Introduction
Colorectal cancer (CRC) is a common malignancy in
the colon or rectum, which has a high morbidity and mortality
worldwide. In the United States, CRC has become the third most
common cancer among the top leading causes of cancer death
(1). Great efforts have been made
to improve prognosis and treatment outcome of CRC, such as the
flexible sigmoidoscopy screening method, various prognostic
biomarkers and histopathological classification criteria (2–4).
Current therapy for CRC relies on surgery and adjuvant radiotherapy
for rectal cancer or chemotherapy for colon cancer (5). Unfortunately, the resistance of CRC
cells to chemotherapy impedes the expected outcome of CRC treatment
(6,7).
Chemotherapeutics usually utilize the cytotoxicity
from synthetic chemical drugs to inhibit cancer cell growth and
accelerate apoptosis. For instance, bleomycin (BLM) is widely
applied in cancer treatment for its induction of DNA damage in
proliferating cells (8). Several
factors are revealed to strongly affect the resistance of cancer
cells to chemotherapy, including v-akt murine thymoma viral
oncogene (AKT), whose inhibitor reduces resistance to
chemotherapeutic drugs (9).
Moreover, some microRNAs are considered to be involved in the
regulation of drug resistance, such as microRNA-374b (miR-374b),
which can restore cisplatin sensitivity of pancreatic cancer cells
(10), suggesting avenues for
enhancing effects of chemotherapy.
Substantial evidence supports that tumor protein p53
is a tumor suppressor in various diseases including CRC. Expression
of p53 can be induced by anticancer agents to speed up CRC cell
apoptosis and improve general health of patients (11,12).
Numerous studies have shown the participation of p53 in cancer cell
response to DNA damage (13–15),
however, there are still much needs to be revealed about the
specific function and mechanism of p53 in CRC cells in response to
DNA damage.
The present study aimed to elucidate the mechanism
of p53 in regulating CRC cell apoptosis in response to DNA damage.
The above-mentioned factors in chemotherapy, AKT1 and miR-374b were
also involved in this study in order to depict a more detailed
frame for p53 mechanism. CRC cell line HCT116 and HT29 were treated
by BLM to induce DNA damage. Cell transfection with p53-specific
small interfering RNA (siRNA), p53 overexpression vector or
miR-374b inhibitor were performed to reveal the effect of these
factors. These experiments were expected to provide general
knowledge for p53 and its related factors in CRC cells upon DNA
damage.
Materials and methods
Cell culture
Human colorectal cancer cell lines HCT116 and HT29
were purchased from ATCC (Manassas, VA, USA). Knockout of p53 was
performed by Genloci Biotechnologies (Nanjing, China) to generate
cell lines HCT116-p53−/− and
HT26-p53−/−. The cells were separately cultured
in ATCC-formulated McCoy's 5A medium modified (ATCC) supplemented
with 10% fetal bovine serum (FBS). The medium was changed every
three days and the cells were incubated at 37°C in humidified
atmosphere with 5% CO2.
Cell transfection
The complete coding sequence of human p53
(GenBank accession no. AB082923) was inserted into pcDNA3-flag
vector (Novagen, Darmstadt, Germany), and the correct plasmid was
screened by sequencing. The specific siRNA for p53 (si-p53), the
negative control (si-control), miR-374b inhibitor and inhibitor
control were produced by Ribobio (Guangzhou, China). Cell
transfection was performed with the assistance of
Lipofectamine-2000 (Invitrogen, Carlsbad, CA, USA) according to the
manufacturer's instructions. Before transfection, the cells were
seeded in FBS-free medium in 24-well plates (2×105 cells
per well) to reach the confluence of approximately 90%. The
transfection complex was prepared and added to each well to a
certain concentration (1 µg/ml for Flag-p53 and blank
vector, 50 nM for si-p53 and si-control, and 100 nM for miR-374b
inhibitor and inhibitor control). The plates were incubated at 37°C
for 24 h and then cells were used in further treatment and
detection.
BLM treatment
BLM (Invitrogen) was dissolved in phosphate-buffered
saline (PBS) before use. After 24 h of incubation for transfection,
the cells were incubated in fresh medium containing BLM of 10
µM, and then incubated for another 24 h (15). Cells with PBS treatment were set as
control groups. After BLM treatment, cells of each group were
collected for further analysis.
Cell apoptosis assay
Four groups of cells were detected for cell
apoptosis: cells transfected with miR-374b inhibitor + BLM
treatment, cells transfected with inhibitor control + BLM
treatment, and two control groups without BLM treatment. Cell
apoptosis assay was performed by Annexin V-fluorescein
isothiocyanate (FITC) and propidium iodide (PI) staining followed
by flow cytometry. Annexin V-FITC Apoptosis Detection Kit I
(Univ-Bio, Shanghai, China) was used to stain the cells according
to the manufacturer's instructions. Briefly, the density of cells
was adjusted to 1×106/ml, and 200 µl was used for
each assay. The cells were washed in PBS two times and resuspended
in 100 µl binding buffer with 2 µl Annexin V-FITC,
and then incubated in the dark on ice for 15 min. PBS (400
µl) and 1 µl PI solution was added and the samples
were immediately detected by a flow cytometry FACSCanto II (BD
Biosciences, San Jose, CA, USA). FITC-positive and PI-negative
cells were considered as apoptotic cells.
