Introduction
Ovarian cancer is a leading cause of cancer
mortality from gynecological malignancies worldwide and, in the
United States and Japan, accounts for ~14,000 and 4,600 deaths
annually (1,2). Ovarian cancer predominantly consists
of four major histological types: serous, clear cell, endometrioid
and mucinous adenocarcinomas. The incidence of each of these
subtypes varies geographically: serous carcinoma is the most common
type in Western and Asian countries; clear cell adenocarcinoma is
prevalent (the second-most common) in Japan, but not in most of the
other Asian countries, and is not prevalent in European countries
(2,3). More than 70% of ovarian cancers are
diagnosed as advanced stage cancers (3). The majority of these patients with
advanced ovarian cancer show an initial response to platinum-based
chemotherapy; however, most of these patients relapse (3). Consequently, the 5-year overall
survival rate for ovarian cancer patients remains <50% (3). Personalized therapy using molecular
targeting drugs based on gene aberrations in tumor cells is a
promising option to improve the therapeutic efficacy of the
treatment for advanced ovarian cancer (4).
Recent genome-wide analysis has revealed alterations
of ovarian cancer genomes (5–8). The
most frequent alterations found are inactivating mutations in tumor
suppressor genes, such as TP53, PTEN, BRCA1, BRCA2, and
RB1, and in a SWI/SNF chromatin remodeling gene,
ARID1A. Other studies have detected activating mutations in
the oncogenes KRAS, BRAF, PIK3CA and ERBB2,
indicating that a subset of patients with ovarian cancer could
benefit from therapy using existing molecular targeting drugs
(5–7,9–11).
However, the prevalence and specificity of such oncogene
aberrations by clinicopathological factors, such as histological
subtype, and whether the aberrations are present in a mutually
exclusive manner, have not been fully examined in a defined
population. Thus, we constructed a profile of actionable
aberrations of 46 cancer-related genes in a cohort of 72 Japanese
ovarian cancer patients. The cohort was chosen from 267 consecutive
patients who had received surgery for ovarian cancer. Of these
patients, 72 patients with ovarian cancer were surgically treated
without prior chemotherapy, and the carcinomas in this cohort
included all four histological tumor types and tumors at various
stages.
Materials and methods
Patient cohort
Seventy-two patients with ovarian cancer (study
cohort subjects) were selected from 267 consecutive patients with
ovarian cancer (original cohort) who received surgery for ovarian
cancer in the Department of Obstetrics and Gynecology, Jikei
University School of Medicine, Tokyo, Japan, between 2000 and 2009.
The 72 subjects were surgically treated patients with ovarian
cancer who had not had prior chemotherapy. The selection procedure
ensured that all four major histological types and stages of tumor
were included in proportions similar to those found in the original
cohort and were also representative of the proportions found in all
Japanese ovarian cancer patients (Fig.
1) (2,3). Written informed consent was obtained
from all patients. This study was approved by the Institutional
Review Board of the contributing institutions.
The tumors and the adjacent non-cancerous tissues
were macro-dissected and flash-frozen after surgery. All tumor
tissues were resected from solid components without necrotic tissue
in each tumor. Several tumor tissues were randomly selected for
making paraffin sections and their cellularity was confirmed as
being >80%.
Clinical information for each patient, including
age, stage, histology, grade, residual tumor, treatment
information, and survival time from primary surgery, was collected
retrospectively. Tumors were staged in accordance with the
International Federation on Gynecology and Obstetrics (FIGO)
system. For each patient, the size of the residual tumor was
recorded at the end of surgery. Tumors resistant to
platinum-containing adjuvant chemotherapy (i.e., platinum
resistance) were defined as those in patients who exhibited
progression-free survival for <6 months after the completion of
chemotherapy.
Cell lines
Fourteen ovarian cancer cell lines were used in this
study. JHOC-5, JHOC-7, JHOC-8, and JHOC-9 were obtained from Riken
BioResource Center (Tsukuba, Japan). HAC-2 was provided by Dr M.
Nishida (Tsukuba University, Tsukuba, Japan). RMG-I and RMG-II were
provided by Dr D. Aoki (Keio University, Tokyo, Japan). A2780
(undifferentiated carcinoma) was provided by Dr E. Reed (NCI,
Bethesda, MD, USA) and 2008 was provided by Dr S.B. Howell (UCSD,
San Diego, CA, USA). SKOV3, MCAS, TYK-nu, Ov-1063, and SW626 were
obtained from ATCC (Rockville, MD, USA).
Deep sequencing of 46 cancer-related
genes
Genomic DNA was extracted using a QIAamp DNA mini
kit according to the manufacturer’s instructions (Qiagen, Limburg,
The Netherlands). Purified genomic DNA obtained from tumor tissues
and cell lines (10 ng) was used for the library construction using
the Ion AmpliSeq Cancer primer pool (cat. no. 4471262, Life
Technologies, Rockville, MD, USA) that targets 739 mutational
hotspot regions of 46 cancer-related genes and, additionally, a set
of custom primers for the E17K mutation hotspot in the AKT1
gene. Sequencing was run on the Ion Proton/PGM platform (Life
Technologies). The median depth of coverage for aligned reads was
3,024 x (2,010–35,534) by map quality ≥20. Data analysis, including
the hg19 human reference genome and variant calling, was carried
out using the Torrent Suite Software v3.2 (Life Technologies).
Sanger sequencing
Genomic DNA (10 ng) was amplified by PCR using KAPA
Taq DNA Polymerase (KAPA Biosystems, Woburn, MA, USA). PCR products
were directly sequenced in both directions using the BigDye
Termination kit and an ABI 3130xl DNA Sequencer (Applied
Biosystems, Foster City, CA, USA).
