Introduction
Renal cell carcinoma (RCC) is identified as the
seventh most common cancer for adult humans and account for 2% of
all cancers worldwide (1,2). The lack of efficient clinical
diagnostic and curative plan is one of the major reasons for RCC
lethality (3,4). Among all the subtypes of RCC,
clear-cell renal cell carcinoma (ccRCC) is the most frequently
discovered subtype. Patients with ccRCC usually have the lowest
probability of recovery compared with other RCC subtypes (5).
ccRCC is a metabolic disease characterized by the
dysregulation of many metabolic pathways, including abnormal oxygen
and energy consumption, as well as aberrant nutrient sensing
(6). During ccRCC progression,
inactivation of certain genes (such as von Hippel-Lindau, VHL) and
signaling pathway activation (such as MYC pathway) were reported to
take part in the abnormal metabolic profiles (7–10).
Recently, a large number of abnormally expressed miRNAs were
reported to be involved in the progression of ccRCC (11). Among these miRNAs, miR-184 is
widely dysregulated in various human cancers (12–15).
Previous studies have demonstrated that miR-184 was downregulated
and functions as a tumor suppressor in RCC (16,17).
However, the molecular mechanism of miR-184 in regulating RCC
progression is still unknown. It is worth mentioning that c-Myc, a
well-known oncogene that promotes cell cycle, cell differentiation
and apoptosis (18), is reported
to be a target of miR-184 in many human malignancies, including
non-small cell lung cancer (19,20),
nasopharyngeal carcinoma (14),
tongue squamous cell carcinoma (12) and hepatocellular carcinoma
(15). Notably, the expression of
c-Myc was indeed upregulated in ccRCC (10,21).
However, whether downregulated miR-184 is related to upregulated
c-Myc in ccRCC is still unknown.
In this study, we indentified PKM2 as a new target
of miR-184. Furthermore, the expression of miR-184 can be
negatively regulated by a well-known oncogene c-Myc. PKM2 is highly
expressed in human ccRCC samples. Remarkably, PKM2 knockdown leads
to metabolism reprogramming and growth inhibition in ccRCC cell
lines.
Materials and methods
Patient samples
Fifty pairs of human ccRCC and normal kidney samples
were collected from our institution, where the samples were
evaluated by a comprehensive clinical assessment. All the
paraffin-embedded sections and frozen samples used in the present
study were approved by ethics license of RJ2014N017.
Cell culture and transient
transfection
The 786-O and RCC4 cell lines were from the American
Type Culture Collection (ATCC; Manassas, VA, USA). The basic medium
for 786-O and RCC4 cells were RPMI and Dulbecco’s modified Eagle’s
medium (DMEM) supplemented with 2 mM glutamine, respectively. The
culture medium consisted of basic medium supplemented with 10%
fetal bovine serum (FBS), 1X penicillin/streptomycin (both from
Invitrogen, Grand Island, NY, USA). Cells were incubated at 37°C in
a humidified atmosphere of 5% CO2 and 95% air. For
transfection, cells were plated subconfluently onto each well of
24-well tissue culture plates 24 h before transfection. Transient
transfection of pre-miR-184 or control plasmid at a final
concentration of 20 nM per well was accomplished with Lipofectamine
2000 (Invitrogen) according to the manufacturer’s protocol. The
human PKM2 siRNA and control siRNA were purchased from OriGene. The
human c-Myc plasmid was generated according to standard molecular
biology techniques and verified by sequencing. c-Myc plasmids were
transfected with Lipofectamine 2000 according to the manufacturer’s
instructions.
RNA isolation and miRNA detection
Total RNA of cells and tissues were extracted using
TRIzol reagent (Invitrogen) according to the manufacturer’s
instructions. For isolation of small RNAs the mirVana miRNA
isolation kit (Ambion) was used. Detecting the miRNA mature form
was performed using the mirVana qRT-PCR miRNA detection kit
(Ambion). qRT-PCR primer sets were according to the manufacturer’s
instructions. We chose U6 small nuclear RNA as an internal control.
The real-time RT-PCR assays were performed using the 7500 Fast
Real-Time PCR System for quantitative mRNA detection and with iTaq
Fast SYBR-Green Supermix (Bio-Rad Laboratories). The primers for
real-time PCR were human actin: 5′-AAGGAGCCCCACGAGAAAAAT-3′
(forward) and 5′-ACCGAACTTGCATTGATTCCAG-3′ (reverse); human c-Myc:
5′-TCAAGAGGTGCCACGTCTCC-3′ (forward) and
5′-TCTTGGCAGCAGGATAGTCCTT-3′ (reverse); human PKM2:
5′-ATAACGCCTACATGGAAAAGTGT-3′ (forward) and
5′-TAAGCCCATCATCCACGTAGA-3′ (reverse).
MTT experiments
Cells were seeded in 96-well microtitre plates with
1×103 cells/well and incubated for 24 h in 100 μl
culture medium. MTT [100 μl (5 g/l)] was added to the cells, which
were then cultured for a further 4 h. Following the removal of the
supernatant fluid, 100 μl/well DMSO was added to the cells which
were agitated for 15 min. The absorbance was measured at 570 nm by
a microplate reader. Each assay was repeated three times.
