Introduction
The cell-killing action of radiotherapy is purposed
not only at tumor cells but also at endothelial cells (ECs) of the
tumor vasculature that provides solid tumors with blood (1,2). The
tumor vessel system, and in turn ECs, constitute a sensitive and
critical target for tumor radiotherapy, resulting from the
induction of radiation damage to the vasculature.
Targeting angiogenesis is an attractive therapeutic
strategy to inhibit tumor growth, particularly since this approach
has less probability of resulting in the development of drug
resistance. Radiation sensitization of tumors has the ability to
increase local tumor control and disease-free survival (3). The combination of radiotherapy and
targeted drugs with anti-angiogenic or anti-vascular effects has
been verified as an important aim for improving the therapeutic
efficiency in cancer treatment (4). Recently, several compounds were
investigated in preclinical studies and have since started clinical
cancer trials that combine anti-angiogenic agents with ionizing
radiation to improve the anticancer effect of radiation (5–9). One
of such candidates, SU5416, is a selective inhibitor of the
tyrosine kinase activity of the vascular endothelial growth factor
(VEGF) receptor Flk-1/KDR and is currently in phase III clinical
trials for the treatment of advanced malignancies by decreasing
vascularization and growth of various human cancers (10,11).
Single agent phase II clinical trials of SU5416 in patients with
metastatic melanoma resulted in potential inhibitory effects on
tumor vascularity (12), and an
earlier study reported that the treatment reduced tumor growth and
vascularization in an animal model of neuroblastoma (13). VEGF is known to be upregulated in
various human tumors (1). The
concept of combining anti-vascular compounds or angiogenesis
inhibitors has also been widely studied for the combination of VEGF
signaling inhibitors concurrently or sequentially together with
radiotherapy (14–22).
In this study, we investigated the mechanism by
which SU5416 increased radiation-induced antitumor and
anti-angiogenic effects by using endothelial cells as a
representative model of the tumor vasculature. Along with the
observed inhibition of VEGF signaling, we found that combination of
SU5416 with radiation therapy markedly enhanced the therapeutic
efficacy in endothelial cells.
Materials and methods
Antibodies and chemicals
Anti-cyclin B, anti-cyclin A, anti-cyclin E,
anti-extracellular signal-regulated kinase (ERK), anti-Akt,
anti-c-Jun N-terminal kinase (JNK), anti-p38 and anti-β-actin were
purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA). Anti-cleaved PARP1 antibody, anti-cleaved caspase-3,
anti-phospho-ERK, anti-phospho-Akt, anti-phospho-p38 and
anti-phospho-JNK were purchased from Cell Signaling Technology
(Danvers, MA, USA). Anti-γ-H2AX antibody was from Millipore. The
angiogenesis inhibitor SU5416 was synthesized at Sugen Inc., as
described previously (23). For
in vitro experiments, it was dissolved in DMSO to make a 10
mmol/l stock solution and stored at −20°C.
Cell culture
Human umbilical vein endothelial cells (HUVECs) were
maintained in endothelial cell basal medium (EGM-2) containing
EGM-2 SingleQuot growth supplements (both from Cambrex) and
maintained for no more than eight culture passages. The murine
endothelial cell line 2H11 was maintained in DMEM plus 10% fetal
bovine serum (FBS) in a humidified 10% CO2
environment.
Irradiation
Cells were plated in 60-mm dishes and incubated at
37°C under humidified conditions and 5% CO2 to 70–80%
confluence. Cells were irradiated with a 137Cs gamma-ray
source (Atomic Energy of Canada, Ltd., Ontario, ON, Canada) at a
dose rate of 3.81 Gy/min.
Colony-forming assay
SU5416 (1 μmol/l) was preincubated for 6 h before
radiation exposure and then incubated for a total of 72 h. After
14–20 days, colonies were stained with 0.4% crystal violet (Sigma,
St. Louis, MO, USA). The plating efficiency (PE) represents the
percentage of seeded cells that grew into colonies under the
specific culture conditions of the given cell line. The survival
fraction, expressed as a function of irradiation, was calculated as
follows: survival fraction, colonies counted/(cells seeded ×
PE/100). The plating efficiency of HUVEC and 2H11 were 0.72±0.18
and 0.79±0.15. To evaluate the radio-sensitizing effects of SU5416,
the ratio of radiation alone to radiation plus SU5416 was
calculated as the dose (Gy) for radiation alone divided by the dose
for radiation plus SU5416 at a surviving fraction of 10%.
Detection of apoptotic cells by Annexin V
staining
After SU5416 preincubation, radiation was added to
the cells, and the cells were subsequently incubated for 48 h.
Cells were then washed with ice-cold PBS, trypsinized, and
resuspended in 1X binding buffer [10 mm HEPES/NaOH (pH 7.4), 140 mm
NaCl, and 2.5 mm CaCl2] at 1×106 cells/ml.
Aliquots (100 μl) of cell solution were mixed with 5 μl Annexin V
FITC (BD Pharmingen) and 10 μl propidium iodide (PI) stock solution
(50 μg/ml in PBS) by gentle vortexing, followed by 15 min
incubation at room temperature in the dark. Buffer (400 μl, 1X) was
added to each sample and analyzed on a FACScan flow cytometer
(Becton-Dickinson, Franklin Lakes, NJ, USA). A minimum of 10,000
cells was counted for each sample, and data analysis was performed
in CellQuest software (BD Biosciences).
Immunohistochemistry
Immunohistochemistry was performed to determine the
nuclear distribution of γ-H2AX in individual cells. Cells were
grown on chamber slides for one day prior to irradiation or SU5416
treatments. After SU5416 exposure, cells were irradiated and
treated for various time-points. All treatments were performed
while cells remained attached to the slides, followed by fixing
with 4% paraformaldehyde and permeabilization with 0.2% Triton
X-100 in PBS. Detection was performed after blocking the slides in
10% FBS/1% bovine serum albumin (BSA) for 1 h with a 1:1,000
dilution of fluorescein isothiocyanate (FITC)-labeled mouse
monoclonal antibody against γ-H2AX, in the background-reducing
antibody diluent (DAKO plus S3022) (both from Millipore, Billerica,
MA, USA).