Real-time genomic PCR
Copy number variations suggested by deep sequencing
analysis were validated by real-time genomic PCR using a TaqMan
Copy Number Assay and the ABI 7900HT real-time PCR system (Applied
Biosystems). All TaqMan probes were purchased from Thermo Applied
Biosystems: ERBB2 (ID Hs01932585_cn), PTEN (ID
Hs05128032_cn), RB1 (ID Hs00331762_cn and a set of custom
primers), TP53 (ID Hs06424630_cn), and FGFR1 (ID
Hs02422066_cn) with RPPH1 (cat. no. 4403328) as a reference.
Data were analyzed using ABI PRISM 7900HT Sequence Detection
Software v2.3 for copy number analysis.
Statistical analysis
Statistical analyses were performed using JMP
software (SAS Institute, New York, NY, USA). Associations of the
gene alterations with clinicopathological factors were evaluated
using Fisher’s exact test. For survival analysis, the Cox
proportional hazard model was used for the univariate and
multivariate analyses.
Results
Profiling of aberrations in 46
cancer-related genes in 72 ovarian cancers
Clinical and histological characteristics of the
study subjects are provided in Table
I. The frequency of clear cell adenocarcinoma in this cohort
was higher than that found in other countries, reflecting known
prevalence of ovarian cancer in Japan (2,3).
 | Table ICharacteristics of 72 Japanese
patients with ovarian cancer. |
Table I
Characteristics of 72 Japanese
patients with ovarian cancer.
| Clinicopathological
variables | No. of patients
with ovarian cancer | Frequency (%) |
|---|
| Age |
| <60 | 49 | 68 |
| ≥60 | 23 | 32 |
| Stage |
| I | 31 | 43 |
| II | 7 | 10 |
| III | 27 | 37 |
| IV | 7 | 10 |
| Histology |
| Clear cell | 27 | 37 |
| Endometrioid | 10 | 14 |
| Mucinous | 3 | 4 |
| Other | 7 | 10 |
| Serous | 25 | 35 |
| Grade |
| G1 | 11 | 15 |
| G2 | 15 | 21 |
| G3 | 11 | 15 |
| Unknown/not
graded | 35 | 49 |
| Residual tumor
(cm) |
| ≤1 | 55 | 76 |
| >1 | 17 | 24 |
| Adjuvant
chemotherapy |
| Platinum | 1 | 1 |
| Platinum +
Taxane | 45 | 63 |
| Platinum +
Irinotecan | 18 | 25 |
| None | 8 | 11 |
| Platinum
resistance |
| Sensitive | 52 | 72 |
| Resistant | 11 | 15 |
| Not evaluated | 9 | 13 |
We sequenced genomic DNAs from 72 ovarian cancer
tissues, with a mean sequencing depth >2,000 in all cases,
followed by Sanger sequencing validation (representative results in
Fig. 2A). The results revealed 115
single-nucleotide variations (SNVs), but no insertions/deletions,
at 740 hotspot sites in 46 cancer-related genes. The 115 SNVs were
of 50 distinct types, and included 64 SNVs (43 types) that were
deduced as somatic according to data from the Catalogue of Somatic
Mutation in Cancer (COSMIC) database (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/).
The somatic nature of these mutations was verified in some samples
by Sanger sequencing of DNAs from corresponding non-cancerous
tissues (Table II). In addition,
another SNV detected in a single case, PIK3CA-N345H, caused
an amino acid change in the PIK3CA protein, which is recurrently
mutated in human cancers. Therefore, these 65 SNVs (44 types) were
considered to represent somatic mutations. Furthermore, Sanger
sequencing of DNAs from non-cancerous tissue revealed that the
remaining 50 SNVs (six types) consisted of two missense mutations
and 48 single-nucleotide polymorphisms (four types). Thus, in
total, 67 somatic missense mutations (46 types) were detected in
the study cohort (Table II).
| Table IISomatic SNVs detected in 72 patients
with ovarian cancer. |
Table II
Somatic SNVs detected in 72 patients
with ovarian cancer.