qRT-PCR
RNA samples of cells were isolated with TRIzol
(Invitrogen) according to the manufacturer's instructions and
purified with RNA purification kit (Tiangen, Beijing, China). The
quantification of miRNAs were performed based on a previous study
(16). Reverse transcription for
pri-miRNAs was performed with 1 µg RNA for each sample under
the catalysis of SuperScript III Reverse Transcriptase
(Invitrogen). Reverse transcription for pre-miRNAs and mature
miRNAs were performed by One Step PrimeScript miRNA cDNA Synthesis
(Takara, Dalian, China). qRT-PCR was conducted on QuantStudio 6
Flex Real-time PCR System (Applied Biosystems, Carlsbad, CA, USA),
and the reaction conditions were: pre-denaturation at 95°C for 10
min, 40 cycles of 95°C for 30 sec, 64°C for 30 sec and 72°C for 30
sec, followed by a melting curve of 95°C for 15 sec, 60°C for 1 min
and 95°C for 15 sec. Each reaction sample contained 20 ng
complementary DNAs and a pair of specific primers for mature
miR-374b-5p (forward: 5′-CACTC CAGCT GGGAT ATAAT ACAAC CTGC-3′ and
reverse: 5′-TGGTG TCGTG GAGTC G-3′), pre-miR-374b (forward:
5′-CCTAT ATTAT GTTGG ACG-3′ and reverse: 5′-CTGTG CCTGT TACTA
TTATG-3′), pri-miR-374b (forward: 5′-ACCAT CTGCT CTCGG TATG-3′ and
reverse: 5′-CCTGG AGTGG TGCTC CTCTG-3′), pre-miR-34 (forward:
5′-GGCCT CGACA CTCAC AAA-3′ and reverse: 5′-GGTGT TGCAC GTCGT
GAA-3′) or pri-miR-34 (forward: 5′-GAGGC CTACG GCACC TGG-3′ and
reverse: 5′-GAGCG AAGTA GAAGG GAGAA-3′). Results were normalized by
GAPDH (forward: 5′-GGTGA TCCTG GTGAA GGAGA-3′ and reverse:
5′-CTTAA TGTGC CCGTC CTTGT-3′) for pri-miRNAs or U6 (forward:
5′-CTCGC TTCGG CAGCA CATAT ACT-3′ and reverse: 5′-ACGCT TCACG AATTT
GCGTG TC-3′) for miR-374b or pre-miRNAs. Similarly, AKT homolog 1
(AKT1) mRNA level was detected with its specific primers
(forward: 5′-GAAGG TGAAG GTCGG AGTC-3′ and reverse: 5′-GAAGA TGGTG
ATGGG ATTTG-3′) and normalized by GAPDH. Data were
calculated with the 2−ΔΔCt method.
Western blotting
Protein samples from cells were isolated with M-PER
mammalian protein extraction reagent (Thermo Scientific, Carlsbad,
CA, USA) and quantified with Bio-Rad Protein Assay (Bio-Rad,
Hercules, CA, USA). Protein was separated by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and blotted to
polyvinylidene fluoride membranes. The blots were blocked in 5%
skim milk overnight at 4°C, and then incubated in rabbit monoclonal
antibodies against AKT1 or p53 (1:1000, ab32505 and ab32049, Abcam,
Cambridge, UK) for 2 h at room temperature. Anti-GAPDH antibodies
(1:1000, ab9485) were used as an internal control. After washed in
PBS 3 times (5 min each time), the blots were incubated in
horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary
antibodies (1:2000, ab7090) for 1 h at room temperature. Signals
were developed by EasyBlot ECL kit (Sangon Biotech, Shanghai,
China) and the relative density was analyzed by ImageJ 1.49
(National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
All experiments were performed in triplicate.
Results are presented as mean ± standard deviation. Data were
analyzed with one-way analysis of variance (ANOVA) and t-test by
SPSS 20 (IBM, New York, NY, USA). Difference between groups was
considered significant at P<0.05.