Western blotting
After SU5416 exposure, endothelial cells were
irradiated and cultured for 1 and 24 h. Protein from treated cells
was extracted with RIPA buffer, separated by SDS-polyacrylamide gel
electrophoresis (PAGE), and transferred to nitrocellulose
membranes. Membranes were blocked with 1% (v/v) nonfat dry milk in
Tris-buffered saline with 0.05% Tween-20 and incubated with the
indicated antibodies. Blots were reacted with primary antibodies at
1:1,000 dilutions and secondary antibodies at 1:5,000 dilutions.
Immunoreactive protein bands were visualized by Enhanced
Chemiluminescence (Amersham Biosciences) and scanned.
Wound healing scratch assay
Human endothelial cells were seeded onto 6-well
plates (Corning) at 2.5×104 cells/well with 3 ml of
medium. At two days, the monolayers were mechanically disrupted
with a sterile 200 μl pipette tip. The assay was performed in
duplicate and wells were photographed every 48 h prior to staining
with 0.2% crystal violet. Cell migration was monitored using a
Nikon Eclipse Ti microscope with a DS-Fi1 camera. The cells were
counted using ImageJ software (US National Institutes of Health,
Bethesda, MD, USA).
Invasion assay
The invasive ability in vitro was measured by
using Transwell chambers, according to the manufacturer's protocol.
Briefly, cells were seeded onto the membrane of the upper chamber
of the Transwell at a concentration of 4×105/ml in 150
μl of medium and were left untreated or treated with the indicated
doses of SU5416, radiation, or combine treatment for 24 h. The
medium in the upper chamber was serum-free, whereas the lower
chamber medium contained 10% FBS as a source of chemo-attractants.
Cells that passed through the Matrigel-coated membrane were stained
with Cell Stain Solution containing crystal violet supplied in the
Transwell invasion assay (Chemicon, Millipore, GA, USA) and
photographed after a 24-h incubation period.
Matrigel in vitro endothelial tube
formation assay
Endothelial cell tube formation was carried out on
Matrigel-coated chamber slides as described (24). The results of each assay were
photographed (Nikon Eclipse Ti microscope with DS-Fi1 camera) at
×40 magnification. Tube formation was quantified by counting the
number of connected cells in randomly selected fields at ×400
magnification with a microscope, and dividing that number by the
total number of cells in the same field. Tube formation was
quantified by counting the number of connected cells in randomly
selected fields at ×400 magnification with a microscope, and
dividing that number by the total number of cells in the same
field.
Statistical analysis
All data were plotted as the mean ± standard error
of the mean (SEM). Results of colony forming assays were analyzed
using paired t-test with SPSS 18.0 software (SPSS, Inc., Chicago,
IL, USA). All other data were analyzed by parametric repeated
measure one-way ANOVA followed by Tukey's-HSD test (SPSS 18.0).
Statistical significance was set at p<0.05.
Results
SU5416 radiosensitized ECs in vitro
To examine the effects of SU5416 on human
endothelial cell radiosensitivity, we selected two endothelial cell
lines, HUVEC and 2H11. We treated the EC cells with 1 μmol/l of
SU5416 for 24 h prior to receiving different doses of irradiation.
Using the concentration of SU5416 that showed a 20% decrease in
cell survival (1 μM) after a 6 h pretreatment (data not shown), we
further investigated the effects of SU5416 pretreatment on
radiation-induced cell death. The results of colony forming assay
of γ-irradiated EC cells with and without SU5416 pretreatment in
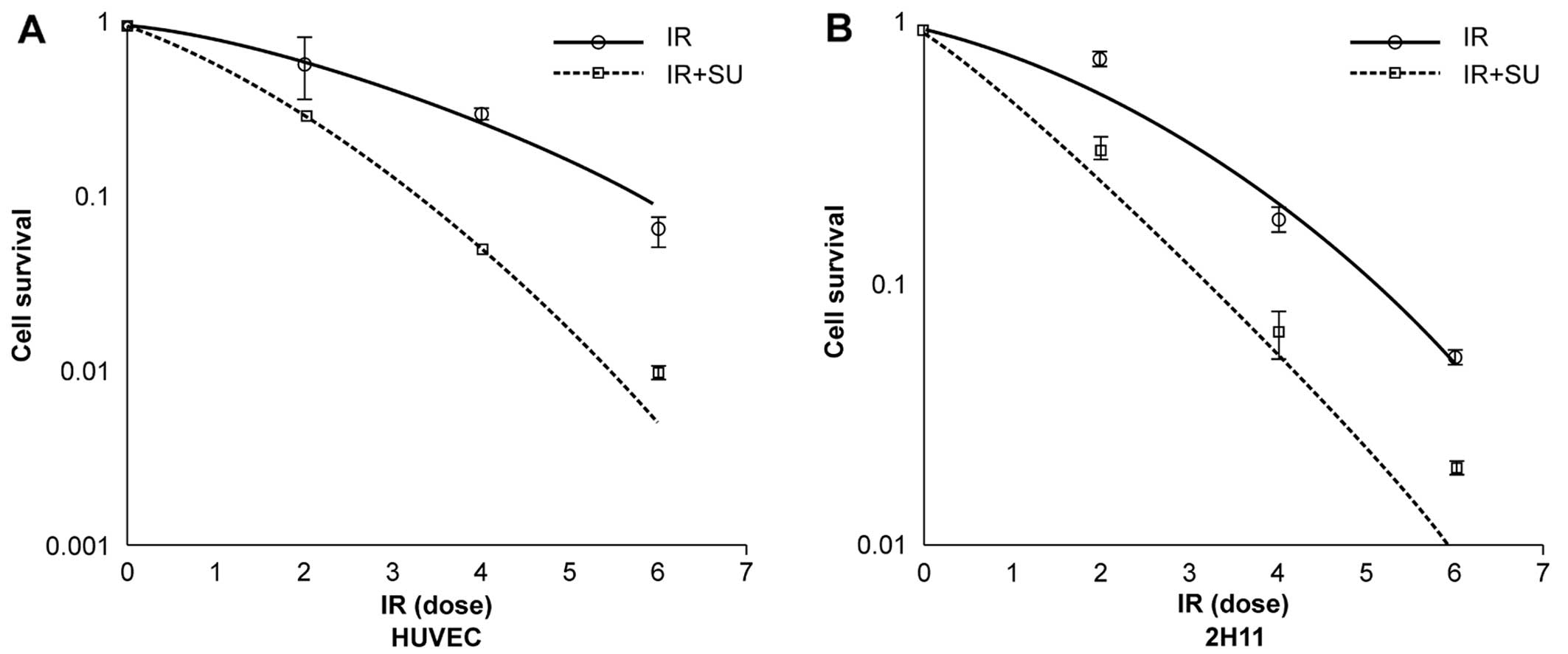
form of a survival curves are shown in Fig. 1. The experimental survival fraction
(S/S0) data points for γ-irradiated EC cells were fitted with
linear quadratic dose (D) dependent relation given by
S/S0=exp−(αD+βD2) where α and β are
constants. The fitted values of α and β for γ-irradiated EC cells
are given in Table I. The values
from the fitted curves show that there is a significant decrease in
survival fraction on combined treatment of SU5416 and radiation in
comparison to those cells exposed to radiation only for 90% cell
killing as shown in Table II.