| Gene | Position (ch:
bp) | Nucleotide
change | AA change | COSMIC ID | Variant rate
(%) | SNV type | Validated as
somatic change | Mutated sample |
|---|
| TP53 | 17: 7,579,882 | G31C | E11Q | COSM11606 | 52.9 | Non-synonymous | C | T12 |
| 17: 7,579,358 | G329C | R110P | COSM11250 | 48.6 | Non-synonymous | C | T55 |
| 17: 7,578,535 | A395G | K132R | COSM11582 | 32.9 | Non-synonymous | C | T3 |
| 17: 7,578,534 | G396C | K132N | COSM43963 | 83.9 | Non-synonymous | C, S | T44 |
| 17: 7,578,526 | G404A | C135Y | COSM10801 | 70.5 | Non-synonymous | C | T8 |
| 17: 7,578,461 | G469T | V157F | COSM10670 | 64.4 | Non-synonymous | C, S | T22 |
| 17: 7,578,454 | C476T | A159V | COSM11148 | 59.0 | Non-synonymous | C, S | T33 |
| 17: 7,578,406 | G524A | R175H | COSM10648 | 32.1–76.4 | Non-synonymous | C, S | T23, T50, T57,
T59 |
| 17: 7,578,271 | A578T | H193L | COSM11066 | 58.5–73.2 | Non-synonymous | C, S | T16, T39 |
| 17: 7,578,269 | C580T | L194F | COSM10995 | 67.3 | Non-synonymous | C | T65 |
| 17: 7,578,263 | C586T | R196X | COSM10705 | 65.6 | Stop-gain | C, S | T48 |
| 17: 7,578,257 | G592T | E198X | COSM44241 | 50.8 | Stop-gain | C, S | T30 |
| 17: 7,578,212 | C637T | R213X | COSM10654 | 52.3 | Stop-gain | C, S | T21 |
| 17: 7,578,203 | G646A | V216M | COSM10667 | 71.8 | Non-synonymous | C | T13 |
| 17: 7,578,203 | G646T | V216L | COSM11210 | 81.3 | Non-synonymous | C, S | T67 |
| 17: 7,578,196 | T653G | V218G | COSM44198 | 71.5 | Non-synonymous | C | T43 |
| 17: 7,577,538 | G743A | R248Q | COSM10662 | 76.1 | Non-synonymous | C | T46 |
| 17: 7,577,120 | G818A | R273H | COSM10660 | 51.1–67.1 | Non-synonymous | C | T49, T51 |
| 17: 7,577,114 | G824T | C275F | COSM10701 | 59.5 | Non-synonymous | C, S | T69 |
| 17: 7,577,106 | C832A | P278T | COSM43697 | 22.1 | Non-synonymous | C | T36 |
| 17: 7,577,094 | C844T | R282W | COSM10704 | 94.3 | Non-synonymous | C, S | T25 |
| 17: 7,577,022 | C916T | R306X | COSM10663 | 55.1 | Stop-gain | C | T58 |
| 17: 7,574,003 | C1024T | R342X | COSM11073 | 83.1 | Stop-gain | C | T68 |
| PIK3CAa | 3: 178,916,876 | G263A | R88Q | COSM746 | 32.5 | Non-synonymous | C | T14 |
| 3: 178,921,551 | A1033C | N345H | | 65.6 | Non-synonymous | | T45 |
| 3: 178,936,074 | C1616G | P539R | COSM759 | 45.5 | Non-synonymous | C, S | T71 |
| 3: 178,936,082 | G1624A | E542K | COSM760 | 10.1–22.2 | Non-synonymous | C, S | T6, T42, T57,
T63 |
| 3: 178,936,091 | G1633A | E545K | COSM763 | 18.8–71.6 | Non-synonymous | C | T53, T60 |
| 3: 178,936,094 | C1636A | Q546K | COSM766 | 92.8–93.4 | Non-synonymous | C | T27, T28 |
| 3: 178,952,085 | A3140T | H1047L | COSM776 | 31.6 | Non-synonymous | C | T20 |
| 3: 178,952,085 | A3140G | H1047R | COSM775 | 12.5–42.4 | Non-synonymous | C, S | T4, T7, T11, T12,
T35, T36 |
| KRASa | 12: 25,398,284 | G35C | G12A | COSM522 | 6.8–22.1 | Non-synonymous | C | T15, T63 |
| 12: 25,398,284 | G35A | G12D | COSM521 | 29.1–95.7 | Non-synonymous | C, S | T10, T54, T72 |
| 12: 25,398,284 | G35T | G12V | COSM520 | 6.4–58.3 | Non-synonymous | C, S | T5, T29, T37,
T58 |
| 12: 25,398,285 | G34C | G12R | COSM518 | 46.1 | Non-synonymous | C, S | T4 |
| CDKN2Aa | 9: 21,971,161 | A197G | H66R | COSM14253 | 36.5 | Non-synonymous | C | T50 |
| 9: 21,971,203 | T155G | M52R | COSM608436 | 50.0 | Non-synonymous | C | T51 |
| FGFR2 | 10:
123,279,677 | C755G | S252W | COSM36903 | 21.3 | Non-synonymous | C | T36 |
| 10:
123,258,036 | A1645C | N549H | COSM250083 | 38.7 | Non-synonymous | C, S | T14 |
| PTENa | 10: 89,711,902 | T520G | Y174D | COSM28897 | 73.6 | Non-synonymous | C | T20 |
| 10: 89,720,799 | T950G | V317G | | 41.9 | Non-synonymous | S | T44 |
| AKT1a | 14:
105,246,551 | G49A | E17K | COSM33765 | 23.0 | Non-synonymous | C | T11 |
| CTNNB1a | 3: 41,266,103 | G100C | G34R | COSM5684 | 16.8 | Non-synonymous | C | T31 |
| KIT | 4: 55,593,464 | A1621C | M541L | COSM28026 | 56.2 | Non-synonymous | C | T2 |
| MET | 7: 116,411,966 | T3005C | V1002A | | 52.2 | Non-synonymous | S | T65 |
| NRASa | 1: 115,258,747 | G35A | G12D | COSM564 | 60.4 | Non-synonymous | C | T20 |
TP53 (38.9%), PIK3CA (25.0%), and
KRAS (13.9%) were the three most frequently mutated genes
(Table III). The other genes in
which mutations were found were PTEN, FGFR2, CDKN2A, AKT1,
CTNNB1, NRAS, MET and KIT. The frequency of the TP53,
PIK3CA and KRAS mutations were different in each
histological subtype. TP53 was more frequently mutated in
serous carcinomas (56.0%) than in the other subtypes (P=0.042 by
Fisher’s exact test, compared with non-serous patients with ovarian
cancer; 29.8%); PIK3CA was more frequently mutated in clear
cell carcinomas (48.1%) than in the other subtypes (P<0.001 by
Fisher’s exact test, compared with non-clear cell carcinoma
patients; 11.1%), as previously indicated (7,10,12–14).