Metabolism assays
786-O and RCC4 cells were seeded into 24-well plate
with 200 ml of medium in each well. To determine the levels of
glucose and lactate, the supernatants of cell culture media were
collected and assayed for glucose and lactate levels by using
glucose assay kit and lactate assay kit (BioVision Inc., Milpitas,
CA, USA) according to the manufacturer’s instructions. Glucose
consumption and lactate production were calculated based on the
standard curve, and normalized to the cell number.
Colony formation assay
Anchorage-independent growth was determined by the
ability of cells to form colonies in soft agar. 786-O and RCC4
cells were diluted in fresh medium, reseeded in 0.3% agar in 6-well
plates. Cell growth was allowed to proceed for 25 days. Formed
colonies were fixed with 10% formaldehyde and counted using the Col
Count instrument (Oxford Optronix Ltd., Abingdon, UK).
Luciferase reporter assay
Luciferase assay was performed as previously
described (22). Briefly, 786-O
cells of 50% confluence in 24-well plates were transfected with
FuGENE HD reagent (Roche). In each well, 200 ng pRL-TK-PKM2-3′-UTR
reporter firefly luciferase construct was co-transfected with 1 ng
Renilla plasmid and pre-miR-184, or Ctrl microRNA (Ambion)
into cells. Cell extracts were prepared 48 h after transfection and
luciferase activity was measured with the Dual-Luciferase reporter
assay system (Promega). PKM2 3′-UTR-luciferase or PKM2
3′-UTR-luciferase mutant activities were normalized by first
dividing the luciferase activity in each well by Renilla
activity.
Western blot analysis
Lysates were prepared from 1×107 cells by
dissolving cell pellets in 100 μl of lysis buffer containing
protease inhibitors. Lysates were centrifuged at 18,000 × g for 15
min and the supernatant was collected. The protein concentration
was estimated with the Bio-Rad protein assay kit (Bio-Rad
Laboratories, Hercules, CA, USA) using bovine serum albumin as a
standard. Sample proteins were resolved by 10% sodium
dodecylsulfate polyacrylamide gel (Bio-Rad Laboratories)
electrophoresis and then electrophoretically transferred to PVDF
membrane (Millipore) and blocked with 5% BSA (Sigma-Aldrich).
Subsequently the primary antibodies PKM2 (1:1,000; Cell Signalling
Technology), c-Myc (1:2,000; Santa Cruz Biotechnology) and actin
(1:10,000; Sigma-Aldrich) were added. After overnight incubation at
4°C the blots were washed, exposed to HRP-conjugated corresponding
secondary antibodies for one hour and finally were visualized by
ECL Advanced Solution (GE Healthcare Life Sciences). Digital images
were captured by Gel Doc™ gel documentation system (Bio-Rad
Laboratories).
Immunocytochemistry and in situ
hybridization
The procedures of immunohistochemical studies for
paraffin-embedded sections were performed as previously described
(23). Anti-human PKM2 antibody
(1:100; GeneTex Inc.) and anti-human c-Myc antibody (1:100; Santa
Cruz Biotechnology) were used as primary antibody. miR-184 in
situ hybridization assay was conducted by using locked nucleic
acid (LNA)-digoxigenin (DIG)-labeled probes (Exiqon, Vedbæk,
Denmark) as previously described (24). Briefly, deparaffinized sections
were treated with Proteinase K for 40 min. The hybridization was
performed at 45°C for 75 min in a hybridization buffer containing
Dual-DIG-labelled LNA probes miR-184 detection probe (probe
sequence: ACCCTTATCAGTTCTCCGTCCA) or scramble-miR (probe sequence:
GTGTAACACGTCTAT ACGCCCA) (Exiqon). Sections were blocked in a
blocking buffer for 30 min and washed in PBS three times.
miR-184 target gene prediction
algorithms
The presence of miR-184 target gene binding sites
was analyzed using miRWalk, which collates data from multiple
prediction programs as previously described (25).
Statistical analysis
Statistical analyses were performed using the SPSS
software package. Results are expressed as mean ± SD of at least
three independent experiments. Statistical significance was
assessed by two-tailed Student’s t-test. Differences were
considered statistically significant at P<0.05. P<0.05,
P<0.01 and P<0.001.