 | Table IFitting parameters α and β for
survival assay data. |
Table I
Fitting parameters α and β for
survival assay data.
| Cell type | Radiation type |
α(Gy−1) |
β(Gy−1) |
|---|
| HUVEC | gamma-ray | 0.169±0.040 | 0.039±0.012 |
| gamma-ray +
SU5416 | 0.463±0.021 | 0.070±0.009 |
| 2H11 | gamma-ray | 0.166±0.610 | 0.054±0.117 |
| gamma-ray +
SU5416 | 0.626±0.610 | 0.025±0.124 |
| Table IIRadiation dose required for 90% cell
killing with and without SU5416. |
Table II
Radiation dose required for 90% cell
killing with and without SU5416.
| Cell type | Radiation type | Dose(Gy) for 90%
cell killing without SU5416 | Dose(Gy) for 90%
cell killing with SU5416 |
|---|
| HUVEC | gamma-ray | 5.84 | 3.33 |
| 2H11 | gamma-ray | 5.16 | 3.25 |
The REF values for γ-irradiation of SU5416 treated
EC cells are shown in Table III.
These data showed that SU5416 had radiosensitizing effects on ECs
in vitro.
| Table IIIREF and dose reduction values. |
Table III
REF and dose reduction values.
| Cell type | Radiation type | REF value | Dose reduction
(%) |
|---|
| HUVEC | gamma-ray | 1.75 | 43 |
| 2H11 | gamma-ray | 1.59 | 38 |
Effect of SU5416 on radiation-induced
apoptosis
Ionizing radiation induces cell death after DNA
damage (25). To investigate the
induction of apoptosis after the combination treatment, we assessed
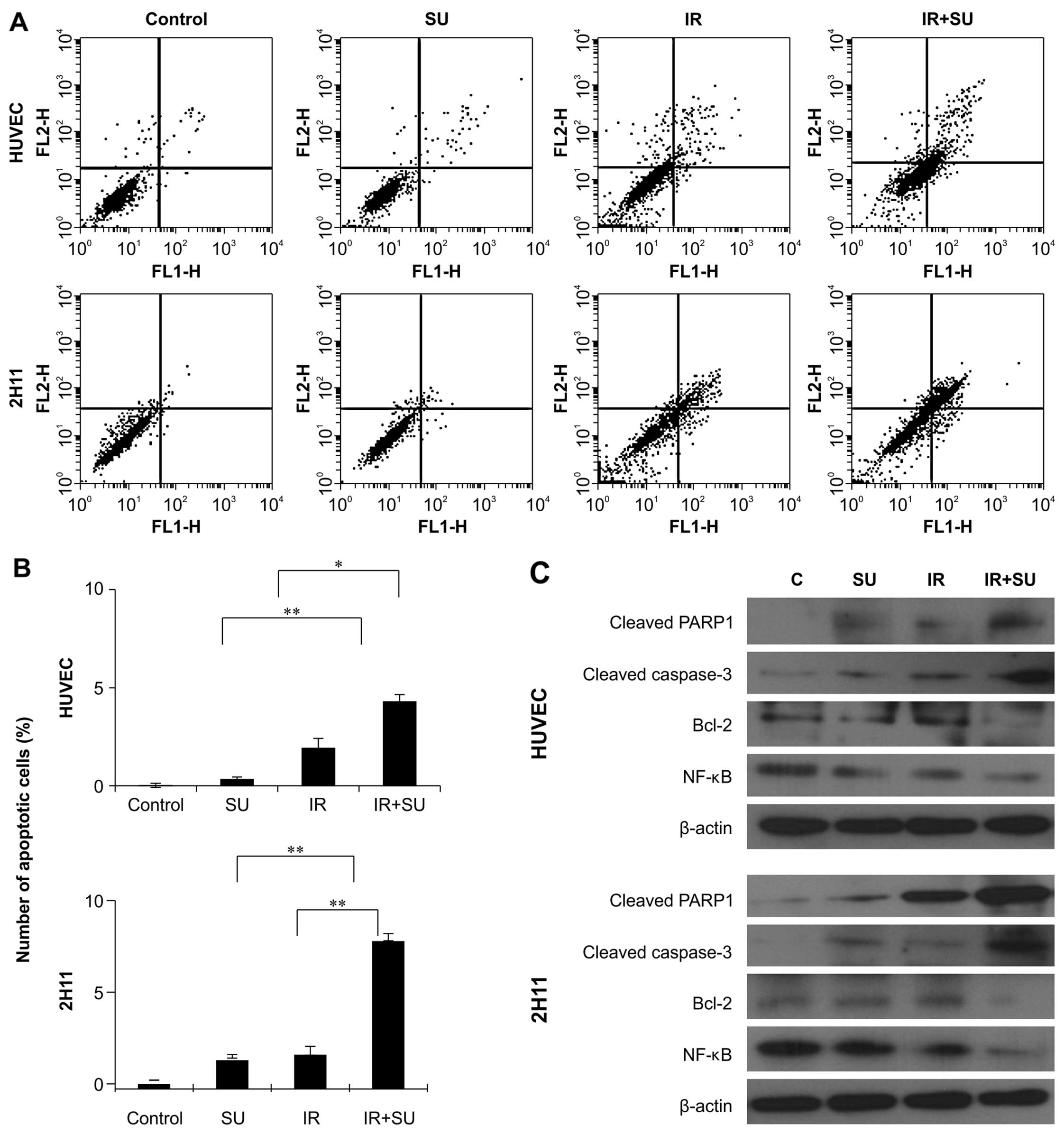
early apoptosis by Annexin V and PI staining. Notably, 48 h of
SU5416 and radiation exposure significantly increased the
percentage of early apoptotic cells in endothelial cell lines
(Fig. 2). We also examined whether
SU5416 enhanced radiation cytotoxicity resulted from the further
activation of the chief executioner of cell death, caspase-3 and
PARP fragmentation, in endothelial cells (Fig. 2C). Our results showed that
caspase-3 activation and PARP cleavage on treatment with SU5416 in
combination with radiation was enhanced in comparison to that
observed in the groups treated with SU5416 alone. We also monitored
the expression of anti-apoptotic Bcl-2 protein and cell survival
protein NF-κB and found that combination treatment clearly
decreased the expression of Bcl-2 and NF-κB protein in both
endothelial cell types (Fig.