Results
p53 promotes miR-374b and inhibits AKT1
protein in response to DNA damage
Since AKT has been found to be involved in
regulation of cell sensitivity to chemotherapy, this study began
with analyzing the relationship between p53 and AKT1 in HCT116
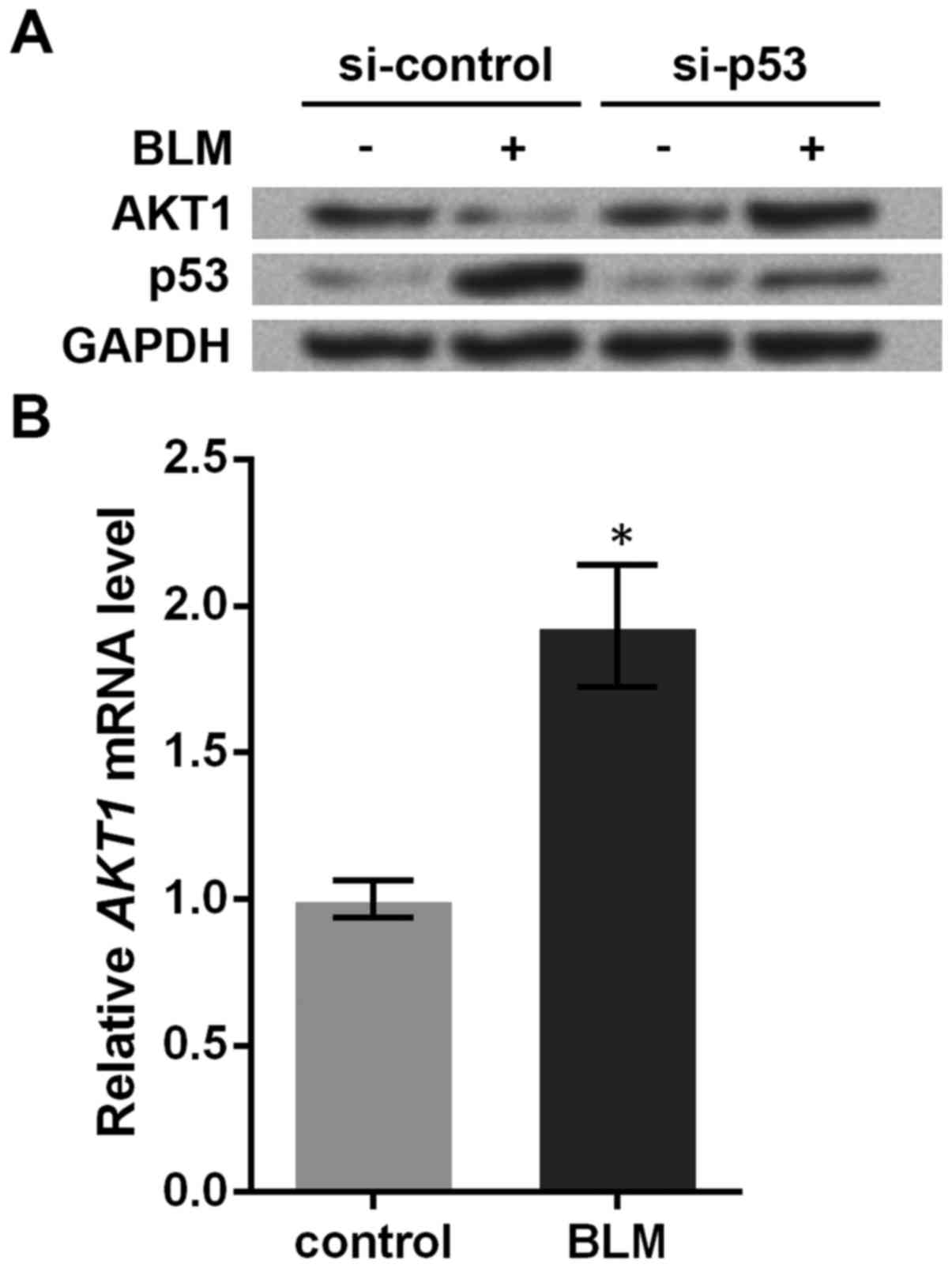
cells. Western blotting showed that BLM treatment decreased AKT1
protein and increased p53 protein (Fig. 1A). However, the effect of BLM
treatment was abrogated and AKT1 protein was increased when p53 was
knocked down, implying that the suppression of AKT1 by BLM
treatment was dependent on p53.
The underlying mechanism between p53 and AKT1 is
unknown. To solve this, we detected AKT1 mRNA levels in
HCT116 cells before and after BLM treatment, and found that BLM
treatment could significantly upregulate AKT1 mRNA levels
(P<0.05, Fig. 1B), which showed
an opposite changing pattern compared to its protein level. In
light of the disparity, it was likely that AKT1 was inhibited
post-transcriptionally. Through searching TargetScanHuman 7.0
(17), we found AKT1 mRNA
could be regulated by several miRNAs, among which miR-374b was
related to cell chemosensitivity, and has been revealed as a direct
up-stream factor of AKT1 mRNA in C2C12 myoblasts (18). Thus, miR-374b was introduced into
this study.
