a Phase 1a trial of dulanermin
(rhApo2L/TRAIL) in patients with advanced tumours. Br J Cancer.
105:1830–1838. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wiezorek J, Holland P and Graves J: Death
receptor agonists as a targeted therapy for cancer. Clin Cancer
Res. 16:1701–1708. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dimberg LY, Anderson CK, Camidge R,
Behbakht K, Thorburn A and Ford HL: On the TRAIL to successful
cancer therapy? Predicting and counteracting resistance against
TRAIL-based therapeutics. Oncogene. 32:1341–1350. 2013. View Article : Google Scholar
|
|
10
|
Zhang L and Fang B: Mechanisms of
resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther.
12:228–237. 2005. View Article : Google Scholar
|
|
11
|
Jin Z, McDonald ER III, Dicker DT and
El-Deiry WS: Deficient tumor necrosis factor-related
apoptosis-inducing ligand (TRAIL) death receptor transport to the
cell surface in human colon cancer cells selected for resistance to
TRAIL-induced apoptosis. J Biol Chem. 279:35829–35839. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim HS, Lee JW, Soung YH, et al:
Inactivating mutations of caspase-8 gene in colorectal carcinomas.
Gastroenterology. 125:708–715. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hernandez A, Wang QD, Schwartz SA and
Evers BM: Sensitization of human colon cancer cells to
TRAIL-mediated apoptosis. J Gastrointest Surg. 5:56–65. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Burns TF and El-Deiry WS: Identification
of inhibitors of TRAIL-induced death (ITIDs) in the TRAIL-sensitive
colon carcinoma cell line SW480 using a genetic approach. J Biol
Chem. 276:37879–37886. 2001.PubMed/NCBI
|
|
15
|
Cummins JM, Kohli M, Rago C, Kinzler KW,
Vogelstein B and Bunz F: X-linked inhibitor of apoptosis protein
(XIAP) is a nonredundant modulator of tumor necrosis factor-related
apoptosis-inducing ligand (TRAIL)-mediated apoptosis in human
cancer cells. Cancer Res. 64:3006–3008. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Murphy JJ, Heptinstall S and Mitchell JR:
Randomised double-blind placebo-controlled trial of feverfew in
migraine prevention. Lancet. 2:189–192. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hehner SP, Heinrich M, Bork PM, et al:
Sesquiterpene lactones specifically inhibit activation of NF-kappa
B by preventing the degradation of I kappa B-alpha and I kappa
B-beta. J Biol Chem. 273:1288–1297. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lyss G, Knorre A, Schmidt TJ, Pahl HL and
Merfort I: The anti-inflammatory sesquiterpene lactone helenalin
inhibits the transcription factor NF-kappaB by directly targeting
p65. J Biol Chem. 273:33508–33516. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang S, Ong CN and Shen HM: Involvement
of proapoptotic Bcl-2 family members in parthenolide-induced
mitochondrial dysfunction and apoptosis. Cancer Lett. 211:175–188.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim SL, Trang KT, Kim SH, et al:
Parthenolide suppresses tumor growth in a xenograft model of
colorectal cancer cells by inducing mitochondrial dysfunction and
apoptosis. Int J Oncol. 41:1547–1553. 2012.PubMed/NCBI
|
|
21
|
Wen J, You KR, Lee SY, Song CH and Kim DG:
Oxidative stress-mediated apoptosis. The anticancer effect of the
sesquiterpene lactone parthenolide. J Biol Chem. 277:38954–38964.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang S, Ong CN and Shen HM: Critical
roles of intracellular thiols and calcium in parthenolide-induced
apoptosis in human colorectal cancer cells. Cancer Lett.
208:143–153. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pajak B, Gajkowska B and Orzechowski A:
Molecular basis of parthenolide-dependent proapoptotic activity in
cancer cells. Folia Histochem Cytobiol. 46:129–135. 2008.PubMed/NCBI
|
|
24
|
Dai Y, Guzman ML, Chen S, et al: The NF
(Nuclear factor)-kappaB inhibitor parthenolide interacts with
histone deacetylase inhibitors to induce MKK7/JNK1-dependent
apoptosis in human acute myeloid leukaemia cells. Br J Haematol.
