Introduction
Cancer remains one of the last bastions unconquered
by medicine. A multitude of risk factors, including chemicals and
viruses, have been identified and many genetic mechanisms
associated with carcinogenesis in different organs have been
elucidated. However, a major breakthrough in the control of cancer
will depend on identifying and addressing decisive cellular
mechanisms associated with invasive growth and metastasis, the
final stages of all types of cancer irrespective of their origin.
Developing effective therapeutic approaches to inhibit these final
stages of the disease is the key to the control of the cancer
epidemic.
Cancer progression is characterized by loss of
extracellular matrix (ECM) integrity which is a precondition for
invasive tumor growth and metastasis (1), and several mechanisms have been
proposed how cancer cells invade tissue and migrate through the
body, including free radical and enzyme related extracellular
matrix degradation. One of the most intriguing mechanisms
facilitating invasion and metastasis is the production of
plasminogen activators by malignant cells. The secretion of these
enzymes leads to the activation of plasminogen with triggering
plasmin-induced cascade of enzymatic degradation of the
extracellular matrix, facilitating cancer cell spread (2,3).
The potential significance of the mechanism of
plasmin-induced proteolysis has been highlighted by the fact that
the unique macromolecule apolipoprotein(a) [apo(a)] has been found
elevated in the blood of cancer patients and deposited in the
vicinity of cancer in tissue. Apo(a) is essentially composed of
structural homologues to kringle IV of the plasminogen molecule and
functions as a competitive inhibitor for the activation of plasmin
in fibrinolysis (4). This
structural homology of apo(a) to plasminogen also explains its
binding affinity to fibrinogen and fibrin.
Apo(a) is a high molecular weight adhesive protein
transported in blood bound to low-density-lipoproteins (LDL), via
apoprotein B-100 (ApoB), thereby forming lipoprotein(a), Lp(a)
(5). Moreover, by means of its
homology to angiostatin, a degradation product of plasminogen,
apo(a) exerts a direct anti-neoplastic effect by inhibiting
angiogenesis (4).
Apo(a) has been proposed as a competitive inhibitor
of plasmin-induced proteolysis in cancer and other diseases
(2). According to this concept,
apo(a) would be deposited at sites of accelerated tissue
degradation caused by the plasminogen/plasmin cascade.
To elucidate this important aspect towards the
control of cancer in humans we used a transgenic mouse model
expressing human apo(a) and lipoprotein(a). Moreover, this animal
model, as in humans, cannot produce endogenous ascorbate (vitamin
C). Thus, by decreasing ascorbate in the diet of these animals,
similar to chronic dietary vitamin C deficiency in humans, the
stability of the extracellular matrix could be experimentally
compromised in order to facilitate cancer spread.
Using this animal model we recently demonstrated
that apo(a) and Lp(a) are deposited in structurally impaired
vascular wall of animals kept on an ascorbate deficient diet
(6). Here we use this unique mouse
model to evaluate the significance of the plasmin-induced
proteolysis pathway and the role of Lp(a) as a key mechanism for
invasion and metastasis that can define new therapeutic approaches
towards the control of cancer.
Materials and methods
Animals generating human Lp(a)+;
Gulo−/− mice
The founder mouse strains used for cross breeding
were BALB/cBy-Gulo−/− mice. The strain,
BALB/cBy-Gulosfx/J was a spontaneous
mutation, mapped to the gulonolactone oxidase locus, a gene
necessary for vitamin C synthesis. The Gulo−/−
strain mouse was generated from heterozygous
Gulo+/− breeders obtained from
the Jackson Laboratory (Sacramento, CA, USA). The human apo(a)
mouse was obtained from the Mutant Mouse Regional Resource Centers
(MMRRC; Columbia, MO, USA). The human Apo B-100 mouse was obtained
from Taconic Biosciences, Inc. (Hudson, NY, USA) under an academic
research agreement.
Cross breeding for Gulo−/−;
Lp(a)+ mice
Human apo(a) and human apoB-100 mice wild-type for
the Gulo locus were bred to Gulo−/− mice
separately to generate two experimental founder mouse strains:
Gulo−/−; human apo(a)+ and
Gulo−/−; human apoB-100+. Subsequently,
the newly generated mouse breeders of both strains were crossed to
generate the new mouse strain: Gulo−/−; human
apo(a)+; human apoB-100+ named as
‘Gulo−/−; Lp(a)+’ strain.
Genotyping
Genotyping for the Gulo locus and its
homozygosity, as well as for the presence of human apoB-100
and human apo(a) was performed via TaqMan FAM Probe
Real-Time PCR at Transnetyx (Cordova, TN, USA) upon tail clip
tissue derived DNA obtained using standard DNA isolation and PCR
techniques.
Female Gulo−/−; Lp(a)+ mice and
wild-type female mice approximately one year of age were acclimated
for a week before treatments, housed in standard separator cages
with bedding on a 24-h light/dark schedule. All animals were cared
for in accordance with institutional guidelines for the care and
use of experimental animals.