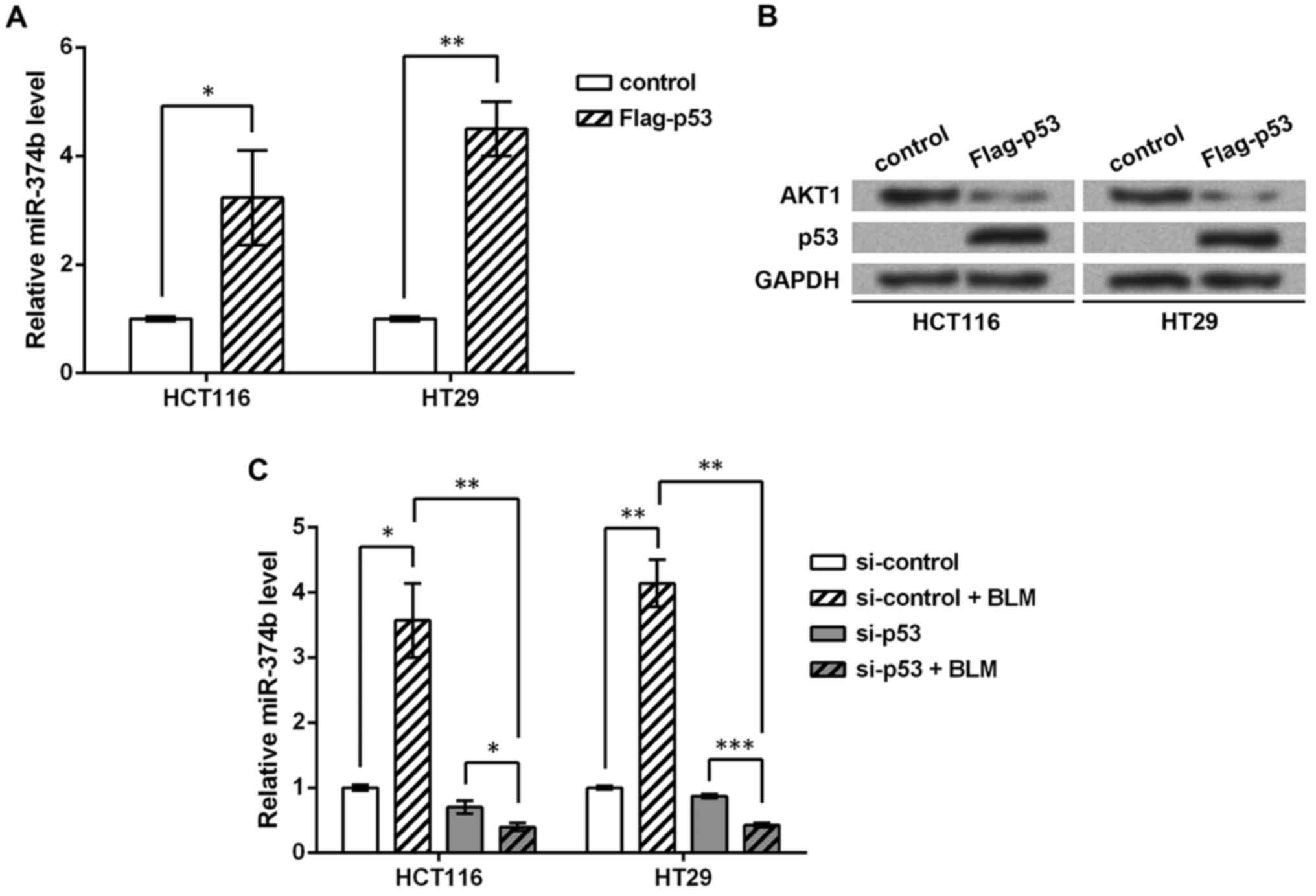
Factors p53, AKT1 and miR-374b were detected in the
CRC cell lines HCT116 and HT29. Overexpression of p53 in the cell
lines (p53−/−) significantly induced miR-374b
level (P<0.05 or P<0.01, Fig.
2A), and AKT1 protein expression was inhibited (Fig. 2B), which further suggested the
reverse regulation between p53 and AKT1 protein. Then knockdown of
p53 and BLM treatment were performed in wild-type cells. miR-374b
level was significantly induced by BLM treatment (P<0.05 or
P<0.01, Fig. 2C), while it was
significantly inhibited when p53 was knocked down (P<0.01),
suggesting that the induction of miR-374b by DNA damage was also
dependent on p53. Taken together, p53 might promote miR-374b to
inhibit AKT1 protein expression in response to DNA damage in CRC
cells.
p53 regulates the processing of
pri-miR-374b
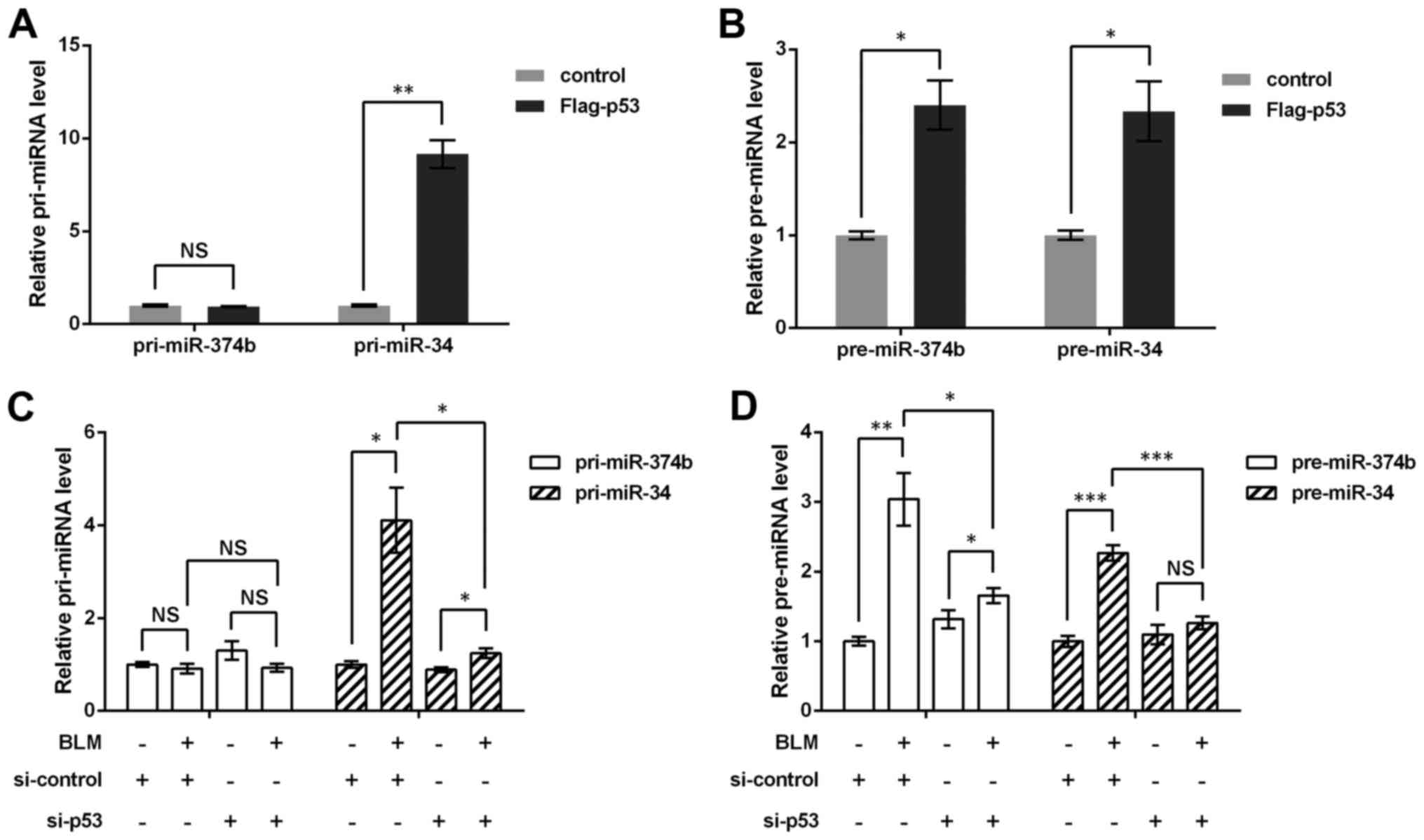
Next, the regulation of miR-374b by p53 was
investigated. miRNA generation is composed of the processing of
pri-miRNA to pre-miRNA, and pre-miRNA to mature miRNA (16), thereby the regulation of
pri-miR-374b and pre-miR-374b by p53 was detected via qRT-PCR
method, respectively. Moreover, miR-34 transcription is modulated
by p53, thus it was used as a positive control, and results showed
both pri-miR-34 and pre-miR-34 were markedly upregulated by p53 in
p53−/− HCT116 cells (P<0.05 or P<0.01,
Fig. 3A and B). Although
pre-miR-374b was elevated by p53 (P<0.05), no significant change
was found in pri-miR-374b level, which implied that p53 did not
impact pri-miR-374b.
Consistently, in the wild-type HCT116 cells with p53
knockdown and BLM treatment, it was found that pri-miR-374b was not
affected by BLM or si-p53 (Fig.
3C), but pre-miR-374b could be upregulated by BLM treatment
(P<0.01) and then suppressed by si-p53 (P<0.05, Fig. 3D). Together with the previous