KRAS was more frequently mutated in clear cell carcinomas (25.9%)
than in the other subtypes, consistent with previous reports
(P=0.034 by Fisher’s exact test, compared with non-clear cell
carcinoma patients; 6.7%) (9,10,15–17).
| Table IIISNVs and CNAs detected in 72 ovarian
cancers. |
Table III
SNVs and CNAs detected in 72 ovarian
cancers.
| No (%) |
|---|
|
|
|---|
| Type of
alteration |
Gene/alteration | Total (n=72) | Clear cell
(n=27) | Serous (n=25) | Endometrioid
(n=10) | Mucinous (n=3) | Others (n=7) |
|---|
| SNV | TP53 | 28 (38.9) | 5 (18.5) | 14 (56.0) | 2 (7.1) | 3 (100) | 4 (14.3) |
|
PIK3CAa | 18 (25.0) | 13 (48.1) | 0 | 2 (20.0) | 1 (33.3) | 2 (28.6) |
| KRASa | 10 (13.9) | 7 (25.9) | 0 | 1 (10.0) | 0 | 2 (28.6) |
| PTENa | 2 (2.8) | 0 | 1 (4.0) | 1 (10.0) | 0 | 0 |
| FGFR2 | 2 (2.8) | 1 (3.7) | 0 | 1 (10.0) | 0 | 0 |
|
CDKN2Aa | 2 (2.8) | 0 | 1 (4.0) | 0 | 1 (33.3) | 0 |
| AKT1a | 1 (1.4) | 1 (3.7) | 0 | 0 | 0 | 0 |
|
CTNNB1a | 1 (1.4) | 1 (3.7) | 0 | 0 | 0 | 0 |
| NRASa | 1 (1.4) | 0 | 0 | 1 (10.0) | 0 | 0 |
| MET | 1 (1.4) | 0 | 0 | 1 (10.0) | 0 | 0 |
| KIT | 1 (1.4) | 0 | 0 | 1 (10.0) | 0 | 0 |
| Total | 67 | 28 | 16 | 10 | 5 | 8 |
| CNAs |
ERBB2a/gain | 3 (4.3) | 0 | 0 | 1 (10.0) | 2 (67.7) | 0 |
| RBa/homozygous deletion | 2 (2.8) | 1 (3.7) | 0 | 0 | 0 | 1 (14.3) |
| PTENa/homozygous deletion | 1 (1.4) | 0 | 1 (4.0) | 0 | 0 | 0 |
| Total | 6 | 1 | 1 | 1 | 2 | 1 |
Copy number aberrations (CNAs) in the 46 genes were
deduced by calculating the ratios of the sequence read fraction in
each tumor compared to the sequence read fraction of a single
non-cancerous tissue subjected to sequencing. Loci that were
potentially affected were selected using the criteria of
>2-times gains and <1/4-times losses (suggesting homozygous
deletion), followed by verification with quantitative genomic PCR
analysis (Fig. 2B). Ten CNAs in
PTEN (1 case), RB1 (4 cases), TP53 (1 case),
ERBB2 (3 cases) and FGFR1 (1 case) were suggested in
10 tumors, and six of them were confirmed by quantitative genomic
PCR analysis; gains of the ERBB2 gene in three patients
(4.2%); and homozygous deletions of the RB1 gene in two
patients (2.8%) and of the PTEN gene in a patient (1.4%)
(Table III).
Profile of cancer-related gene actionable
alterations in the histological subtypes
A profile of therapeutically actionable alterations
was next constructed (Fig. 2C).
Genetic alterations, possibly affecting the gene function and
sensitivity to existing therapeutic drugs or strategies, were
selected here as actionable alterations. ERBB2 amplification
is a well-known actionable alteration (18–20).
All the SNVs detected in the AKT1, CTNNB1, KRAS, PIK3CA and
NRAS genes (31 SNVs in total) affected hotspot amino acids,
and were considered actionable as targets for existing protein
kinase inhibitors (21–25). In addition, the SNVs found in the
CDKN2A and PTEN genes, and the RB and
PTEN homozygous deletions, were considered actionable as
they are linked to responsiveness to several inhibitors (26–28).
In total, 41 actionable gene alterations were
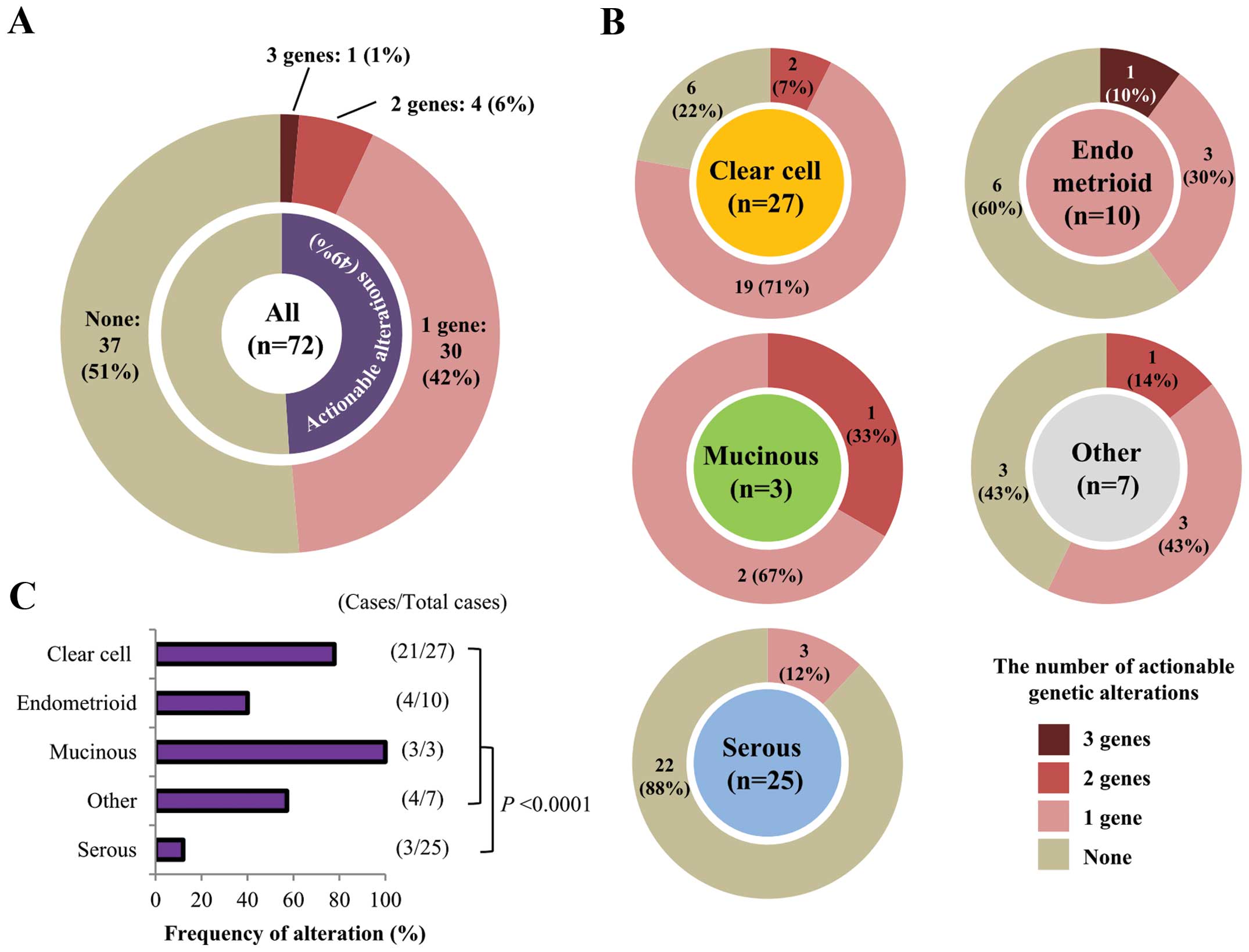
detected in 35 of 72 (48.6%) patients (Fig. 3A). These mutations tended to exist
in a mutually exclusive manner (Fig.
2C). In 30 of the 35 (85.7%) patients there was only one
actionable alteration, while five patients had multiple
alterations: one patient had mutations in three genes and four
patients had mutations in two genes. In these five patients,
fractions of mutant alleles were not evidently different between