151:70–83. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sun Y, St Clair DK, Xu Y, Crooks PA and St
Clair WH: A NADPH oxidase-dependent redox signaling pathway
mediates the selective radiosensitization effect of parthenolide in
prostate cancer cells. Cancer Res. 70:2880–2890. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sobota R, Szwed M, Kasza A, Bugno M and
Kordula T: Parthenolide inhibits activation of signal transducers
and activators of transcription (STATs) induced by cytokines of the
IL-6 family. Biochem Biophys Res Commun. 267:329–333. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kim SL, Lee ST, Trang KT, et al:
Parthenolide exerts inhibitory effects on angiogenesis through the
downregulation of VEGF/VEGFRs in colorectal cancer. Int J Mol Med.
33:1261–1267. 2014.PubMed/NCBI
|
|
28
|
Carlisi D, D’Anneo A, Angileri L, et al:
Parthenolide sensitizes hepatocellular carcinoma cells to TRAIL by
inducing the expression of death receptors through inhibition of
STAT3 activation. J Cell Physiol. 226:1632–1641. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fang LJ, Shao XT, Wang S, Lu GH, Xu T and
Zhou JY: Sesquiterpene lactone parthenolide markedly enhances
sensitivity of human A549 cells to low-dose oxaliplatin via
inhibition of NF-kappaB activation and induction of apoptosis.
Planta Med. 76:258–264. 2010. View Article : Google Scholar
|
|
30
|
Gao ZW, Zhang DL and Guo CB: Paclitaxel
efficacy is increased by parthenolide via nuclear factor-kappaB
pathways in in vitro and in vivo human non-small cell lung cancer
models. Curr Cancer Drug Targets. 10:705–715. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yun BR, Lee MJ, Kim JH, Kim IH, Yu GR and
Kim DG: Enhancement of parthenolide-induced apoptosis by a
PKC-alpha inhibition through heme oxygenase-1 blockage in
cholangiocarcinoma cells. Exp Mol Med. 42:787–797. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee CS, Kim YJ, Lee SA, Myung SC and Kim
W: Combined effect of Hsp90 inhibitor geldanamycin and parthenolide
via reactive oxygen species-mediated apoptotic process on
epithelial ovarian cancer cells. Basic Clin Pharmacol Toxicol.
111:173–181. 2012.PubMed/NCBI
|
|
33
|
Yip-Schneider MT, Nakshatri H, Sweeney CJ,
Marshall MS, Wiebke EA and Schmidt CM: Parthenolide and sulindac
cooperate to mediate growth suppression and inhibit the nuclear
factor-kappa B pathway in pancreatic carcinoma cells. Mol Cancer
Ther. 4:587–594. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kim SL, Kim SH, Trang KT, et al:
Synergistic antitumor effect of 5-fluorouracil in combination with
parthenolide in human colorectal cancer. Cancer Lett. 335:479–486.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fulda S and Debatin KM: Death receptor
signaling in cancer therapy. Curr Med Chem Anticancer Agents.
3:253–262. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fearon ER and Vogelstein B: A genetic
model for colorectal tumorigenesis. Cell. 61:759–767. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bates RC, Edwards NS, Burns GF and Fisher
DE: A CD44 survival pathway triggers chemoresistance via lyn kinase
and phosphoinositide 3-kinase/Akt in colon carcinoma cells. Cancer
Res. 61:5275–5283. 2001.PubMed/NCBI
|
|
38
|
Woynarowski JM and Konopa J: Inhibition of
DNA biosynthesis in HeLa cells by cytotoxic and antitumor
sesquiterpene lactones. Mol Pharmacol. 19:97–102. 1981.PubMed/NCBI
|
|
39
|
Nakshatri H, Rice SE and Bhat-Nakshatri P:
Antitumor agent parthenolide reverses resistance of breast cancer
cells to tumor necrosis factor-related apoptosis-inducing ligand
through sustained activation of c-Jun N-terminal kinase. Oncogene.