result that p53 could regulate mature miR-374b, it was possible
that the regulation of miR-374b by p53 started with the processing
of pri-miR-374b to pre-miR-374b, rather than the transcription of
pri-miR-374b.
Inhibiting miR-374b reduces BLM-induced
CRC cell apoptosis
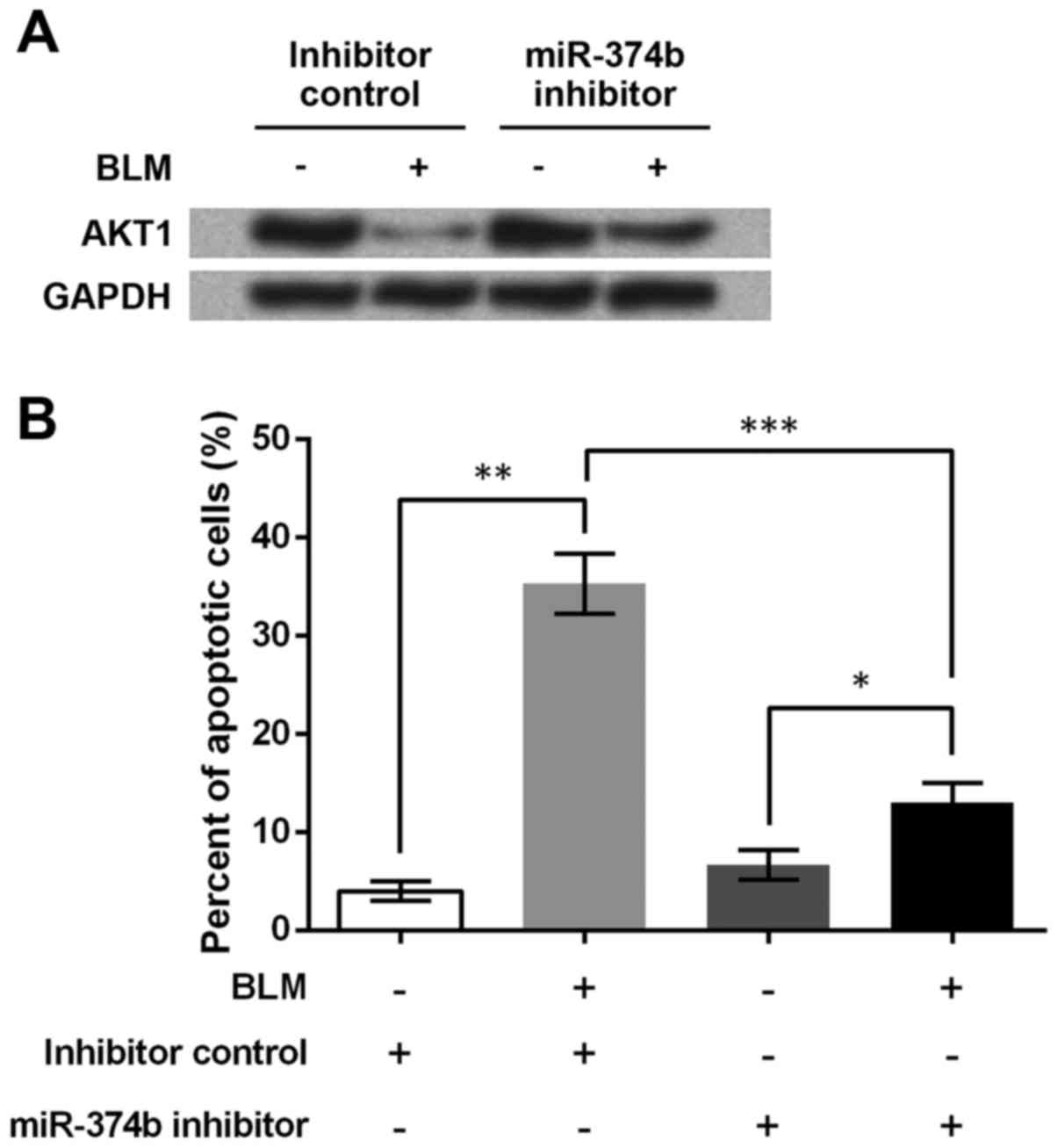
Finally, the regulation of AKT1 and HCT116 cell
apoptosis by miR-374b was analyzed. Western blotting showed that
miR-374b inhibitor clearly relieved the BLM-induced inhibition on
AKT1 protein (Fig. 4A), implying
that miR-374b might help to suppress AKT1 expression. Since
AKT1 mRNA was predicted to be a direct target of miR-374b,
it was possible that miR-374b directly suppressed the translation
of AKT1 protein.
HCT116 cell apoptosis was detected by flow cytometry
after miR-374b transfection and BLM treatment. Changes in apoptotic
cell percent indicated that BLM induced cell apoptosis (P<0.01,
Fig. 4B). Cell apoptosis was also
elevated by BLM when miR-374b was inhibited (P<0.05), which,
however, was significantly weakened because of the inhibited
miR-374b (P<0.001). Thus suppressing miR-374b was able to
inhibit HCT116 cell apoptosis upon DNA damage, which might be the
result of the decreased sensitivity of HCT116 cells to BLM.
Discussion
Cancer cells respond differently to
chemotherapy-induced DNA damage. In this study, BLM treatment
induces p53 expression and upregulation of AKT1 mRNA, but
AKT1 protein levels is downregulated in CRC HCT116 cells. Further
analyses indicated p53 regulates the generation of pre-miR-374b,
but did not affect pri-miR-374b level. When miR-374b is inhibited,
AKT1 protein level arises and the HCT116 cell apoptosis is
suppressed.
After pri-miRNAs are transcribed from the genome,
they go through various procedures before generating mature miRNAs,
two main steps among which are the cropping of pri-miRNAs to