mutated genes, therefore, these mutations were likely to have
occurred in similar fractions of cancer cells in each tissue. Of
the different histological subtypes, clear cell carcinomas showed
the highest frequency of actionable alterations (21/27; 77.8%),
whereas serous carcinomas had the lowest frequency (3/25; 12.0%;
Fig. 3B and C). This was largely
due to the differential occurrence of PIK3CA mutations in
the different histological subtypes (Fig. 2C). In non-serous carcinomas, the
majority (32/47; 68.1%) had at least one actionable mutation
(P<0.0001 by Fisher’s exact test, compared with serous
carcinomas).
We proceeded to investigate the associations between
the actionable alterations and clinicopathological factors
(Fig. 4). However, we identified
no significant associations between any of the actionable
alterations and age, stage, differentiation grade, presence/absence
of residual tumor, or therapeutic response to platinum therapy.
Actionable gene alterations were not significantly associated with
prognosis, either among all cases (data not shown) or among
non-serous carcinomas in particular.
Discussion
We constructed an actionable gene alteration profile
of ovarian cancer of a Japanese population using the deep genome
sequencing method. The majority (48.6%) of ovarian cancers, in
particular non-serous carcinomas (68.1%), were found to carry at
least one actionable alteration in the 46 cancer-related genes
examined. The TP53, PIK3CA and KRAS genes were top
three mutation genes. Consistent with previous reports (13,29),
TP53 and PIK3CA were preferentially mutated in serous
and clear cell carcinoma, respectively, while KRAS was
preferentially mutated in clear cell carcinoma (Table III). Distinct molecular features
of Japanese ovarian cancers were suggested. Frequency of
KRAS mutation in clear cell carcinoma (25.9%) was higher
than that in cases described previously (7%) (13), while frequency of hotspot
TP53 mutations (57.8%) in high-grade serous carcinoma were
less than that in Caucasian cases (>80%) (7,12).
These results provide basic information for the understanding of
ovarian carcinogenesis by different ethnicity.
According to this profile, therapeutic strategy
using molecularly targeted drugs can be considered (Fig. 5). Tumors with PIK3CA, AKT1
and PTEN mutations, which cause activation of the PI3K-AKT-mTOR
pathway, are targetable by PI3K/AKT/mTOR inhibitors (21,24,27).
Indeed, the results of clinical trials have demonstrated that
ovarian tumors with PIK3CA mutations exhibit a high response
rate to these inhibitors (30).
Tumors with ERBB2 amplification are targetable by ERBB2
inhibitors or antibodies, as evidenced by observations that ovarian
cancer cases with ERBB2 amplification exhibit high response
rates to an anti-ERBB2 antibody drug (20,31,32).
Tumors with KRAS and NRAS mutations
can be targeted by MAPK inhibitors, although in many cancers
therapeutic responses are less than expected based on clinical
trials (33). Sorafenib, which
targets RAF and other kinases and inhibits the RAS-RAF-ERK pathway,
has been shown to be an effective treatment for two Japanese
patients with recurrent ovarian clear cell carcinoma (34). Selumetinib, a MEK1/MEK2 inhibitor,
significantly suppressed the growth of a mouse xenograft of a human
ovarian clear cell adenocarcinoma (35). In addition, molecular targeted
therapies for tumors with CTNNB1 mutations and CDKN2A
inactivation have been indicated (22,28),
while tumors with RB1 mutations are treatable by TORC
inhibition based on synthetic lethality (28).