Results
c-Myc negatively regulates miR-184
expression in ccRCC
In order to investigate the relationship between
c-Myc and miR-184, we first examined their expression in 50 matched
pairs of human ccRCC samples and tumor adjacent normal kidney
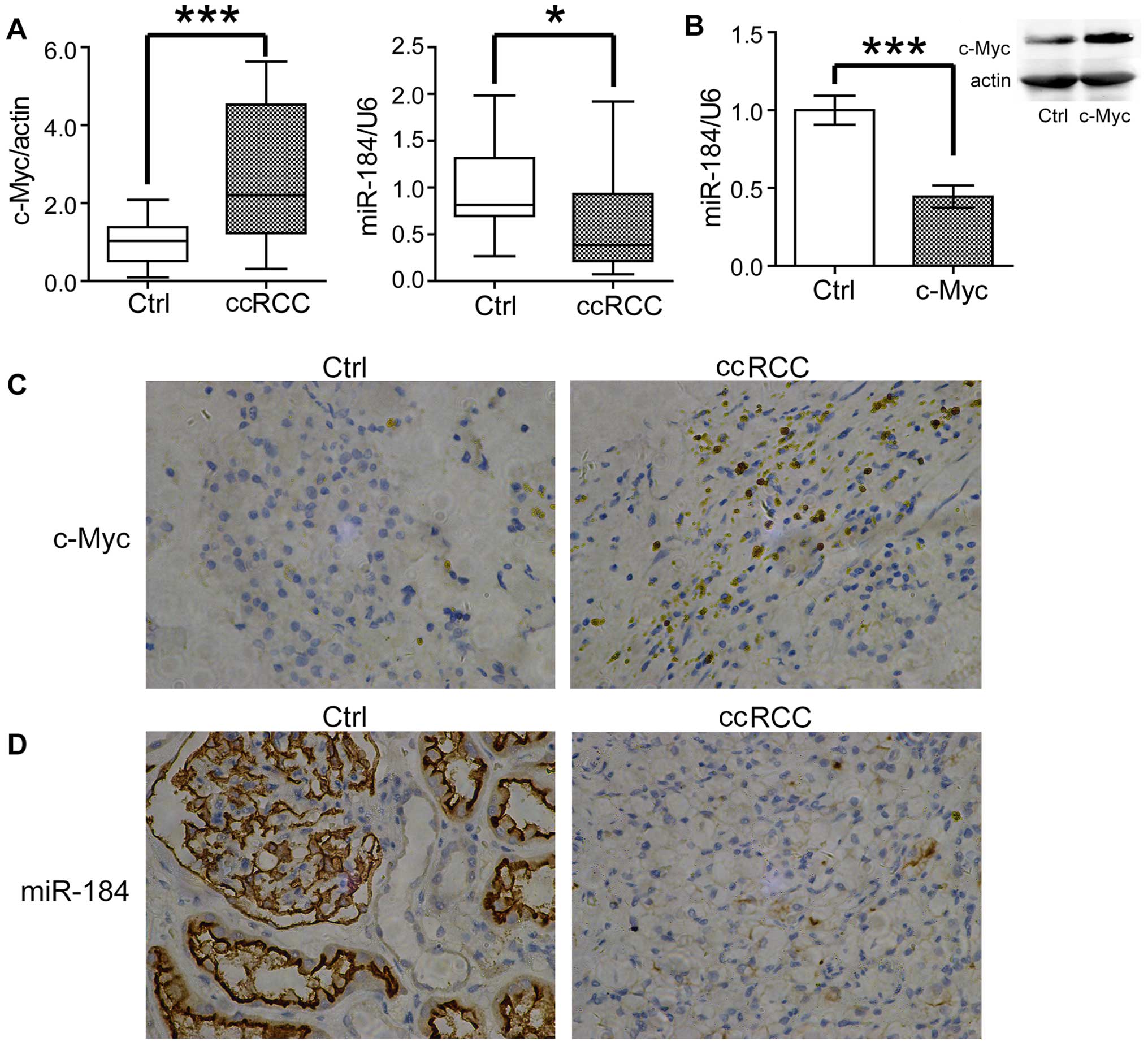
tissue. As shown in Fig. 1A,
consistent with previous reports, the expression of c-Myc was
greatly increased in ccRCC samples, whereas miR-184 was decreased
comparing with the normal tissues. To confirm these results, we
detected c-Myc and miR-184 expression by immunohistochemistry and
in situ hybridization, respectively. Consistently, increased
c-Myc and decreased miR-34a were observed in ccRCC samples
(Fig. 1C and D). As c-Myc was
reported to be a target of miR-184 (12,14,15,19,20),
we assume that the opposite expression profile between c-Myc and
miR-184 may be a result of miR-184 targeting. However, we also
noted that inhibition of MYCN, another member of MYC gene family,
causes significantly upregulation of miR-184 (26). It is known that c-Myc functions as
a transcription factor to influence the expression of a broad range
of human genes involved in the progression of cell carcinogenesis
(18). We wondered whether the
downregulated expression of miR-184 could also be regulated by
c-Myc. To test this possibility, we transfected c-Myc into the
ccRCC cell line 786-O. Surprisingly, overexpression of c-Myc
significantly downregulated the expression of miR-184 (Fig. 1B), which indicated miR-184 is a
probable target of c-Myc in ccRCC cells.