Diet
The transgenic and control mice were divided into 4
different dietary groups in respect to dietary vitamin C intake: i)
low ascorbate intake for 6 weeks; ii) high ascorbate intake for 6
weeks; iii) low ascorbate intake for 3 weeks followed by high
ascorbate for 3 weeks; iv) high ascorbate intake for 3 weeks
followed by low ascorbate for 3 weeks. Control groups of Lp(a)+;
Gulo(−/−) mice without tumor inoculation were put on the same
vitamin C regimens. Wild-type controls, which included mice without
and with 4T1 inoculation, were kept on regular mouse chow
(Laboratory Rodent Diet 5001 from Test Diet; Purina Mills, LLC,
Richmond, IN, USA) and distilled water for 6 weeks. The high
vitamin C-supplemented diet was composed of the regular mouse chow
supplemented with 500 ppm L-ascorbyl-2-polyphosphate and distilled
water with 150 mg/l ascorbic acid, 0.01 mM EDTA. The low vitamin
C-supplemented diet was composed of the regular mouse chow
supplemented with 500 ppm L-ascorbyl-2-polyphosphate and distilled
water with 30 mg/l ascorbic acid, 0.01 mM EDTA. The
ascorbate-supplemented nutrient mix diet was milled and pressed by
Purina Mills (see Table I for
Lp(a)+; Gulo(−/−) and wild-type mouse group names and
treatments).
 | Table ILp(a)+; Gulo(−/−) and wild-type group
names and treatments. |
Table I
Lp(a)+; Gulo(−/−) and wild-type group
names and treatments.
| Group name (n=) | Mouse genotype | 4T1 Tumor
injection | Diet post 4T1 or PBS
injection |
|---|
| GAHCI (n=6) | Gulo(−/−);
Lp(a)+ | + | 6 weeks high vitamin
C |
| GAHC2 (n=6) | Gulo(−/−);
Lp(a)+ | + | 3 weeks high then 3
weeks low vitamin C |
| GALC1 (n=6) | Gulo(−/−);
Lp(a)+ | + | 6 weeks low vitamin
C |
| GALC2 (n=6) | Gulo(−/−);
Lp(a)+ | + | 3 weeks low then 3
weeks high vitamin C |
| GBHC1(n=6) | Gulo(−/−);
Lp(a)+ | − | 6 weeks high vitamin
C |
| GBHC2 (n=6) | Gulo(−/−);
Lp(a)+ | − | 3 weeks high then 3
weeks low vitamin C |
| GBLC1 (n=6) | Gulo(−/−);
Lp(a)+ | − | 6 weeks low vitamin
C |
| GBLC2 (n=6) | Gulo(−/−);
Lp(a)+ | − | 3 weeks low then 3
weeks high vitamin C |
| WT injected
(n=6) | Wild-type | + | Regular mouse
chow |
| WT Control (n=6) | Wild-type | − | Regular mouse
chow |
Experimental design
Lp(a)+; Gulo (−/−) mice were divided into two
groups: those receiving breast cancer cell injections and those
receiving mock injections. 4T1 breast cancer cells
(5×105 4T1 in 0.2 ml PBS) were injected into the mammary
pad of test mice and PBS mock injection into control group of mice.
After injections, mice were randomly assigned to 4 different
dietary regimens for 6 weeks (Table
I). Wild-type mice were divided into those receiving 4T1
injections and those receiving mock injections. Wild-type mice were
placed on regular mouse chow. After 6 weeks the mice were
sacrificed, their blood was drawn for serum analysis and tumors
were measured, excised, weighed, photographed and processed for
histology and immunohistochemistry. All procedures were conducted
under protocols approved by the Internal Animal Care and Use
Committee (IACUC).
Metastasis to lung
Evaluation of metastasis to the lung was done by
nodule count of photographed dorsal and ventral surfaces of freshly
harvested lung pairs kept in PBS prior to fixation in 10% neutral
buffered formalin.
Histology and immunohistochemistry of
tumors
Histopathology readings of primary tumors embedded,
cut and stained for elastic Van Gieson at IDEXX Reference
Laboratory (Sacramento, CA, USA) was conducted by Dr A.
DePaoli.
Formalin-fixed, paraffin-embedded primary tumors
from wild-type and Lp(a)+; Gulo−/− mice were
sectioned and immunostained for human apo(a), human ApoB, mouse
ApoB with respective negative and positive controls at HistoTox
Labs, Inc. (Boulder, CO, USA). The human apo(a) primary antibody
used does not cross-react with plasminogen.
Serum cholesterol profiling
Lipoprotein cholesterol gel electrophoresis,
staining, and analyses, including HDL, LDL and Lp(a), were provided
by Health Diagnostic Laboratory, Inc. (Richmond, VA, USA).
Quantitative values in mg/dl were derived by applying the provided
relative lipoprotein fraction cholesterol cargo data to total
cholesterol values. Serum total cholesterol was determined using
the Cholesterol/Cholesteryl Ester Quantitation Colorimetric kit II
from BioVision (Mountain View, CA, USA).
Serum apolipoprotein determinations
Human ApoB protein determinations in mouse sera were
made with the AssayMax Human ApoB ELISA kit from Assaypro LLC (St.
Charles, MO, USA). Human apo(a) protein determinations were made
with the Lipoprotein(a) ELISA kit from IBL International Gmbh
(Hamburg, Germany).
Serum ascorbate determination
Serum was processed from whole blood and stored at
−80°C until analyzed. Serum ascorbate analysis was performed using
the Ferric Reducing Ascorbate (FRASC) assay kit from BioVision.