2C).
Effects of SU5416 and radiation on cell
cycle phase distribution
To investigate what cellular mechanisms may underlie
the enhanced ionizing radiation-induced cell death following the
combined treatment of SU5416, we examined the cell cycle profiles
(Fig. 3A and B). Results showed
that combination treatments caused alterations in distribution of
cells in different phases of cell cycle in both endothelial cell
lines. SubG1 phase, indicating the apoptotic population, was only
moderately changed in endothelial cells following treatment with
SU5416; however, the combined treatment caused accumulation of
cells in subG1 phase in endothelial cells. Combination therapy
caused the most accumulation of cells at G2/M phases, suggesting
the highest increase of cell cycle arrest at G2/M phase in both
endothelial cell lines. We also studied the expression of cell
cycle regulator following combined treatment with SU5416 and
radiation (Fig. 3C). Western
blotting showed that radiation alone showed an accumulation of
cyclin B1 involved in the G2/M transition, whereas SU5416 alone
reduced the expression of the regulator. In contrast, cyclin B1
expression was attenuated by 24 h of SU5416 treatment, regardless
of the radiation dose (Fig.
3C).
Influence of SU5416 on radiation-induced
DNA damage and DNA repair activity
We further evaluated the DNA damage response by
analyzing the expression of the damage-responsive protein H2AX.
Histone H2AX is phosphorylated at Ser139 (γ-H2AX) in
response to double-strand break (DSB) processing. As expected,
SU5416-pretreated cells showed a higher level of ionizing
radiation-induced γ-H2AX (Fig. 4),
since it is known that SU5416 increases ionizing radiation-induced
DSB. The expression of γ-H2AX was consistently observed until 24 h
after radiation exposure in the presence of SU5416. Both
endothelial cells after the combined treatment with SU5416 and
radiation showed damaged DNA foci, which appeared at 1 h after
radiation exposure and remained even until 24 h (Fig. 4A). Furthermore, SU5416 treatment
itself did not alter the induction or subsequent disappearance of
foci at any time point examined.
Effects of SU5416 and radiation on
mitogen-activated protein kinase (MAPK) expression in ECs
Members of the MAPK, ERK and JNK are well known to
be involved in DNA damage responses (26,27).
We performed western blotting to assess the levels of the
phosphorylated form of pro-survival marker ERK and the
pro-apoptotic marker JNK (Fig. 5).
The level of p-ERK in IR irradiated cells was significantly
increased, whereas, sharply decreased in the combination treatment
cells.
Since the radioprotective response of ECs includes
Akt activation (28,29), we theorized that the enhanced
irradiation-induced apoptosis observed in SU5416-treated ECs was
caused by the inhibition of Akt activation. In line with this, an
increase of the active phosphorylated form of Akt protein (p-Akt)
was found in EC cells subjected to IR or SU5416 treatment; however,
combination treatment reduced the level of the induced p-Akt
(Fig. 5). This inhibition of p-Akt
may be responsible for the radiosensitizing effects of SU5416 by
enhancing the susceptibility of G2/M arrested cells to undergo
apoptosis in response to the IR induced DNA damage.
Effect of SU5416 and radiation on cell
motility and cell invasion
We next evaluated the effects of SU5416 on the
invasive and migratory capacities of endothelial cells using a
scratch assay. Notably, compared with treatment with SU5416 or
radiation alone, combination treatment significantly inhibited cell
migration (Fig. 6A). We also
performed Matrigel invasion assay to examine the effect of SU5416
combination treatment on tumor cell invasiveness and found that
combination treatment was highly effective at inhibiting tumor cell
invasion in both endothelial lines (Fig. 6B).