23:7330–7344. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Vasilevskaya IA and O’Dwyer PJ:
17-Allylamino-17-demethoxygeldanamycin overcomes TRAIL resistance
in colon cancer cell lines. Biochem Pharmacol. 70:580–589. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Galligan L, Longley DB, McEwan M, Wilson
TR, McLaughlin K and Johnston PG: Chemotherapy and TRAIL-mediated
colon cancer cell death: the roles of p53, TRAIL receptors, and
c-FLIP. Mol Cancer Ther. 4:2026–2036. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Saturno G, Valenti M, De Haven Brandon A,
et al: Combining trail with PI3 kinase or HSP90 inhibitors enhances
apoptosis in colorectal cancer cells via suppression of survival
signaling. Oncotarget. 4:1185–1198. 2013.PubMed/NCBI
|
|
43
|
Ashkenazi A and Herbst RS: To kill a tumor
cell: the potential of proapoptotic receptor agonists. J Clin
Invest. 118:1979–1990. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Eberle J, Fecker LF, Forschner T, Ulrich
C, Rowert-Huber J and Stockfleth E: Apoptosis pathways as promising
targets for skin cancer therapy. Br J Dermatol. 156(Suppl 3):
18–24. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Pennarun B, Meijer A, de Vries EG,
Kleibeuker JH, Kruyt F and de Jong S: Playing the DISC: turning on
TRAIL death receptor-mediated apoptosis in cancer. Biochim Biophys
Acta. 1805:123–140. 2010.
|
|
46
|
Jung YH, Heo J, Lee YJ, Kwon TK and Kim
YH: Quercetin enhances TRAIL-induced apoptosis in prostate cancer
cells via increased protein stability of death receptor 5. Life
Sci. 86:351–357. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Prasad S, Ravindran J, Sung B, Pandey MK
and Aggarwal BB: Garcinol potentiates TRAIL-induced apoptosis
through modulation of death receptors and antiapoptotic proteins.
Mol Cancer Ther. 9:856–868. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kim YH, Lee DH, Jeong JH, Guo ZS and Lee
YJ: Quercetin augments TRAIL-induced apoptotic death: involvement
of the ERK signal transduction pathway. Biochem Pharmacol.
75:1946–1958. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Psahoulia FH, Drosopoulos KG, Doubravska
L, Andera L and Pintzas A: Quercetin enhances TRAIL-mediated
apoptosis in colon cancer cells by inducing the accumulation of
death receptors in lipid rafts. Mol Cancer Ther. 6:2591–2599. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sung B, Park B, Yadav VR and Aggarwal BB:
Celastrol, a triterpene, enhances TRAIL-induced apoptosis through
the down-regulation of cell survival proteins and up-regulation of
death receptors. J Biol Chem. 285:11498–11507. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Park MH, Jo M, Won D, Song HS, Song MJ and
Hong JT: Snake venom toxin from Vipera lebetina turanica sensitizes
cancer cells to TRAIL through ROS- and JNK-mediated upregulation of
death receptors and downregulation of survival proteins. Apoptosis.
17:1316–1326. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Fernandes-Alnemri T, Litwack G and Alnemri
ES: CPP32, a novel human apoptotic protein with homology to
Caenorhabditis elegans cell death protein Ced-3 and mammalian
interleukin-1 beta-converting enzyme. J Biol Chem. 269:30761–30764.
1994.PubMed/NCBI
|
|
53
|
Teitz T, Wei T, Valentine MB, et al:
Caspase 8 is deleted or silenced preferentially in childhood
neuroblastomas with amplification of MYCN. Nat Med. 6:529–535.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Mignotte B and Vayssiere JL: Mitochondria
and apoptosis. Eur J Biochem. 252:1–15. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Reed JC: Double identity for proteins of
the Bcl-2 family. Nature. 387:773–776. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
De Angelis PM, Stokke T, Thorstensen L,
Lothe RA and Clausen OP: Apoptosis and expression of Bax, Bcl-x,
and Bcl-2 apoptotic regulatory proteins in colorectal carcinomas,
and association with p53 genotype/phenotype. Mol Pathol.
51:254–261. 1998. View Article : Google Scholar
|
|
57
|
Maurer CA, Friess H, Buhler SS, et al:
Apoptosis inhibiting factor Bcl-xL might be the crucial member of
the Bcl-2 gene family in colorectal cancer. Dig Dis Sci.
43:2641–2648. 1998. View Article : Google Scholar
|
|
58
|
Rampino N, Yamamoto H, Ionov Y, et al:
Somatic frameshift mutations in the BAX gene in colon cancers of
the microsatellite mutator phenotype. Science. 275:967–969. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Sionov RV and Haupt Y: The cellular
response to p53: the decision between life and death. Oncogene.
18:6145–6157. 1999. View Article : Google Scholar : PubMed/NCBI
|