Statistical analysis
The results were expressed as means + SD, as
indicated in the results, for the groups. Data was analyzed by
independent sample t-test.
Results
Primary tumor development in groups of
mice
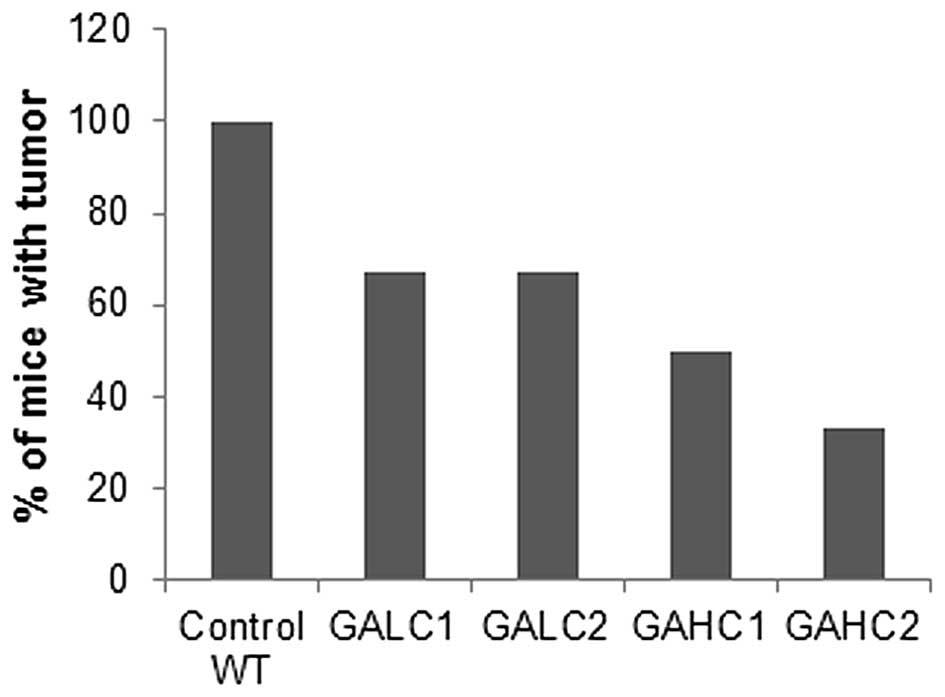
As shown in Fig. 1,
six weeks after injection of 4T1 breast cancer cells, 100% of
wild-type mice developed tumors, while primary tumor incidence in
the Lp(a)+; Gulo(−/−) mice kept on high ascorbate diet (GAHC1) was
reduced by 50% and in mice kept on low ascorbate (GALC1) for 6
weeks by 33%. Mice kept on low ascorbate for 3 weeks and then
switched to high ascorbate for another 3 weeks (GALC2) had 50%
lower incidence of tumors compared to wild-type mice and equal to
mice fed continuously low ascorbate diet for 6 weeks. Dietary
change from high ascorbate to low ascorbate (GAHC2) resulted in
further reduction in tumor incidence compared to mice on continuous
6-week high ascorbate diet, but the difference was not
significant.
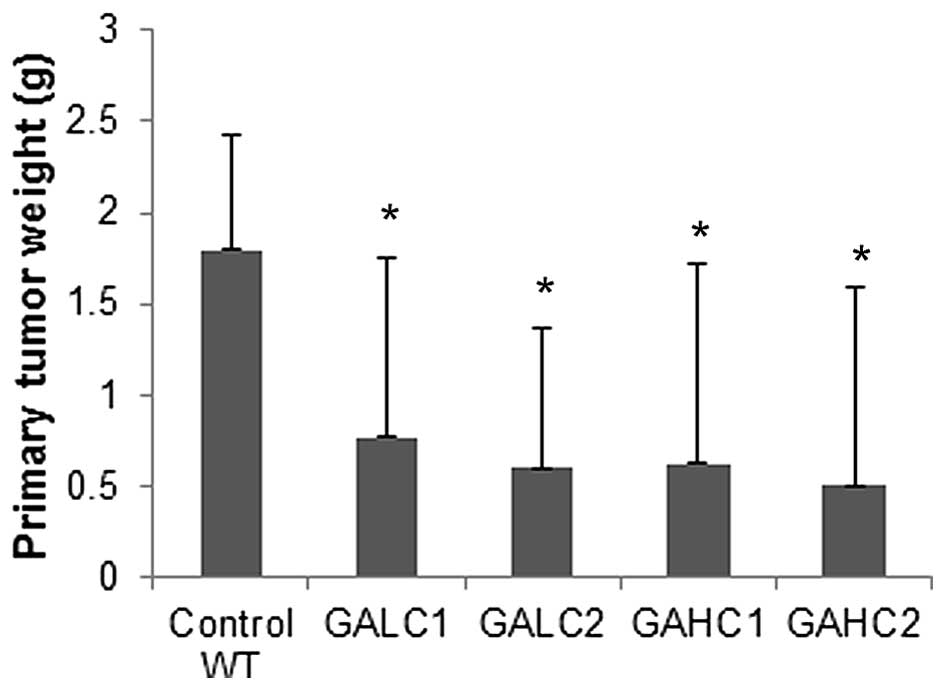
In Lp(a)+; Gulo(−/−) mice on low (GALC1) and high
(GAHC1) ascorbate diet for 6 weeks, the mean tumor mass was reduced
to 42.7% (P=0.05) and 35% (P=0.045) respectively, of control
wild-type mice tumors, as shown in Fig. 2. Tumors from Lp(a)+; Gulo(−/−) mice
on low then high ascorbate (GALC2) and those on high then low
ascorbate (GAHC2) had their mass reduced to 33.3% (P=0.01) and
28.3% (P=0.003), respectively, of the wild-type mice tumor mass. In
a small number of these mice with no primary tumor development,
residual tumor cells or inflammatory infiltrates found in the lungs
confirmed the viability of the injected cells and evidence of a
true rejection response
Histopathology
Histology of the tumors from wild-type and Lp(a)+;
Gulo (−/−) mice did not significantly differ by H&E staining
except for size; established primary tumors ranged from very large
and typical solid tumors to very small dense cysts with caseous
necrotic cores. Thus, no H&E figures are provided. These tumors
did differ significantly when immunostained, as shown in Fig. 7. Viable tumor tissue was
characterized by sheets of irregularly round, pleomorphic cells
with large, irregularly round nuclei and minimal to mild amounts of
eosinophilic cytoplasm, with 50–75% of the tumor being
necrotic.