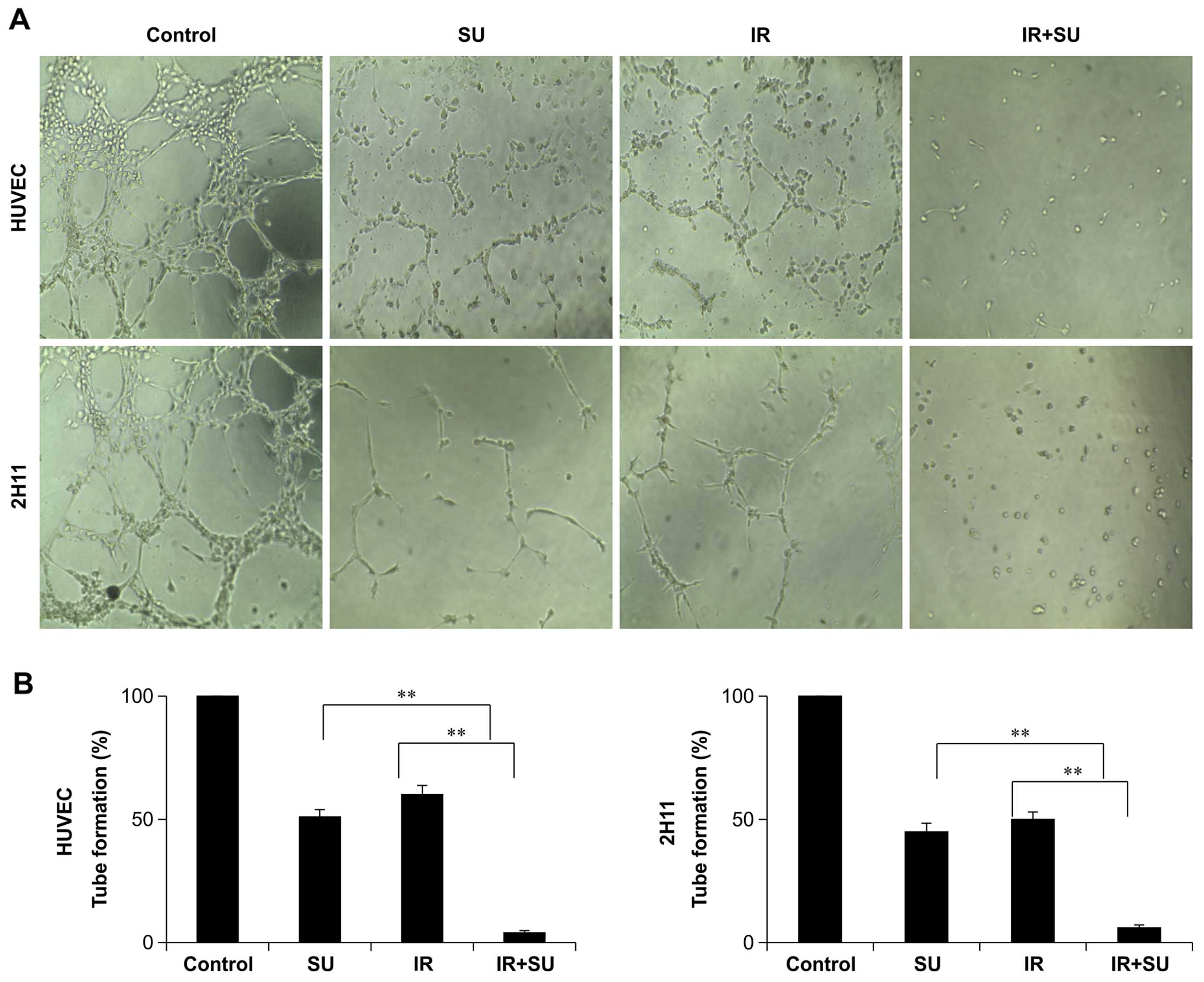
Combination treatment significantly
inhibits angiogenesis
The production of tubular structures is another
critical process in angiogenesis; therefore, we investigated the
effects of SU5416 on HUVEC and 2H11 tube formation. SU5416
treatment not only quantitatively reduced the number of formed
tubes in EC cultures, but also altered the morphology of the cells
(Fig. 7). These effects were
significantly greater in combination-treated cells as compared to
each treatment alone, suggesting at least an additive inhibition of
tube formation (Fig. 7).
Discussion
Combinations of radiotherapy with drugs are being
investigated to enhance the cure rates and decrease side effects.
In this regard, drugs with anti-angiogenic effects are of great
interest. The importance of angiogenesis in the progression of
human tumors has been presented by many outstanding academic
studies describing the relationship between angiogenic tumor
phenotypes and patient survival (30). These studies suggested that the
number of microvessels in primary tumors possibly predict the
prognosis of various tumors such as lung (31,32),
breast (33,34), bladder (35) and colon (36) carcinomas, and in tumors of the oral
cavity (37). Thus, control of
angiogenesis has been identified as an critical approach for the
therapeutic applications of human cancers (38,39).
The vascular endothelium is very resistant to the effects of
radiation, so VEGF may cause enhanced resistance to
radiation-induced damage (40).
Therefore, preclinical studies propose that anti-angiogenic agents
increase tumor control in response to radiation (6). Many such compounds are already in
clinical studies or have been approved for the treatment of certain
malignancies (41). However, it is
still unclear which combinations of signaling inhibitors would be
the most effective as an anticancer regimen and which of these
would benefit from a combination with radiotherapy. In this
context, we showed in ECs that the combination of VEGF signaling
inhibition increases the anticancer effects of each monotherapy. We
also found that radiotherapy in combination with VEGF signaling
inhibition highly increases anti-angiogenic and antitumor effects
of the respective drug therapies. In these studies, we used SU5416
as an inhibitor of VEGF signaling.
Recently, clinical studies have shown that the
combination of SU5416 and radiation may offer clinical gains in
patients with human cancer cell types (14); however, the mechanism underlying
this effect appears to be somewhat more complex than that predicted
in previous studies. Here, we provide a scientific rationale for
the clinical application of SU5416 as a radiosensitizer in ECs. We
have studied the various mechanisms by which SU5416 may increase
the therapeutic efficacy of radiation by inhibiting tumor cell
survival, cell cycle regulation, DNA repair activity, tumor cell
invasiveness and angiogenesis in ECs.
Previous studies show that SU5416 combined with
radiation significantly decrease the clonogenic survival abilities
and enhance the radiosensitivity of ECs by activating apoptosis
through PARP and caspase-3 (Figs.
1 and 2). When SU5416 was
treated after radiation, ECs failed to undergo mitosis seemingly
due to a block in transition from G2 to M phase that may result
from the downregulation of cyclin B1 (Fig. 3). The irradiation of cells leads to
electron scattering through the Compton effect, which subsequently
causes DNA breaks (42). H2AX
phosphorylation, a marker of DNA DSBs, was estimated as an
indication of the radiation-induced DNA damage response (43–45).
The results show that the combined treatment delayed the clearance
of γ-H2AX, suggesting that SU5416 maintains DNA damage, thus
increasing radiosensitivity (Fig.
4).
Migration of cancer cells is also a major event in
meta-static cascade of cancers. Cell migration is a highly
integrated process that is important for the growth of cancer cells
in various organs of the body; therefore, we examined the effect of
our combination therapy in inhibiting the migratory capacity of
ECs. VEGF-2 expression plays an important role in cell migration by
initiating many intracellular signaling pathways (46). Our data showed that decreases in
VEGF-2 expression are associated with a decrease in the migratory
potential of ECs following exposure to combination of SU5416 and
radiation (Fig. 6). In ECs,
ionizing radiation leads to phosphatidylinositol 3-kinase
(PI3K)/Akt pathway activation that contributes to post-irradiation
cell survival (28,29). Some tumor-derived growth factors,
particularly VEGF and basic fibroblast growth factor (bFGF), can
enhance the radioresistance of ECs (47), but this can also occur through
PI3K/Akt signaling (48).