Importantly, the ability to prescribe a personalized
therapy based on genetic alterations, guided by the single
sequencing test described here, would be useful in clinical
settings. Ovarian cancers in Japan include more non-serous cases
than in other countries due to a higher fraction of clear cell
cancers. Our study indicates that frequencies of the actionable
alterations do not differ significantly by clinicopathological
factors, therefore, analysis of all non-serous ovarian cancers at
progressive stages will be an effective way to perform precision
medicine of ovarian cancers based on actionable gene aberrations.
In the strategy above, patients with serous ovarian cancers will
not benefit from the therapy. However, recent studies have also
suggested therapeutic approaches targeting p53 mutant proteins
(36). Such approaches will
benefit ovarian cancer patients with serous type ovarian cancer due
to frequent TP53 mutations.
The present study has limitations. First, the
utility of the above inhibitors has not been biologically proved.
Gene aberration profiles for the same 46 genes were also obtained
for 14 commonly used ovarian cancer cell lines (Table IV and Fig. 6). The results are consistent with
those deposited in the COSMIC database (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/).
PIK3CA and KRAS mutations are present in a subset of
ovarian cancer cell lines, and consistent in vitro
therapeutic responses by PI3K and MAPK inhibitors have been
reported in a few cell lines (35–37).
However, cell lines with other infrequent alterations were not
detected in these cell lines. Thus, more sets of cultured ovarian
cancer cells are needed to investigate the therapeutic significance
of such gene alterations. Second, tumor suppressor and chromatin
remodeling genes that lack mutation hotspots but are actionable for
synthetic lethality therapy were not examined in the present study:
BRCA1, BRCA2 and ARID1A are examples (5,6,9).
Tumors with BRCA1 and BRCA2 inactivation are
susceptible to PARP1 inhibitors, while therapeutic strategies
against tumors with ARID1A inactivation are also proposed
(38,39). A more comprehensiveprofiling study
including these genes are ongoing in our laboratory.
| Table IVSNVs detected in 14 ovarian cancer
cell lines. |
Table IV
SNVs detected in 14 ovarian cancer
cell lines.
| Sample name | Gene | Position (ch:
bp) | Nucleotide
change | AA change | Variant rate
(%) | SNV type |
|---|
| 2008 | PIK3CA | 3: 178,936,091 | G1633A | E545K | 47.9 | Non-synonymous |
| A2780 | ATM | 11:
108,123,551 | C1810T | P604S | 25.7 | Non-synonymous |
| Hac2 | PIK3CA | 3: 178,952,085 | A3140G | H1047R | 70.8 | Non-synonymous |
| JHOC7 | KIT | 4: 55,593,461 | G1606C | V540L | 58.5 | Non-synonymous |
| JHOC7 | PIK3CA | 3: 178,936,082 | G1624A | E542K | 35.8 | Non-synonymous |
| JHOC8 | FBXW7 | 4: 153,247,366 | G1196A | R399Q | 75.3 | Non-synonymous |
| JHOC8 | KIT | 4: 55,593,464 | A1621C | M541L | 99.8 | Non-synonymous |
| JHOC8 | TP53 | 17: 7,578,406 | G524A | R175H | 99.6 | Non-synonymous |
| JHOC9 | PIK3CA | 3: 178,936,082 | G1624A | E542K | 96.1 | Non-synonymous |
| MCAS | KRAS | 12: 25,398,284 | G35A | G12D | 80.7 | Non-synonymous |
| MCAS | PIK3CA | 3: 178,952,085 | A3140G | H1047R | 48.2 | Non-synonymous |
| MCAS | SMAD4 | 18: 48,604,690 | T1512A | S504R | 95.1 | Non-synonymous |
| OV-1063 | RB1 | 13: 49,037,903 | A2143T | K715X | 99.2 | Stop-gain |
| OV-1063 | TP53 | 17: 7,578,181 | C272T | P91L | 22.0 | Non-synonymous |
| OV-1063 | TP53 | 17: 7,577,118 | G820T | V274F | 59.0 | Non-synonymous |
| RMG-1 | CDKN2A | 9: 21,971,184 | A217C | S73R | 100.0 | Non-synonymous |
| SKOV-3 | FBXW7 | 4: 153,247,288 | G1274T | R425L | 44.8 | Non-synonymous |
| SKOV-3 | PIK3CA | 3: 178,952,085 | A3140G | H1047R | 46.0 | Non-synonymous |
| SW626 | KIT | 4: 55,593,464 | A1621C | M541L | 48.3 | Non-synonymous |
| SW626 | KRAS | 12: 25,398,284 | G35T | G12V | 51.0 | Non-synonymous |
| SW626 | SMAD4 | 18: 48,591,888 | G1051C | D351H | 99.6 | Non-synonymous |
| TYK-nu | KIT | 4: 55,593,464 | A1621C | M541L | 99.6 | Stop-gain |
| TYK-nu | NRAS | 1: 115,258,747 | G35A | G12D | 33.7 | Non-synonymous |
| TYK-nu | NRAS | 1: 115,256,530 | C181A | Q61K | 63.5 | Non-synonymous |
| TYK-nu | TP53 | 17: 7,578,406 | G524A | R175H | 99.3 | Non-synonymous |
Acknowledgements
This study was supported by Grants-in-Aid from the
Ministry of Health, Labor, and Welfare for Research for the 3rd
Term Comprehensive 10-Year Strategy for Cancer Control and
Promotion of Cancer Control Programs; and Extramural Collaborative
Research Grant of Cancer Research Institute, Kanazawa University.