miR-184 inhibits glucose metabolism in
ccRCC cells
As ccRCC is featured by abnormal cell proliferation
and metabolism, including increased glucose consumption, lactate
production (6), we next wondered
whether the abnormal proliferative and metabolic features in ccRCC
cells could be improved by miR-184 overexpression. We transfected
either the pre-miR negative control or the pre-miR-184 into the
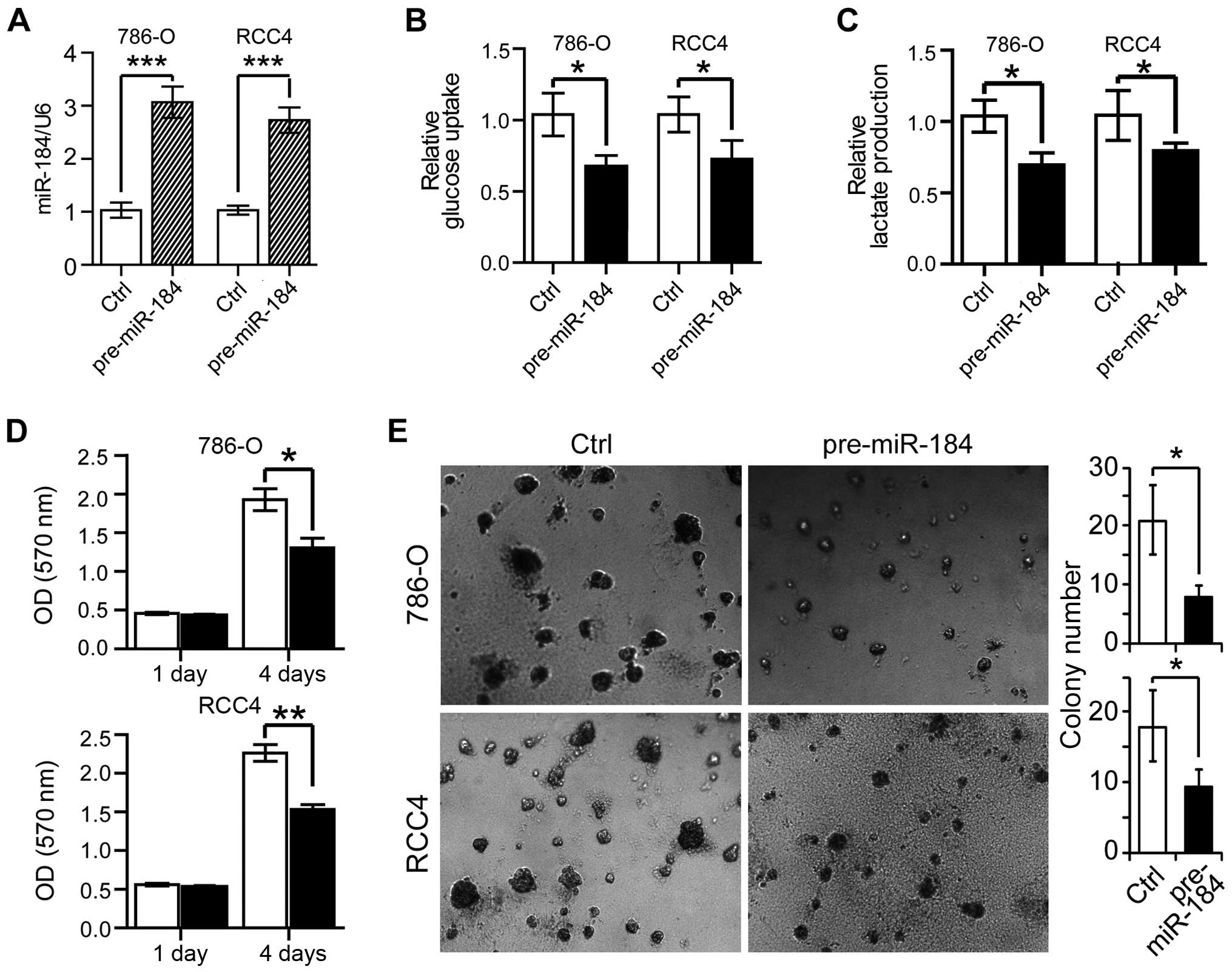
ccRCC cell lines 786-O and RCC4. As shown in Fig. 2A, transfection of pre-miR-184 led
to a ~3-fold increase of the miR-184 expression level both in 786-O
and RCC4 cells. Next, we measured glucose consumption and lactate
production in these cells. As shown Fig. 2B, we observed significant glucose
uptake reduction in pre-miR-184 transfected 786-O and RCC4 cells;
in addition, lactate production were also decreased ~30% in 786-O
cells and ~20% in RCC4 cells with miR-184 over-expression (Fig. 2C), indicating the metabolic
features were improved by miR-184 overexpression. The effect of
miR-184 on RCC cell proliferation was then analyzed. As shown in
the MTT assay of Fig. 2D, the cell
proliferation was markedly reduced by miR-184 overexpression in
786-O and RCC4 cells on the fourth day after transfection.
Similarly, miR-184 overexpression impaired colony formation of both
786-O and RCC4 cells (Fig. 2E).
Thus, miR-184 inhibited ccRCC cell proliferation, glucose uptake
and lactate production.
miR-184 specifically represses PKM2
expression
To uncover the mechanism of miR-184 contribution to
ccRCC progression, we investigated putative miR-184 targets that
directly regulate ccRCC metabolism and proliferation using
bioinformatics methods. The bioinformatics analysis revealed that
PKM2 (M2 isoform of pyruvate kinase) contains a potential miRNA
response element (MRE) for miR-184 in its 3′-untranslated region
(3′-UTR) (Fig. 3A). PKM2 is a
‘glycolytic valve’ in deciding the direction of glucose flux in
glycolysis (27). It catalyzes the
conversion of phosphoenolpyruvate (PEP) to pyruvate with
concomitant production of ATP (28), which is independent of oxygen
consumption. Thus, the high expression of PKM2 in tumor tissues
helps tumor cells to grow rapidly even in hypoxic conditions
(29). To determine whether PKM2
is indeed a target that is directly affected by miR-184, we
examined the luciferase activity driven by PKM2-3′UTR containing
the MRE sequence. As shown in Fig.