pre-miRNAs by RNase III enzyme Drosha and the dicing of pre-miRNAs
to mature miRNAs by RNase III enzyme Dicer (19). The transcription of pri-miRNAs from
the genome is also regulated by various factors in their promoter
(20,21). However, pri-miR-374b level was not
affected by up- or down-regulation of p53 in HCT116 cells,
suggesting that p53 has no dominant effect on the transcription of
pri-miR-374b, although the possibility of its direct binding to the
miR-374b promoter cannot be ruled out according to previous
findings (22). pre-miR-374b and
mature miR-374 levels could be promoted by p53, implying that p53
may promote the cropping step of miR-374b generation. Since p53 is
a nuclear protein (23), it is
less possible to regulate the dicing step. Moreover, existing
evidence supports that p53 can modulate Drosha, and mutant p53
severely affected Drosha activity (24,25).
Thereby the results in this study demonstrate that p53 regulates
the processing from pri-miR-374b to pre-miR-374b.
AKT1 mRNA was conjectured to be a direct
target of miR-374b according to the prediction on TargetScanHuman
7.0 database. AKT1 mRNA level was elevated, while its
protein level is obviously downregulated in response to BLM
treatment, suggesting a potential post-transcriptional regulation
on AKT1 expression. miRNAs are powerful modulators of the
translational status (26);
moreover, this study indicated that AKT1 protein level was promoted
by miR-374b inhibitor, implying a reverse regulation between the
two factors. A previous study found evidence that miR-374b directly
targets and inhibits AKT1 mRNA, thus suppressing cell
proliferation (27). Thus it is
reasonable to speculate that miR-374b can also inhibit the
translation from AKT1 mRNA to AKT1 protein in HCT116 cells,
which constitutes the downstream part of the p53 signaling.