M.T. and R.I. are awardees of the Research Resident Fellowship from
the Foundation for Promotion of Cancer Research for the 3rd Term
Comprehensive 10-Year Strategy for Cancer Control, Japan.
References
|
1
|
American Cancer Society. Cancer Facts
& Figures 2013. American Cancer Society Inc. http://www.cancer.org.
Atlanta: 2013
|
|
2
|
Haruta S, Furukawa N, Yoshizawa Y, Tsunemi
T, Nagai A, Kawaguchi R, Tanase Y, Yoshida S and Kobayashi H:
Molecular genetics and epidemiology of epithelial ovarian cancer
(Review). Oncol Rep. 26:1347–1356. 2011.PubMed/NCBI
|
|
3
|
Heintz AP, Odicino F, Maisonneuve P, Quinn
MA, Benedet JL, Creasman WT, Ngan HY, Pecorelli S and Beller U:
Carcinoma of the ovary. FIGO 26th Annual Report on the Results of
Treatment in Gynecological Cancer. Int J Gynaecol Obstet Off Organ
Int Fed Gynaecol Obstet. 95:S161–S192. 2006. View Article : Google Scholar
|
|
4
|
Vaughan S, Coward JI, Bast RC Jr, Berchuck
A, Berek JS, Brenton JD, Coukos G, Crum CC, Drapkin R,
Etemadmoghadam D, et al: Rethinking ovarian cancer: recommendations
for improving outcomes. Nat Rev Cancer. 11:719–725. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jones S, Wang TL, Shih IeM, Mao TL,
Nakayama K, Roden R, Glas R, Slamon D, Diaz LA Jr, Vogelstein B, et
al: Frequent mutations of chromatin remodeling gene ARID1A in
ovarian clear cell carcinoma. Science. 330:228–231. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wiegand KC, Shah SP, Al-Agha OM, Zhao Y
and Al E: ARID1A mutations in endometriosis-associated ovarian
carcinomas. N Engl J Med. 363:1532–1543. 2011. View Article : Google Scholar
|
|
7
|
Bell D, Berchuck A, Birrer M, Chien J,
Cramer D, Dao F, Dhir R, Di Saia P, Gabra H, Glenn P, et al:
Integrated genomic analyses of ovarian carcinoma. Nature.
474:609–615. 2011. View Article : Google Scholar
|
|
8
|
Kanchi KL, Johnson KJ, Lu C, McLellan MD,
Leiserson MD, Wendl MC, Zhang Q, Koboldt DC, Xie M, Kandoth C, et
al: Integrated analysis of germline and somatic variants in ovarian
cancer. Nat Commun. 5:31562014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kalamanathan S, Bates V, Lord R and Green
JA: The mutational profile of sporadic epithelial ovarian
carcinoma. Anticancer Res. 31:2661–2668. 2011.PubMed/NCBI
|
|
10
|
Kurman RJ and Shih I-M: Molecular
pathogenesis and extraovarian origin of epithelial ovarian cancer -
shifting the paradigm. Hum Pathol. 42:918–931. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tuefferd M, Couturier J, Penault-Llorca F,
Vincent-Salomon A, Broët P, Guastalla JP, Allouache D, Combe M,
Weber B, Pujade-Lauraine E, et al: HER2 status in ovarian
carcinomas: A multicenter GINECO study of 320 patients. PLoS One.
2:e11382007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ahmed AA, Etemadmoghadam D, Temple J,
Lynch AG, Riad M, Sharma R, Stewart C, Fereday S, Caldas C, Defazio
A, et al: Driver mutations in TP53 are ubiquitous in high grade
serous carcinoma of the ovary. J Pathol. 221:49–56. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kuo KT, Mao TL, Jones S, Veras E, Ayhan A,
Wang TL, Glas R, Slamon D, Velculescu VE, Kuman RJ and Shih IeM:
Frequent activating mutations of PIK3CA in ovarian clear cell
carcinoma. Am J Pathol. 174:1597–1601. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yamamoto S, Tsuda H, Takano M, Iwaya K,
Tamai S and Matsubara O: PIK3CA mutation is an early event in the
development of endometriosis-associated ovarian clear cell
adenocarcinoma. J Pathol. 225:189–194. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Singer G, Oldt R, Cohen Y, Wang BG,
Sidransky D, Kurman RJ and Shih I-M: Mutations in BRAF and KRAS
characterize the development of low-grade ovarian serous carcinoma.
J Natl Cancer Inst. 95:484–486. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gemignani ML, Schlaerth AC, Bogomolniy F,
Barakat RR, Lin O, Soslow R, Venkatraman E and Boyd J: Role of KRAS
and BRAF gene mutations in mucinous ovarian carcinoma. Gynecol
Oncol. 90:378–381. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mayr D, Hirschmann A, Löhrs U and Diebold
J: KRAS and BRAF mutations in ovarian tumors: a comprehensive study
of invasive carcinomas, borderline tumors and extraovarian
implants. Gynecol Oncol. 103:883–887. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen JS, Lan K and Hung MC: Strategies to
target HER2/neu overexpression for cancer therapy. Drug Resist
Updat. 6:129–136. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ross JS: Disease-free survival according
to degree of HER2 amplification for patients treated with adjuvant
chemotherapy with or without 1 year of trastuzumab: the HERA trial.