Inhibitors of PI3K, wortmannin, and LY294002, are well known to
enhance the radiosensitivity of ECs (28); therefore, appropriate pharmacologic
blockers of the PI3K/Akt pathway could be applied to radiosensitize
the tumor vasculature. Akt is a pro-survival gene required for the
G2/M transition (49,50), and oncogenic Akt can enhance the
survival of cells after DNA damage by overcoming this checkpoint
block, as well as the apoptosis induced by DNA damage (51). Recent evidence shows that many
radiosensitizers possess anticancer effects through the inhibition
of Akt (52), and VEGF is known as
an important target of SU5416. VEGF dominantly acts on angiogenesis
via the PI3K/Akt and MAPK signaling cascade (53). We observed that the VEGF2
phosphorylation raised by irradiation was downregulated by both
combinations of SU5416 and radiation (Fig. 5). ERK is a key downstream component
of the RAF/MEK/ERK signaling pathway and aberrant signaling through
the ERK pathway was able to increase cell immortalization,
proliferation and resistance to radiation (54). Western blot analysis demonstrated
that radiation led to ERK activation, which was suppressed by the
post-irradiation treatment of SU5416. Active form of Akt has been
studied to indirectly inhibit JNK, a kinase known to control
apoptosis (55). Consistent with
previous studies, we observe that IR- and combination-treated EC
cells show a negative correlation between p-Akt and p-JNK protein
levels. ERK pathway activation is rescued with radioresistance
(56), and the observable decrease
in ERK activation after combination treatment likely sensitizes the
cells to apoptosis. Our data are consistent with a model whereby
combination treatment sensitizes the cells by relieving IR-induced
resistance to apoptosis in ECs by first limiting the Akt survival
pathway, and subsequently inducing JNK activation, thus committing
the cells to an apoptotic fate. Moreover, VEGF treatment of
endothelial cells significantly enhanced the tube formation on
growth factor reduced Matrigel (Fig.
7). Our data demonstrate that SU5416 in combination treatment
also inhibited VEGF-mediated endothelial cells tube formation
(in vitro angiogenesis assay).
In conclusion, our investigation demonstrated that
the anti-angiogenic compound SU5416 had an effective therapeutic
radiosensitizing potential in endothelial cells. This
radiosensitizing effect was associated with an inhibition of cell
survival, cell cycle regulation, DNA repair activity, tumor cell
invasiveness, and angiogenesis. We have provided evidence for the
molecular basis of chemoradiation treatment; however, in
vivo mouse model experiments should be carried out to minimize
the possible complications in clinical applications. Furthermore,
as the radiosensitizing effect of SU5416 in photon beam treatment
is well known, it will be necessary to compare its sensitizing
effect to proton or carbon beams in particle radiation to enhance
the biological efficiency and safety of these forms of
radiotherapy.
Acknowledgements
This study was supported by the National Research
Foundation of Korea (NRF) funded by the Korean government (MSIP;
no.NRF-2014R1A1A3053958,NRF2015R1A2A1A05001823).
References
|
1
|
Jubb AM, Pham TQ, Hanby AM, Frantz GD,
Peale FV, Wu TD, Koeppen HW and Hillan KJ: Expression of vascular
endothelial growth factor, hypoxia inducible factor 1α, and
carbonic anhydrase IX in human tumours. J Clin Pathol. 57:504–512.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Garcia-Barros M, Paris F, Cordon-Cardo C,
Lyden D, Rafii S, Haimovitz-Friedman A, Fuks Z and Kolesnick R:
Tumor response to radiotherapy regulated by endothelial cell
apoptosis. Science. 300:1155–1159. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Huber PE, Bischof M, Jenne J, Heiland S,
Peschke P, Saffrich R, Gröne HJ, Debus J, Lipson KE and Abdollahi
A: Trimodal cancer treatment: Beneficial effects of combined
antiangiogenesis, radiation, and chemotherapy. Cancer Res.
65:3643–3655. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Poggi MM, Coleman CN and Mitchell JB:
Sensitizers and protectors of radiation and chemotherapy. Curr
Probl Cancer. 25:334–411. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chan LW and Camphausen K: Angiogenic tumor
markers, anti-angiogenic agents and radiation therapy. Expert Rev
Anticancer Ther. 3:357–366. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wachsberger P, Burd R and Dicker AP: Tumor
response to ionizing radiation combined with antiangiogenesis or
vascular targeting agents: Exploring mechanisms of interaction.
Clin Cancer Res. 9:1957–1971. 2003.PubMed/NCBI
|
|
7
|
Gorski DH, Beckett MA, Jaskowiak NT,
Calvin DP, Mauceri HJ, Salloum RM, Seetharam S, Koons A, Hari DM,
Kufe DW, et al: Blockage of the vascular endothelial growth factor
stress response increases the antitumor effects of ionizing
radiation. Cancer Res. 59:3374–3378. 1999.PubMed/NCBI
|
|
8
|
Lee CG, Heijn M, di Tomaso E,
Griffon-Etienne G, Ancukiewicz M, Koike C, Park KR, Ferrara N, Jain
RK, Suit HD, et al: Anti-vascular endothelial growth factor
treatment augments tumor radiation response under normoxic or
hypoxic conditions. Cancer Res. 60:5565–5570. 2000.PubMed/NCBI
|
|
9
|
Geng L, Donnelly E, McMahon G, Lin PC,
Sierra-Rivera E, Oshinka H and Hallahan DE: Inhibition of vascular
endothelial growth factor receptor signaling leads to reversal of
tumor resistance to radiotherapy. Cancer Res. 61:2413–2419.