3B, in cells transfected with promoter containing PKM2 WT MRE
sequences, pre-miR-184 could significantly reduce the luminescence
comparing with the control group. However, when the PKM2 MRE was
mutated, miR-184 could not change the luciferase activity (Fig. 3A and B). To directly monitor the
target effect of miR-184 on PKM2, we tested PKM2 expression both at
mRNA level and protein expression level. In accordance, pre-miR-184
decreased PKM2 expression by 50% at mRNA level (Fig. 3C), and repressed PKM2 protein
expression obviously (Fig. 3D). We
also examined the effects of c-Myc overexpression on PKM2
expression. As shown in Fig. 3E,
PKM2 expression was enhanced by c-Myc over-expression. However,
when c-Myc and pre-miR-184 were co-expressed, the upregulated PKM2
by c-Myc overexpression was abolished in response to miR-184
overexpression, suggesting that c-Myc indeed increased PKM2
expression by inhibiting miR-184 expression. In brief, our data
indicated PKM2 is a new target of miR-184 in ccRCC cells.
Knockdown of PKM2 suppressed the
glycolysis and cell proliferation
After discovering PKM2 as a direct target of
miR-184, we next asked whether the altered metabolic and
proliferative features in ccRCC were the results of increased PKM2
level, as the miR-184 was downregulated. To test this hypothesis,
we transfected PKM2 siRNA into 786-O cells to lower PKM2 expression
level (Fig. 4A). Firstly we
analyzed the proliferation of PKM2 knockdown cells, and found a
profound decrease in proliferative cell numbers by MTT on the
fourth day post-PKM2 siRNA transfection (Fig. 4B). Furthermore, consistent with our
expectations, knockdown of PKM2 remarkably decreased glucose uptake
and lactate production in 786-O cells (Fig. 4C and D). Furthermore, we found
pre-miR-184 overexpression in 786-O cells resulted in impaired
glucose uptake, which could be rescued by PKM2 overexpression. This
result, therefore, suggested that miR-184 inhibited glycolysis in a
PKM2-dependent manner (Fig. 4E and
F). These data supported the conclusion that knockdown of PKM2
inhibits abnormal cell proliferation and metabolism in ccRCC cell
line.
Expression of PKM2 is increased in
ccRCC
Finally, we examined the expression of PKM2 in human