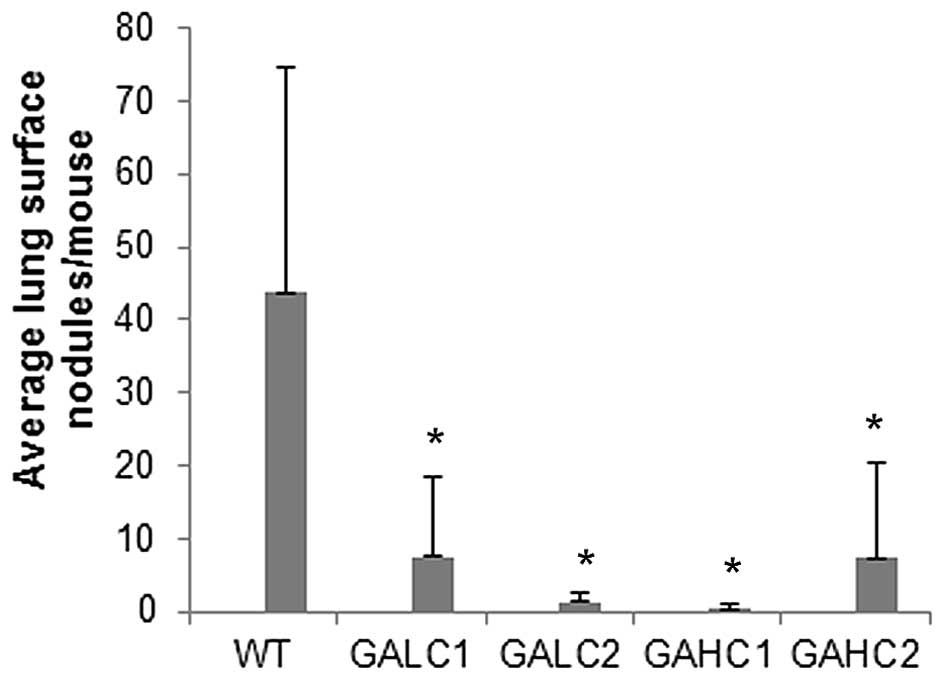
Metastasis
Metastasis to lungs was markedly reduced in Lp(a)+;
Gulo(−/−) mice both on low and high ascorbate diet for 6 weeks
compared to wild-type mice (Fig.
3). Total number of lung surface nodules per group was reduced
by 82.8% in Lp(a)+; Gulo (−/−) mice on low ascorbate, by 99.2% on
those on high ascorbate, by 97.3% in mice on low then high
ascorbate and by 83.2% on mice on high then low ascorbate compared
to control wild-type mice, as shown in Fig. 4.
Mean lung surface nodules per mouse were reduced by
83% (P=0.022) in mice on low ascorbate diet and by 94% (P=0.006) in
high ascorbate supplemented mice compared to control wild-type
mice, as shown in Fig. 5. Mean
lung surface nodules per mouse in mice on low then high ascorbate
diet were reduced by 97% (P=0.007) and in high then low ascorbate
diet by 83.3% (P=0.02) compared to control wild-type mice.
Average lung mass per mouse was reduced by 50%
(P=0.02) in animals on 6-week low ascorbate diet and by 55%
(P=0.01) in mice supplemented with high ascorbate for 6 weeks
compared to control wild-type mice, as shown in Fig. 6. Mean lung mass per mouse in mice
on low then high ascorbate diet was reduced by 52% (P=0.01) and in
high then low ascorbate diet by 55% (P=0.01) compared to control
wild-type mice.
Tumor apo(a), human ApoB-100 and mouse
ApoB protein immunostains in groups
Neither apo(a) nor human ApoB-100 were detected in
4T1 primary tumors from WT mice (Fig.
7D and E). In contrast, apo(a) and human ApoB-100 were
abundantly present in tumors from Lp(a)+; Gulo(−/−) mice
co-localized predominantly in the center of the tumor (Fig. 7A and B). Lp(a) was also detected in
the tumor capsule and peripheral tumor vasculature. Mouse ApoB was
found located outside areas of Lp(a) deposition in all mouse
tumors, mostly in areas of non-necrotic cells (Fig. 7C and F).