Cell apoptosis was detected when miR-374b was
inhibited, and miR-374b inhibitor was able to suppress the
BLM-induced cell apoptosis, implying that miR-374b may promote
HCT116 cell apoptosis. A similar role of miR-374b has been
reported: miR-374b elevates chemotherapeutic agent-induced
apoptosis in T-cell lymphoblastic lymphoma cells (27). Besides miR-374b, p53 and AKT1 are
also greatly involved in the regulation of cell apoptosis, albeit
playing different roles. As a tumor suppressor, p53 induces cell
apoptosis through multiple pathways when DNA damage occurs or
oncogene is overexpressed (28,29),
while AKT1 is a major AKT isoform that protects against apoptosis,
as reported in various cell types including CRC cells (30–32).
In this study, the induced changes in p53, miR-374b, AKT1 and cell
apoptosis were consistent with existing studies on these factors,
implying the p53, miR-374b and AKT1 may participate in the
regulation of BLM-induced HCT116 cell apoptosis.
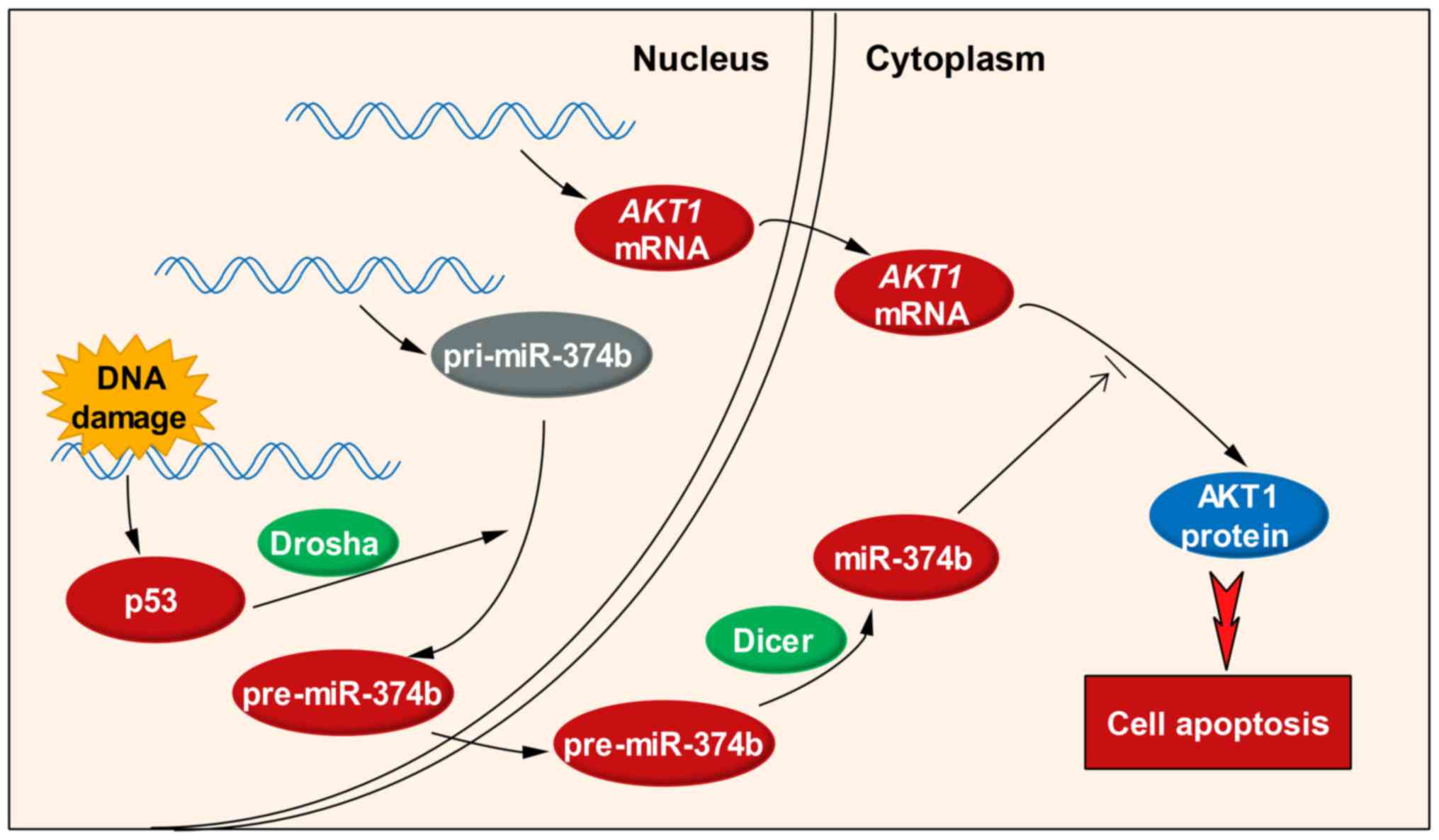
Based on the above results and discussion, a brief
regulatory cascade (Fig. 5)
containing p53, miR-374b and AKT1 can be depicted: in response to
chemotherapy-induced DNA damage, a series of changes may happen in
the nucleus, including the elevated expression of p53 and the
promoted transcription of AKT1 mRNA. The transcription of
pri-miR-374b seems unaffected, but its processing to generate
pre-miR-374b is accelerated, possibly due to the increased Drosha
activity by p53. pre-miR-374b is transferred to the cytoplasm,
where it is diced into mature miR-374b by the Dicer. Although
AKT1 mRNA level is upregulated, it is a potential direct
target of miR-374b and its translation is suppressed, thus leading
to the downregulation of AKT1 protein. Through this regulatory
cascade, p53, miR-374b and AKT1 are capable of modulating HCT116
cell apoptosis in response to DNA damage. More details in the
p53/miR-374b/AKT1 signaling are to be revealed in further
research.