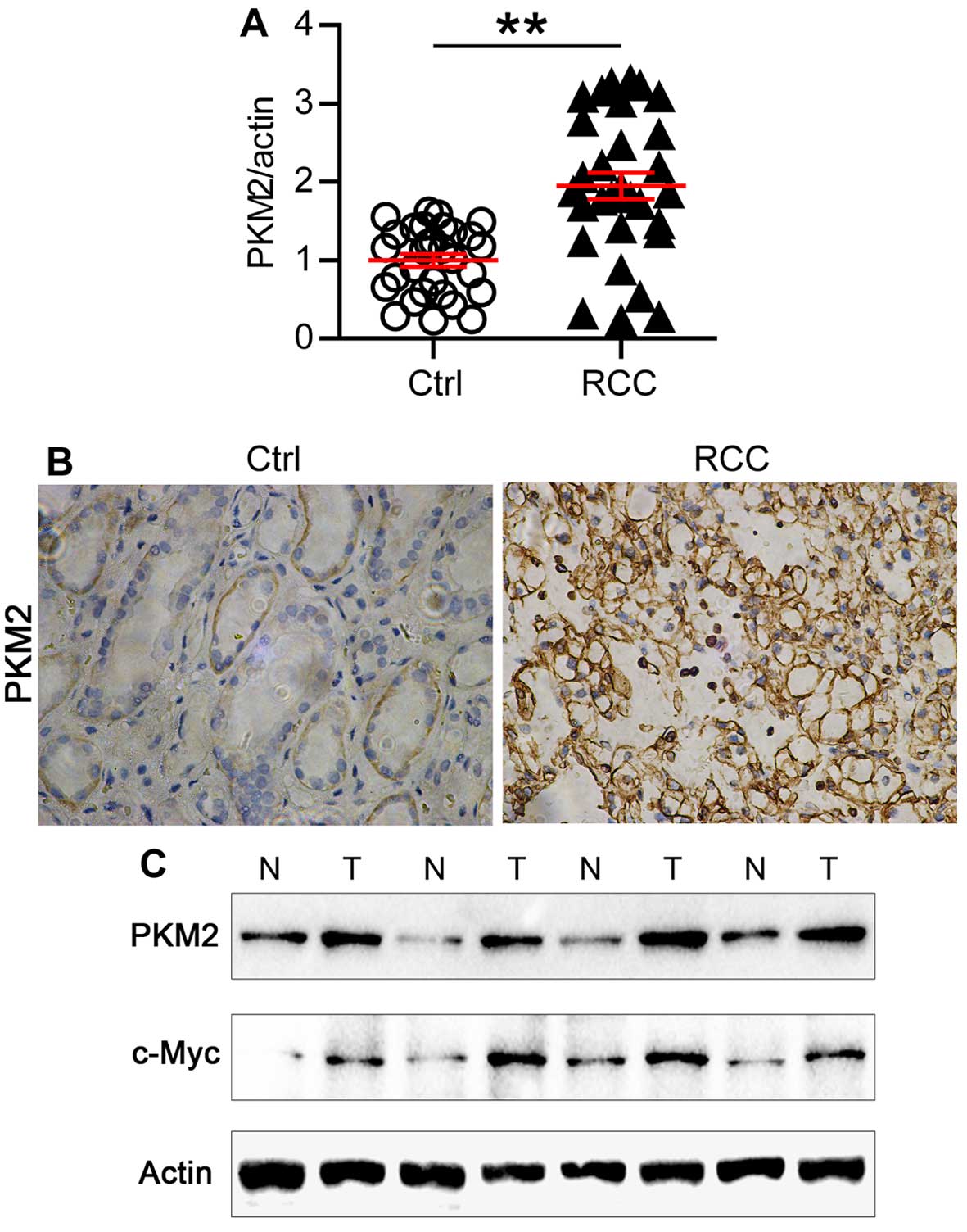
ccRCC samples. As shown in Fig.
5A, the expression of PKM2 mRNA doubled in the ccRCC samples
compared with the normal tissue. Consistently, high expression of
PKM2 protein level was also observed in ccRCC samples examined by
immunohistochemistry (Fig. 5B).
Then we analyzed the protein expression level of PKM2 and c-Myc in
paired ccRCC samples. Strikingly, increased expression of both PKM2
and c-Myc were observed in one same ccRCC sample comparing with
paired normal tissue (Fig. 5C).
Thus, our results indicated that the high expression of PKM2 in
ccRCC tissues might be a reason leading to hyperproliferation and
metabolic disorder of ccRCC.
Discussion
ccRCC is one of the most malignant tumors worldwide.
It consists of several histologic subtypes, including clear cell,
papillary, chromophobe and collecting duct (3). Each subtype is different in molecular
profiling, and ccRCC is the most common and aggressive one. One of
the most important difficulties for ccRCC treatment is its
resistance to traditional therapies (4,30).
Therefore, the mechanism study in ccRCC progression is critical for
uncovering the putative molecular targets for ccRCC therapy.
To date, many proteins and RNAs have been reported
to take part in the progression of ccRCC, such as c-Myc, VHL,
miR-34 and miR-184 (7,10,21,31).
However, the exact mechanism was not clarified. In the present
study, we identified that miR-184 could be regulated by c-Myc,
which is contrary to general opinion that c-Myc is a target of
miR-184.
miR-184 has been reported to be downregulated in
many cancer types, including ccRCC. In this study, we found
over-expression of pre-miR-184 could reduce ccRCC cell glucose
consumption, lactate production and cell proliferation. Further
analysis by computer bioinformatics revealed that PKM2 is a target
of miR-184. Both the mRNA and protein level of PKM2 was markedly
reduced in response to miR-184 overexpression. Most importantly,
the PKM2 expression level was indeed increased in ccRCC samples,
which is totally reverse to the decreased miR-184 expression level.
This result indirectly verified that PKM2 is a target of miR-184 in
human ccRCC samples.