Serum lipoprotein cholesterol profiling
in mice
Cholesterol profile from mice representing different
dietary groups and wild-type mice is presented on Fig. 8. Size of primary tumors from
Lp(a)+; Gulo(−/−) mice immunostained positively for Lp(a) was in
general in inverse proportion to the intensity of Lp(a) cholesterol
bands as evaluated using equal amounts of serum electrophoretically
separated for lipoproteins and subsequently stained for particle
cholesterol cargo. Lp(a)+; Gulo(−/−) mice with no primary tumor
development consistently showed the presence of more intensive
Lp(a) than LDL band staining regardless of the type of ascorbate
diet. Lp(a) could not be detected in tumors from wild-type mice.
This observation also confirms the specificity of the antibodies
used previously in the present study.
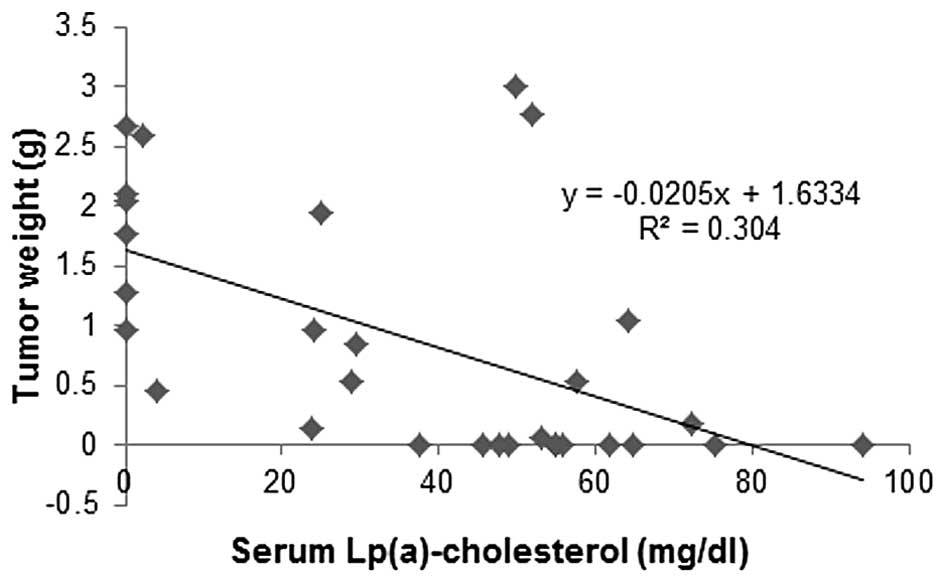
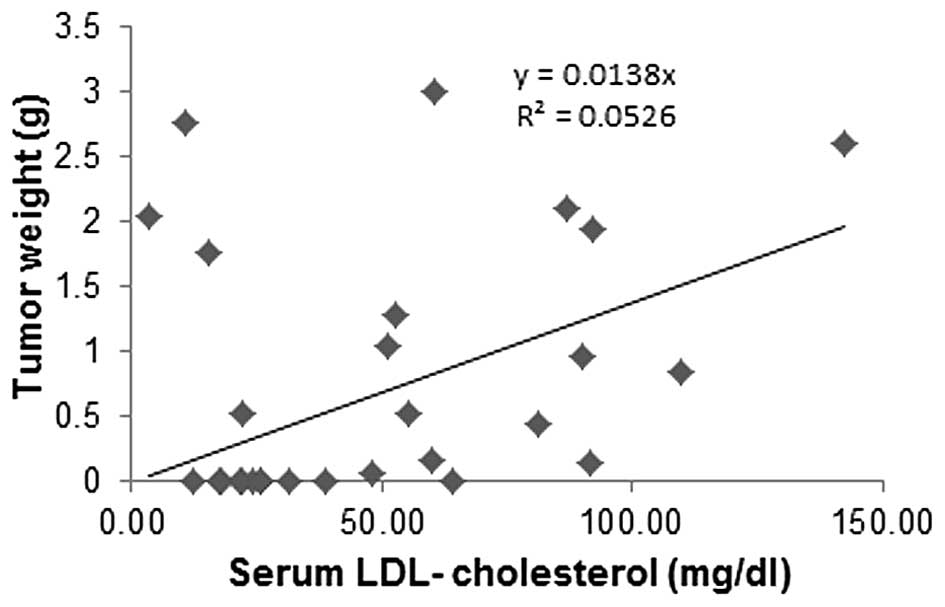
Correlation of tumor mass to Lp(a)
cholesterol and LDL cholesterol in mouse serum
Primary tumor weights from the experimental groups
carrying tumors were plotted against serum Lp(a) cholesterol and
LDL cholesterol cargo concentrations. Tumor mass from animals in
different dietary groups was inversely correlated to respective
serum Lp(a) cholesterol concentrations (Fig. 9), while a trend was towards
increased tumor mass with an increase in respective LDL cholesterol
concentrations (Fig. 10). There
was no correlation between HDL cholesterol and tumor mass (data not
shown).