2001.PubMed/NCBI
|
|
10
|
Fong TA, Shawver LK, Sun L, Tang C, App H,
Powell TJ, Kim YH, Schreck R, Wang X, Risau W, et al: SU5416 is a
potent and selective inhibitor of the vascular endothelial growth
factor receptor (Flk-1/KDR) that inhibits tyrosine kinase
catalysis, tumor vascularization, and growth of multiple tumor
types. Cancer Res. 59:99–106. 1999.PubMed/NCBI
|
|
11
|
Mendel DB, Laird AD, Smolich BD, Blake RA,
Liang C, Hannah AL, Shaheen RM, Ellis LM, Weitman S, Shawver LK, et
al: Development of SU5416, a selective small molecule inhibitor of
VEGF receptor tyrosine kinase activity, as an anti-angiogenesis
agent. Anticancer Drug Des. 15:29–41. 2000.PubMed/NCBI
|
|
12
|
Peterson AC, Swiger S, Stadler WM, Medved
M, Karczmar G and Gajewski TF: Phase II study of the Flk-1 tyrosine
kinase inhibitor SU5416 in advanced melanoma. Clin Cancer Res.
10:4048–4054. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bäckman U, Svensson A and Christofferson
R: Importance of vascular endothelial growth factor A in the
progression of experimental neuroblastoma. Angiogenesis. 5:267–274.
2002. View Article : Google Scholar
|
|
14
|
Timke C, Zieher H, Roth A, Hauser K,
Lipson KE, Weber KJ, Debus J, Abdollahi A and Huber PE: Combination
of vascular endothelial growth factor receptor/platelet-derived
growth factor receptor inhibition markedly improves radiation tumor
therapy. Clin Cancer Res. 14:2210–2219. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Abdollahi A, Lipson KE, Han X, Krempien R,
Trinh T, Weber KJ, Hahnfeldt P, Hlatky L, Debus J, Howlett AR, et
al: SU5416 and SU6668 attenuate the angiogenic effects of
radiation-induced tumor cell growth factor production and amplify
the direct anti-endothelial action of radiation in vitro. Cancer
Res. 63:3755–3763. 2003.PubMed/NCBI
|
|
16
|
Wachsberger PR, Burd R, Marero N,
Daskalakis C, Ryan A, McCue P and Dicker AP: Effect of the tumor
vascular-damaging agent, ZD6126, on the radioresponse of U87
glioblastoma. Clin Cancer Res. 11:835–842. 2005.PubMed/NCBI
|
|
17
|
Ning S, Laird D, Cherrington JM and Knox
SJ: The antiangiogenic agents SU5416 and SU6668 increase the
antitumor effects of fractionated irradiation. Radiat Res.
157:45–51. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Griffin RJ, Williams BW, Wild R,
Cherrington JM, Park H and Song CW: Simultaneous inhibition of the
receptor kinase activity of vascular endothelial, fibroblast, and
platelet-derived growth factors suppresses tumor growth and
enhances tumor radiation response. Cancer Res. 62:1702–1706.
2002.PubMed/NCBI
|
|
19
|
Lu B, Geng L, Musiek A, Tan J, Cao C,
Donnelly E, McMahon G, Choy H and Hallahan DE: Broad spectrum
receptor tyrosine kinase inhibitor, SU6668, sensitizes radiation
via targeting survival pathway of vascular endothelium. Int J
Radiat Oncol Biol Phys. 58:844–850. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zips D, Eicheler W, Geyer P, Hessel F,
Dörfler A, Thames HD, Haberey M and Baumann M: Enhanced
susceptibility of irradiated tumor vessels to vascular endothelial
growth factor receptor tyrosine kinase inhibition. Cancer Res.
65:5374–5379. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Edwards E, Geng L, Tan J, Onishko H,
Donnelly E and Hallahan DE: Phosphatidylinositol 3-kinase/Akt
signaling in the response of vascular endothelium to ionizing
radiation. Cancer Res. 62:4671–4677. 2002.PubMed/NCBI
|
|
22
|
Schueneman AJ, Himmelfarb E, Geng L, Tan
J, Donnelly E, Mendel D, McMahon G and Hallahan DE: SU11248
maintenance therapy prevents tumor regrowth after fractionated
irradiation of murine tumor models. Cancer Res. 63:4009–4016.
2003.PubMed/NCBI
|
|
23
|
Krystal GW, Honsawek S, Kiewlich D, Liang
C, Vasile S, Sun L, McMahon G and Lipson KE: Indolinone tyrosine
kinase inhibitors block Kit activation and growth of small cell
lung cancer cells. Cancer Res. 61:3660–3668. 2001.PubMed/NCBI
|
|
24
|
Kumar P, Benedict R, Urzua F, Fischbach C,
Mooney D and Polverini P: Combination treatment significantly
enhances the efficacy of antitumor therapy by preferentially
targeting angiogenesis. Lab Invest. 85:756–767. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lea DE: Actions of Radiations on Living
Cells. 2nd edition. Cambridge University Press; New York, NY:
1955
|
|
26
|
Tang D, Wu D, Hirao A, Lahti JM, Liu L,
Mazza B, Kidd VJ, Mak TW and Ingram AJ: ERK activation mediates
cell cycle arrest and apoptosis after DNA damage independently of
p53. J Biol Chem. 277:12710–12717. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang J, Yu Y and Duerksen-Hughes PJ:
Protein kinases and their involvement in the cellular responses to
genotoxic stress. Mutat Res. 543:31–58. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kabakov AE, Makarova YM and Malyutina YV:
Radio-sensitization of human vascular endothelial cells through
HSP90 inhibition with 17-N-allilamino-17-demethoxygeldanamycin. Int
J Radiat Oncol Biol Phys. 71:858–865. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tan J and Hallahan DE: Growth
factor-independent activation of protein kinase B contributes to
the inherent resistance of vascular endothelium to
radiation-induced apoptotic response. Cancer Res. 63:7663–7667.