PKM2 is a key regulator of the metabolic fate of the
glycolytic intermediates, its high expression in many cancers
favored the cancer cells to proliferate rapidly even in anaerobic
conditions. We found that knockdown of PKM2 in ccRCC cells,
inhibited the rapid proliferation, high glucose consumption and
high lactate production to a certain extent, which indicated
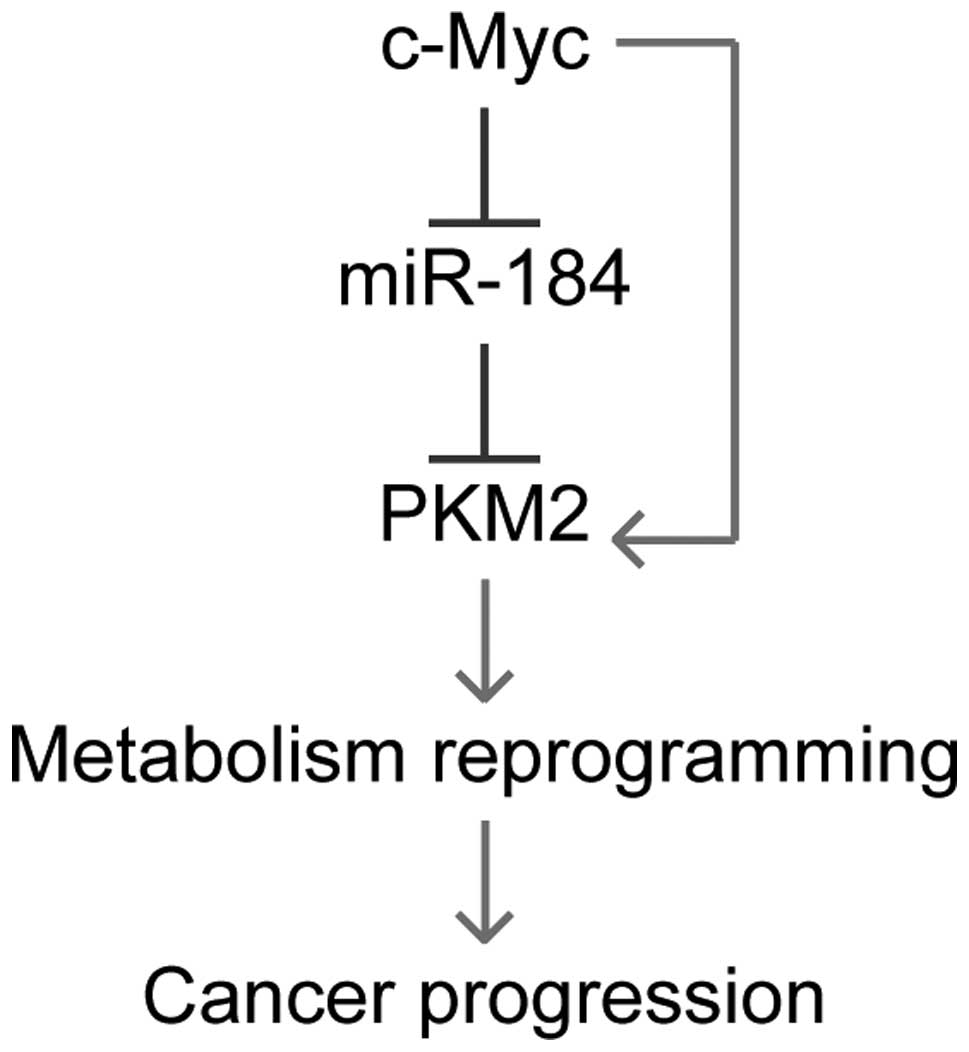
metabolic reprogramming in ccRCC cells. In summary, in the present
study, we identified PKM2 as a new target of miR-184, and that its
expression can be inhibited by c-Myc. As a result, PKM2 expression
can be upregulated by c-Myc overexpression. Knockdown the high PKM2
expression profile in ccRCC leads to metabolism reprogramming in
these cancer cells, and then inhibits cancer progression of ccRCC
(Fig. 6). Our findings shed new
light on ccRCC molecular study and provide a new and solid basis
for developing ccRCC therapy.
Acknowledgements
The design and conduct of the project was supported
by the Award Number 81402084, 81272841, 81472378 from the National
Natural Science Foundation of China and the Award Number
PYXJS16-008 from the Incubating Program for Clinical Research and
Innovation of Renji Hospital.
References
|
1
|
Chow WH, Dong LM and Devesa SS:
Epidemiology and risk factors for kidney cancer. Nat Rev Urol.
7:245–257. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Majer W, Kluzek K, Bluyssen H and Wesoły
J: Potential approaches and recent advances in biomarker discovery
in clear-cell renal cell carcinoma. J Cancer. 6:1105–1113. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cohen HT and McGovern FJ: Renal-cell
carcinoma. N Engl J Med. 353:2477–2490. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Amato RJ: Chemotherapy for renal cell
carcinoma. Semin Oncol. 27:177–186. 2000.PubMed/NCBI
|
|
5
|
Beck SD, Patel MI, Snyder ME, Kattan MW,
Motzer RJ, Reuter VE and Russo P: Effect of papillary and
chromophobe cell type on disease-free survival after nephrectomy
for renal cell carcinoma. Ann Surg Oncol. 11:71–77. 2004.
View Article : Google Scholar
|
|
6
|
Massari F, Ciccarese C, Santoni M,
Brunelli M, Piva F, Modena A, Bimbatti D, Fantinel E, Santini D,
Cheng L, et al: Metabolic alterations in renal cell carcinoma.
Cancer Treat Rev. 41:767–776. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Linehan WM, Srinivasan R and Schmidt LS:
The genetic basis of kidney cancer: A metabolic disease. Nat Rev
Urol. 7:277–285. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gossage L, Eisen T and Maher ER: VHL, the
story of a tumour suppressor gene. Nat Rev Cancer. 15:55–64. 2015.
View Article : Google Scholar
|
|
9
|
Cancer Genome Atlas Research Network.
Comprehensive molecular characterization of clear cell renal cell
carcinoma. Nature. 499:43–49. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tang SW, Chang WH, Su YC, Chen YC, Lai YH,
Wu PT, Hsu CI, Lin WC, Lai MK and Lin JY: MYC pathway is activated
in clear cell renal cell carcinoma and essential for proliferation
of clear cell renal cell carcinoma cells. Cancer Lett. 273:35–43.
2009. View Article : Google Scholar
|
|
11
|
Ma L and Qu L: The function of microRNAs
in renal development and pathophysiology. J Genet Genomics.