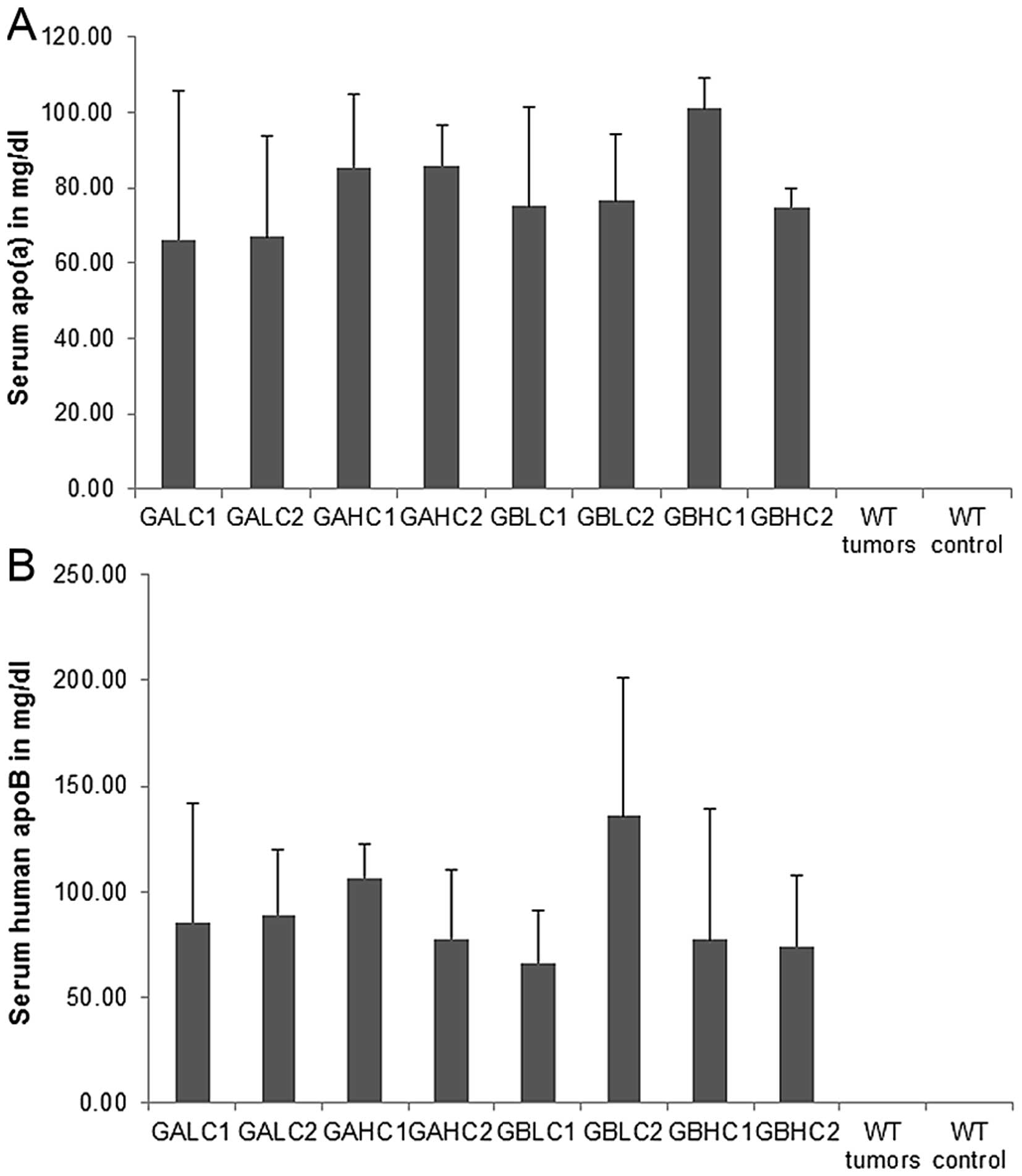
Serum apolipoproteins in mouse
groups
Average values for serum apo(a) levels in different
mouse groups are presented in Fig.
11A. No statistical differences were noted between different
dietary groups of mice and mice with and without 4T1 cell
injection. No serum apo(a) was detected in wild-type mice. There
was no significant difference in human ApoB-100 serum
concentrations between the groups as determined in equal volumes of
serum by ELISA (Fig. 11B). No
apparent trend was observed between groups for serum hApoB other
than confirming the variation of cholesterol cargo seen in the
cholesterol profiling in specimens with similar hApoB mass. Each
particle of Lp(a) has one ApoB molecule. The particles however, can
be more dense (less lipid cargo) or less dense (more lipid cargo)
due to variation in cholesterol, phospholipid and triglyceride
cargo.
Ascorbate levels in mouse groups
Ascorbate serum levels in 4T1 challenged Lp(a)+;
Gulo(−/−) mice on dietary supplementation with high vitamin C for 6
weeks (61.2 μM) were similar to its levels in mice kept initially
on low vitamin C for 3 weeks followed by high vitamin C
supplementation for 3 weeks (59.8 μM) (Fig. 12A). Mice on 6-week continuous low
dietary ascorbate intake had significantly lower serum ascorbate
levels (5.5 μM) compared to mice on a high ascorbate (GAHC1) or on
the last 3-week high ascorbate diet regimen (GALC2). Mice starting
on high vitamin C for 3 weeks followed by 3 weeks low vitamin C
diet had serum ascorbate level significantly reduced to 9.2 μM
compared to GAHC1 and GALC2 mice.