Breast Dis. 21:378–380. 2010.
|
|
20
|
Bang YJ, van Cutsem E, Feyereislova A,
Chung HC, Shen L, Sawaki A, Lordick F, Ohtsu A, Omuro Y, Satoh T,
et al: Trastuzumab in combination with chemotherapy versus
chemotherapy alone for treatment of HER2-positive advanced gastric
or gastro-oesophageal junction cancer (ToGA): a phase 3,
open-label, randomised controlled trial. Lancet. 376:687–697. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Carpten JD, Faber AL, Horn C, Donoho GP,
Briggs SL, Robbins CM, Hostetter G, Boguslawski S, Moses TY, Savage
S, et al: A transforming mutation in the pleckstrin homology domain
of AKT1 in cancer. Nature. 448:439–444. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Anastas JN and Moon RT: WNT signalling
pathways as therapeutic targets in cancer. Nat Rev Cancer.
13:11–26. 2013. View Article : Google Scholar
|
|
23
|
Ascierto PA, Schadendorf D, Berking C,
Agarwala SS, van Herpen CM, Queirolo P, Blank CU, Hauschild A, Beck
JT, St-Pierre A, et al: MEK162 for patients with advanced melanoma
harbouring NRAS or Val600 BRAF mutations: a non-randomised,
open-label phase 2 study. Lancet Oncol. 14:249–256. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Janku F, Wheler JJ, Naing A, Falchook GS,
Hong DS, Stepanek VM, Fu S, Piha-Paul SA, Lee JJ, Luthra R, et al:
PIK3CA mutation H1047R is associated with response to PI3K/AKT/mTOR
signaling pathway inhibitors in early phase clinical trials. Cancer
Res. 73:276–284. 2013. View Article : Google Scholar :
|
|
25
|
Jänne PA, Shaw AT, Pereira JR, Jeannin G,
Vansteenkiste J, Barrios C, Franke FA, Grinsted L, Zazulina V,
Smith P, et al: Selumetinib plus docetaxel for KRAS-mutant advanced
non-small-cell lung cancer: a randomised, multicentre,
placebo-controlled, phase 2 study. Lancet Oncol. 14:38–47. 2013.
View Article : Google Scholar
|
|
26
|
Sheppard KE and McArthur GA: The
cell-cycle regulator CDK4: an emerging therapeutic target in
melanoma. Clin Cancer Res. 19:5320–5328. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hutson TE, Escudier B, Esteban E,
Bjarnason GA, Lim HY, Pittman KB, Senico P, Niethammer A, Lu DR,
Hariharan S, et al: Randomized phase III trial of temsirolimus
versus sorafenib as second-line therapy after sunitinib in patients
with metastatic renal cell carcinoma. J Clin Oncol. 32:760–767.
2014. View Article : Google Scholar
|
|
28
|
Gordon GM, Zhang T, Zhao J and Du W:
Deregulated G1/S control and energy stress contribute to the
synthetic-lethal interactions between inactivation of RB and TSC1
or TSC2. J Cell Sci. 126:2004–2013. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yamamoto S, Tsuda H, Takano M, Tamai S and
Matsubara O: PIK3CA mutations and loss of ARID1A protein expression
are early events in the development of cystic ovarian clear cell
adenocarcinoma. Virchows Arch. 460:77–87. 2012. View Article : Google Scholar
|
|
30
|
Janku F, Wheler JJ, Westin SN, Moulder SL,
Naing A, Tsimberidou AM, Fu S, Falchook GS, Hong DS, Garrido-Laguna
I, et al: PI3K/AKT/mTOR inhibitors in patients with breast and
gynecologic malignancies harboring PIK3CA mutations. J Clin Oncol.
30:777–782. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yan M, Parker BA, Schwab R and Kurzrock R:
HER2 aberrations in cancer: implications for therapy. Cancer Treat
Rev. 40:770–780. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
McAlpine JN, Wiegand KC, Vang R, Ronnett
BM, Adamiak A, Köbel M, Kalloger SE, Swenerton KD, Huntsman DG,
Gilks CB, et al: HER2 overexpression and amplification is present
in a subset of ovarian mucinous carcinomas and can be targeted with
trastuzumab therapy. BMC Cancer. 9:4332009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Miller CR, Oliver KE and Farley JH: MEK1/2
inhibitors in the treatment of gynecologic malignancies. Gynecol
Oncol. 133:128–137. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Koshiyama M, Matsumura N, Baba T,
Yamaguchi K, Yoshioka Y and Konishi I: Two cases of recurrent
ovarian clear cell carcinoma treated with sorafenib. Cancer Biol
Ther. 15:22–25. 2014. View Article : Google Scholar :
|
|
35
|
Bartholomeusz C, Oishi T, Saso H, Akar U,
Liu P, Kondo K, Kazansky A, Krishnamurthy S, Lee J, Esteva FJ, et
al: MEK1/2 inhibitor selumetinib (AZD6244) inhibits growth of
ovarian clear cell carcinoma in a PEA-15-dependent manner in a
mouse xenograft model. Mol Cancer Ther. 11:360–369. 2012.
View Article : Google Scholar :
|
|
36
|
Muller PA and Vousden KH: Mutant p53 in
cancer: new functions and therapeutic opportunities. Cancer Cell.
25:304–317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Honig A, Hahne JC, Meyer S, Kranke P,
Häusler S, Diessner J, Dietl J and Engel JB: PI3K inhibitor
D-116883 is effective in in vitro models of ovarian cancer.
Anticancer Res. 32:2035–2041. 2012.PubMed/NCBI
|
|
38
|
Chan DA and Giaccia AJ: Harnessing
synthetic lethal interactions in anticancer drug discovery. Nat Rev
Drug Discov. 10:351–364. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Helming KC, Wang X, Wilson BG, Vazquez F,
Haswell JR, Manchester HE, Kim Y, Kryukov GV, Ghandi M, Aguirre AJ,
et al: ARID1B is a specific vulnerability in ARID1A-mutant cancers.
Nat Med. 20:251–254. 2014. View Article : Google Scholar : PubMed/NCBI
|