40:143–152. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wong TS, Liu XB, Wong BY, Ng RW, Yuen AP
and Wei WI: Mature miR-184 as potential oncogenic microRNA of
squamous cell carcinoma of tongue. Clin Cancer Res. 14:2588–2592.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Foley NH, Bray IM, Tivnan A, Bryan K,
Murphy DM, Buckley PG, Ryan J, O’Meara A, O’Sullivan M and
Stallings RL: MicroRNA-184 inhibits neuroblastoma cell survival
through targeting the serine/threonine kinase AKT2. Mol Cancer.
9:832010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhen Y, Liu Z, Yang H, Yu X, Wu Q, Hua S,
Long X, Jiang Q, Song Y, Cheng C, et al: Tumor suppressor PDCD4
modulates miR-184-mediated direct suppression of C-MYC and BCL2
blocking cell growth and survival in nasopharyngeal carcinoma. Cell
Death Dis. 4:e8722013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wu GG, Li WH, He WG, Jiang N, Zhang GX,
Chen W, Yang HF, Liu QL, Huang YN, Zhang L, et al: Mir-184
post-transcriptionally regulates SOX7 expression and promotes cell
proliferation in human hepatocellular carcinoma. PLoS One.
9:e887962014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Leng HM, Qian WP, Zhou L, Zhai QN, Li XX,
Guan ZC, Gui YT and Cai ZM: Abnormal expression and significance of
MIR-184 in human renal carcinoma. Beijing Da Xue Xue Bao.
43:509–513. 2011.(In Chinese). PubMed/NCBI
|
|
17
|
Su Z, Chen D, Li Y, Zhang E, Yu Z, Chen T,
Jiang Z, Ni L, Yang S, Gui Y, et al: microRNA-184 functions as
tumor suppressor in renal cell carcinoma. Exp Ther Med. 9:961–966.
2015.PubMed/NCBI
|
|
18
|
Lebofsky R and Walter JC: New Myc-anisms
for DNA replication and tumorigenesis? Cancer Cell. 12:102–103.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lin TC, Lin PL, Cheng YW, Wu TC, Chou MC,
Chen CY and Lee H: MicroRNA-184 deregulated by the microRNA-21
promotes tumor malignancy and poor outcomes in non-small cell lung
cancer via targeting CDC25A and c-Myc. Ann Surg Oncol. 22(Suppl 3):
1532–1539. 2015. View Article : Google Scholar
|
|
20
|
Liu Z, Mai C, Yang H, Zhen Y, Yu X, Hua S,
Wu Q, Jiang Q, Zhang Y, Song X, et al: Candidate tumour suppressor
CCDC19 regulates miR-184 direct targeting of C-Myc thereby
suppressing cell growth in non-small cell lung cancers. J Cell Mol
Med. 18:1667–1679. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shroff EH, Eberlin LS, Dang VM, Gouw AM,
Gabay M, Adam SJ, Bellovin DI, Tran PT, Philbrick WM, Garcia-Ocana
A, et al: MYC oncogene overexpression drives renal cell carcinoma
in a mouse model through glutamine metabolism. Proc Natl Acad Sci
USA. 112:6539–6544. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu W, Cao H, Ye C, Chang C, Lu M, Jing Y,
Zhang D, Yao X, Duan Z, Xia H, et al: Hepatic miR-378 targets p110α
and controls glucose and lipid homeostasis by modulating hepatic
insulin signalling. Nat Commun. 5:56842014. View Article : Google Scholar
|
|
23
|
Jo HS, Kang KH, Joe CO and Kim JW: Pten
coordinates retinal neurogenesis by regulating Notch signalling.
EMBO J. 31:817–828. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jia LF, Wei SB, Gong K, Gan YH and Yu GY:
Prognostic implications of micoRNA miR-195 expression in human
tongue squamous cell carcinoma. PLoS One. 8:e566342013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dweep H, Sticht C, Pandey P and Gretz N:
miRWalk - database: Prediction of possible miRNA binding sites by
‘walking’ the genes of three genomes. J Biomed Inform. 44:839–847.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen Y and Stallings RL: Differential
patterns of microRNA expression in neuroblastoma are correlated
with prognosis, differentiation, and apoptosis. Cancer Res.
67:976–983. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Iqbal MA, Gupta V, Gopinath P, Mazurek S
and Bamezai RN: Pyruvate kinase M2 and cancer: An updated
assessment. FEBS Lett. 588:2685–2692. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gupta V and Bamezai RN: Human pyruvate
kinase M2: a multi-functional protein. Protein Sci. 19:2031–2044.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wong N, Ojo D, Yan J and Tang D: PKM2
contributes to cancer metabolism. Cancer Lett. 356:184–191. 2015.
View Article : Google Scholar
|
|
30
|
Lane BR, Rini BI, Novick AC and Campbell
SC: Targeted molecular therapy for renal cell carcinoma. Urology.
69:3–10. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Redova M, Svoboda M and Slaby O: MicroRNAs
and their target gene networks in renal cell carcinoma. Biochem
Biophys Res Commun. 405:153–156. 2011. View Article : Google Scholar : PubMed/NCBI
|