Serum ascorbate levels in Lp(a)+; Gulo(−/−) mice
with mock-injection (Fig. 12B)
equaled to 75.2 μM in mice on 3-week high ascorbate diet followed
by 3 week low ascorbate (GBLC2) and in mice on 6-week high
ascorbate diet (GBHC1). Mice on low vitamin C for 6 weeks (GBLC1)
had serum ascorbate reduced to 2.43 μM and those starting on high
vitamin C for 3 weeks followed by 3 weeks on low vitamin C (GBHC2)
had it reduced to 7.7 μM. As shown in Fig. 12C, wild-type mice challenged with
4T1 cancer cells had higher vitamin C serum level (63.7 μM) than
control wild-type mice (47.9 μM). The difference was statistically
significant.
Discussion
The main aim of the present study was to confirm key
mechanisms of cancer development and key factors towards its
control in mammals. Its focus was on the stability and integrity of
ECM as a decisive factor on cancer invasion and metastasis. Towards
this end we used the unique molecule, apo(a) and its
blood-transport form Lp(a), as a trace molecule. Due to its unique
structure, apo(a) is uniquely suited for this purpose. Firstly, as
a macromolecule, with a multiple size of collagen, it is formidably
suited to substitute for the connective tissue component at times
of the deficiency. Secondly, due to its structural homology to
plasminogen, it exhibits extreme adhesive properties that allow
apo(a) to function as an effective repair molecule at times of
compromised integrity/instability of the extracellular matrix and
increased need for its repair. Thirdly, as a competitive inhibitor
of plasmin-related pathways, it plays a role as a competitive
inhibitor of plasmin-induced proteolysis of ECM, a mechanism common
to all cancer cells, and, thereby, underscores the significance of
this mechanism in the promotion of cancer.
In the present study we used transgenic mice
expressing human Lp(a). Our first question was whether Lp(a) would
accumulate in cancers. We found Lp(a) to accumulate particularly
inside the core of tumors. Moreover, Lp(a) expressing mice showed a
30–60% reduction in development of primary tumors compared to
wild-type mice, which all developed primary tumors. Furthermore,
Lp(a) serum levels were found to be inversely proportional to tumor
mass. Human apoB-100, the structural core protein of LDL and Lp(a),
was almost exclusively co-localized with apo(a) inside the tumors,
indicating the presence of intact human Lp(a) molecules. In
contrast, mouse apoB, the structural protein of mouse LDL, was
distributed preferentially on the tumor periphery. These data
corroborated with observations that apo(a) has anti-neoplastic
properties (7–9). They are also in accordance with
studies in humans. A cohort study in 10,413 participants determined
that low Lp(a) concentration (defined as 80 mg/l) was associated
with cancer and all-cause deaths (P=0.001 and P=0.03, respectively)
(10).
Apo(a) and Lp(a) are found primarily in species,
including man, that have lost the ability to produce ascorbate
endogenously. Based on this observation, it has been suggested that
Lp(a) would function as a repair molecule for weakened ECM,
compromised by chronic ascorbate deficiency. Thus, we also wanted
to know whether dietary vitamin C deficiency would favor the
occurrence of tumors, giving rise to the deposition of Lp(a). In an
earlier study conducted in mice unable to synthesize vitamin C
endogenously (Gulo−/−), we demonstrated that dietary deficiency of
ascorbate facilitated tumor growth and expansion. Conversely, we
observed that in Gulo−/− mice higher intake of
ascorbate, which is essential for collagen synthesis and optimal
ECM formation, was positively correlated with inhibition of 4T1
tumor growth and metastasis to lungs. Corroborating histological
findings in animals on high ascorbate diet showed thick connective
tissue borders surrounding tumors, thereby confining their growth.
The more vitamin C provided in the diet, the greater the rate of
primary tumor rejection (11).
In the present study, we developed a mouse model
that combined both the inability of endogenous ascorbate synthesis
(Gulo−/−) and the production of human Lp(a) [Lp(a)+]. Here we
demonstrate that firstly, Lp(a) expressing mice had significantly
lower primary tumors as well as metastatic tumors in lungs compared
to wild-type mice inoculated with 4T1 breast cancer cells.
Secondly, among the Lp(a) expressing mice, those with high vitamin
C supplementation had significantly lower incidence of metastatic
tumors in lungs compared to those animals with low amounts of
vitamin C in the diet. In the Lp(a)+; Gulo−/− animals on
high vitamin C diet the metastatic cancers were almost completely
suppressed. In contrast to metastatic cancers, there was no
difference found between the study groups for primary tumors,
suggesting that vitamin C plays a specific inhibitory role in
metastatic stages of cancer.
In summary, the present study shows that vitamin C
is an effective inhibitor of cancer metastasis in mammals lacking
vitamin C synthesis and expressing Lp(a). In this context it is of
interest that the mouse model used in this study mimics human
metabolism with respect to these two important characteristics.
While ECM impairment is a precondition for cancer growth and
expansion, the prevention of ECM degradation and its reconstitution
have not been a focus of therapeutic cancer research. In the
absence of pharmacological stimuli for ECM growth and repair,
vitamin C should be considered as the most effective option in
cancer prevention and therapy. Clinical and epidemiological data
support this approach. Vitamin C deficiency is common in advanced
cancer patients and low plasma levels of this vitamin are
associated with shorter survival of cancer patients (12).