2003.PubMed/NCBI
|
|
30
|
Cherrington JM, Strawn LM and Shawver LK:
New paradigms for the treatment of cancer: The role of
anti-angiogenesis agents. Adv Cancer Res. 79:1–38. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fontanini G, De Laurentiis M, Vignati S,
Chinè S, Lucchi M, Silvestri V, Mussi A, De Placido S, Tortora G,
Bianco AR, et al: Evaluation of epidermal growth factor-related
growth factors and receptors and of neoangiogenesis in completely
resected stage I–IIIA non-small-cell lung cancer: Amphiregulin and
microvessel count are independent prognostic indicators of
survival. Clin Cancer Res. 4:241–249. 1998.PubMed/NCBI
|
|
32
|
Kawaguchi T, Yamamoto S, Kudoh S, Goto K,
Wakasa K and Sakurai M: Tumor angiogenesis as a major prognostic
factor in stage I lung adenocarcinoma. Anticancer Res.
17:3743–3746. 1997.
|
|
33
|
Toi M, Hoshina S, Takayanagi T and
Tominaga T: Association of vascular endothelial growth factor
expression with tumor angiogenesis and with early relapse in
primary breast cancer. Jpn J Cancer Res. 85:1045–1049. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gasparini G and Harris AL: Clinical
importance of the determination of tumor angiogenesis in breast
carcinoma: Much more than a new prognostic tool. J Clin Oncol.
13:765–782. 1995.PubMed/NCBI
|
|
35
|
Dickinson AJ, Fox SB, Persad RA, Hollyer
J, Sibley GN and Harris AL: Quantification of angiogenesis as an
independent predictor of prognosis in invasive bladder carcinomas.
Br J Urol. 74:762–766. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Takahashi Y, Kitadai Y, Bucana CD, Cleary
KR and Ellis LM: Expression of vascular endothelial growth factor
and its receptor, KDR, correlates with vascularity, metastasis, and
proliferation of human colon cancer. Cancer Res. 55:3964–3968.
1995.PubMed/NCBI
|
|
37
|
Williams JK, Carlson GW, Cohen C, Derose
PB, Hunter S and Jurkiewicz MJ: Tumor angiogenesis as a prognostic
factor in oral cavity tumors. Am J Surg. 168:373–380. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ferrara N: Molecular and biological
properties of vascular endothelial growth factor. J Mol Med Berl.
77:527–543. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Boehm-Viswanathan T: Is angiogenesis
inhibition the Holy Grail of cancer therapy? Curr Opin Oncol.
12:89–94. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Brieger J, Kattwinkel J, Berres M,
Gosepath J and Mann WJ: Impact of vascular endothelial growth
factor release on radiation resistance. Oncol Rep. 18:1597–1601.
2007.PubMed/NCBI
|
|
41
|
Jain RK, Duda DG, Clark JW and Loeffler
JS: Lessons from phase III clinical trials on anti-VEGF therapy for
cancer. Nat Clin Pract Oncol. 3:24–40. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chapman JD and Gillespie CJ:
Radiation-induced events and their time-scale in mammalian cells.
Adv Radiat Biol. 9:143–198. 1981. View Article : Google Scholar
|
|
43
|
Rogakou EP, Pilch DR, Orr AH, Ivanova VS
and Bonner WM: DNA double-stranded breaks induce histone H2AX
phosphorylation on serine 139. J Biol Chem. 273:5858–5868. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bonner WM, Redon CE, Dickey JS, Nakamura
AJ, Sedelnikova OA, Solier S and Pommier Y: GammaH2AX and cancer.
Nat Rev Cancer. 8:957–967. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bourton EC, Plowman PN, Smith D, Arlett CF
and Parris CN: Prolonged expression of the γ-H2AX DNA repair
biomarker correlates with excess acute and chronic toxicity from
radio-therapy treatment. Int J Cancer. 129:2928–2934. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Itokawa T, Nokihara H, Nishioka Y, Sone S,
Iwamoto Y, Yamada Y, Cherrington J, McMahon G, Shibuya M, Kuwano M,
et al: Antiangiogenic effect by SU5416 is partly attributable to
inhibition of Flt-1 receptor signaling. Mol Cancer Ther. 1:295–302.
2002.PubMed/NCBI
|
|
47
|
Peña LA, Fuks Z and Kolesnick RN:
Radiation-induced apoptosis of endothelial cells in the murine
central nervous system: Protection by fibroblast growth factor and
sphingomyelinase deficiency. Cancer Res. 60:321–327.
2000.PubMed/NCBI
|
|
48
|
Kumar P, Miller AI and Polverini PJ: p38
MAPK mediates gamma-irradiation-induced endothelial cell apoptosis,
and vascular endothelial growth factor protects endothelial cells
through the phosphoinositide 3-kinase-Akt-Bcl-2 pathway. J Biol
Chem. 279:43352–43360. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Datta SR, Brunet A and Greenberg ME:
Cellular survival: A play in three Akts. Genes Dev. 13:2905–2927.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Dangi S, Cha H and Shapiro P: Requirement
for phosphatidylinositol-3 kinase activity during progression
through S-phase and entry into mitosis. Cell Signal. 15:667–675.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kandel ES, Skeen J, Majewski N, Di
Cristofano A, Pandolfi PP, Feliciano CS, Gartel A and Hay N:
Activation of Akt/protein kinase B overcomes a G(2)/m cell cycle
checkpoint induced by DNA damage. Mol Cell Biol. 22:7831–7841.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Davies SP, Reddy H, Caivano M and Cohen P:
Specificity and mechanism of action of some commonly used protein
kinase inhibitors. Biochem J. 351:95–105. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wilhelm SM, Carter C, Tang L, Wilkie D,
McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, et al:
BAY 43-9006 exhibits broad spectrum oral antitumor activity and
targets the RAF/MEK/ERK pathway and receptor tyrosine kinases
involved in tumor progression and angiogenesis. Cancer Res.
64:7099–7109. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Roberts PJ and Der CJ: Targeting the
Raf-MEK-ERK mitogen-activated protein kinase cascade for the
treatment of cancer. Oncogene. 26:3291–3310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Figueroa C, Tarras S, Taylor J and Vojtek
AB: Akt2 negatively regulates assembly of the POSH-MLK-JNK
signaling complex. J Biol Chem. 278:47922–47927. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Shonai T, Adachi M, Sakata K, Takekawa M,
Endo T, Imai K and Hareyama M: MEK/ERK pathway protects ionizing
radiation-induced loss of mitochondrial membrane potential and cell
death in lymphocytic leukemia cells. Cell Death Differ. 9:963–971.
2002. View Article : Google Scholar : PubMed/NCBI
|