The presence of a Lp(a) in species that have lost
the ability for endogenous vitamin C production has been one of the
unsolved puzzles of science. Particularly compelling is the fact
that the appearance of Lp(a) about 40 million years ago coincided
with the loss of endogenous vitamin C synthesis by the ancestor of
man. Based on this and other observations it has been proposed that
Lp(a) functions as a substitute for vitamin C in stabilizing the
ECM, particularly at times of prolonged nutritional scarcity
(13). If this concept is
confirmed, any pathological condition related to vitamin C
deficiency would involve Lp(a) as compensating factor. In a
previous study we confirmed this concept for cardiovascular
disease, where a prolonged deficiency of dietary vitamin C leads to
the deposition of Lp(a) in the vascular wall and the development of
atherosclerotic plaques (6). Here
we confirm this concept in cancer. In this condition, characterized
by accelerated ECM degradation and associated with vitamin C
deficiency, Lp(a) contributes to protect and reconstitute the
ECM.
In conclusion, our results indicate that the
presence of Lp(a) in this animal model significantly decreased the
development of primary tumors and metastatic tumors in the lung,
suggesting that this molecule has anti-neoplastic properties. The
histological detection of Lp(a) deposits in and around tumors
suggest that this lipoprotein may participate in mitigating ECM
damage during cancer progression, in particular by inhibiting
proteolytic processes characteristic for all types of cancer cells.
The results imply that due to its unique structure, Lp(a) may play
a role in controlling tumor growth and expansion as a competitive
inhibitor of plasmin-induced proteolysis and through its adhesive
properties to ECM components. This study further confirms the
concept that Lp(a) functions as a surrogate for ascorbate in
disease and, thereby, stresses the role of ascorbate in fighting
cancer.
Acknowledgements
Tissues were processed by the IDEXX Reference
Laboratories Inc., and consulting pathologist Alexander DePaoli,
provided the histology slides and analysis. Phil Guadagno, MS at
Health Diagnostic Laboratories, Inc. provided lipoprotein
cholesterol determination expertise. Special thanks to Earl Rainey
for animal colony maintenance and Lei Shi for assistance with the
experiments. The research study was funded by Dr Rath Health
Foundation (Santa Clara, CA, USA), a non-profit organization.
References
|
1
|
Fidler IJ: Molecular biology of cancer:
Invasion and metastasis. Cancer: Principles and Practice of
Oncology. De Vita VT, Hellman S and Rosenberg SA: 5th edition.
Lippincott-Raven; Philadelphia, PA: pp. 135–152. 1997
|
|
2
|
Rath M and Pauling L: Plasmin-induced
proteolysis and the role of apoprotein(a), lysine and synthetic
analogs. Orthomolecular Med. 7:17–23. 1992.
|
|
3
|
Choong PF and Nadesapillai AP: Urokinase
plasminogen activator system: A multifunctional role in tumor
progression and metastasis. Clin Orthop Relat Res. 415(Suppl):
S46–S58. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wahl ML, Kenan DJ, Gonzalez-Gronow M and
Pizzo SV: Angiostatin’s molecular mechanism: Aspects of specificity
and regulation elucidated. J Cell Biochem. 96:242–261. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Utermann G: The mysteries of
lipoprotein(a). Science. 246:904–910. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cha J, Niedzwiecki A and Rath M:
Hypoascorbemia induces atherosclerosis and vascular deposition of
lipoprotein(a) in transgenic mice. Am J Cardiovasc Dis. 5:53–62.
2015.PubMed/NCBI
|
|
7
|
Lippi G, Franchini M, Salvagno GL and
Guidi GC: Lipoprotein[a] and cancer: Anti-neoplastic effect besides
its cardiovascular potency. Cancer Treat Rev. 33:427–436. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kim JS, Chang JH, Yu HK, Ahn JH, Yum JS,
Lee SK, Jung KH, Park DH, Yoon Y, Byun SM, et al: Inhibition of
angiogenesis and angiogenesis-dependent tumor growth by the cryptic
kringle fragments of human apolipoprotein(a). J Biol Chem.
278:29000–29008. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Trieu VN and Uckun FM: Apolipoprotein(a),
a link between atherosclerosis and tumor angiogenesis. Biochem
Biophys Res Commun. 257:714–718. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sawabe M, Tanaka N, Mieno MN, Ishikawa S,
Kayaba K, Nakahara K and Matsushita S; JMS Cohort Study Group. Low
lipoprotein(a) concentration is associated with cancer and
all-cause deaths: A population-based cohort study (the JMS cohort
study). PLoS One. 7:e319542012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cha J, Roomi MW, Ivanov V, Kalinovsky T,
Niedzwiecki A and Rath M: Ascorbate supplementation inhibits growth
and metastasis of B16FO melanoma and 4T1 breast cancer cells in
vitamin C-deficient mice. Int J Oncol. 42:55–64. 2013.
|
|
12
|
Mayland CR, Bennett MI and Allan K:
Vitamin C deficiency in cancer patients. Palliat Med. 19:17–20.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rath M and Pauling L: Hypothesis:
Lipoprotein(a) is a surrogate for ascorbate. Proc Natl Acad Sci
USA. 87:6204–6207. 1990. View Article : Google Scholar : PubMed/NCBI
|