Resistance to chemotherapy of cancer cells can be
modulated by p53 and AKT1. The role of p53 on cancer cell
sensitivity to chemotherapy may be conflicting (33), but most studies demonstrated its
promotive effects on the sensitivity of cancer cells to
chemotherapy (34,35). In addition, AKT1 gene
polymorphism influences neoadjuvant chemotherapeutic sensitivity of
cancer cells (36). In this study,
the p53/miR-374b/AKT1 signaling was demonstrated to play potential
roles in HCT116 cell apoptosis in response to BLM-induced DNA
damage, which implies the possibility of modulating these factors
to improve the sensitivity of CRC cells to chemotherapy.
In summary, this study uncovers the mechanism of p53
regarding the p53/miR-374b/AKT1 signaling in regulating HCT116 cell
apoptosis in response to the BLM-induced DNA damage. These results
demonstrate the p53/miR-374b/AKT1 signaling, as a potential
significant modulator, improves the outcome of chemotherapy in
treating CRC. Further efforts are need to reveal more effects of
this signaling on cancer cells.
References
|
1
|
Siegel R, Desantis C and Jemal A:
Colorectal cancer statistics, 2014. CA Cancer J Clin. 64:104–117.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schoen RE, Pinsky PF, Weissfeld JL,
Yokochi LA, Church T, Laiyemo AO, Bresalier R, Andriole GL, Buys
SS, Crawford ED, et al PLCO Project Team: Colorectal-cancer
incidence and mortality with screening flexible sigmoidoscopy. N
Engl J Med. 366:2345–2357. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sobin LH, Gospodarowicz MK and Wittekind
C: TNM Classification of Malignant Tumours. John Wiley & Sons;
West Sussex: 2011
|
|
4
|
Walther A, Johnstone E, Swanton C, Midgley
R, Tomlinson I and Kerr D: Genetic prognostic and predictive
markers in colorectal cancer. Nat Rev Cancer. 9:489–499. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Brenner H, Kloor M and Pox CP: Colorectal
cancer. Lancet. 383:1490–1502. 2014. View Article : Google Scholar
|
|
6
|
Dylla SJ, Beviglia L, Park IK, Chartier C,
Raval J, Ngan L, Pickell K, Aguilar J, Lazetic S, Smith-Berdan S,
et al: Colorectal cancer stem cells are enriched in xenogeneic
tumors following chemotherapy. PLoS One. 3:e24282008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jensen NF, Stenvang J, Beck MK, Hanáková
B, Belling KC, Do KN, Viuff B, Nygård SB, Gupta R, Rasmussen MH, et
al: Establishment and characterization of models of chemotherapy
resistance in colorectal cancer: Towards a predictive signature of
chemoresistance. Mol Oncol. 9:1169–1185. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Edhemovic I, Gadzijev EM, Brecelj E,
Miklavcic D, Kos B, Zupanic A, Mali B, Jarm T, Pavliha D, Marcan M,
et al: Electrochemotherapy: A new technological approach in
treatment of metastases in the liver. Technol Cancer Res Treat.
10:475–485. 2011.PubMed/NCBI
|
|
9
|
Martelli AM, Tazzari PL, Tabellini G,
Bortul R, Billi AM, Manzoli L, Ruggeri A, Conte R and Cocco L: A
new selective AKT pharmacological inhibitor reduces resistance to
chemotherapeutic drugs, TRAIL, all-trans-retinoic acid, and
ionizing radiation of human leukemia cells. Leukemia. 17:1794–1805.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Schreiber R, Mezencev R, Matyunina LV and
McDonald JF: Evidence for the role of microRNA 374b in acquired
cisplatin resistance in pancreatic cancer cells. Cancer Gene Ther.
23:241–245. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li B, Zhao J, Wang C-Z, Searle J, He TC,
Yuan CS and Du W: Ginsenoside Rh2 induces apoptosis and
paraptosis-like cell death in colorectal cancer cells through
activation of p53. Cancer Lett. 301:185–192. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
He ZY, Shi CB, Wen H, Li FL, Wang BL and
Wang J: Upregulation of p53 expression in patients with colorectal
cancer by administration of curcumin. Cancer Invest. 29:208–213.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mirzayans R, Andrais B, Scott A and Murray
D: New insights into p53 signaling and cancer cell response to DNA
damage: Implications for cancer therapy. J Biomed Biotechnol.
2012:1703252012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ng KW, Khoo SP, Heng BC, Setyawati MI, Tan
EC, Zhao X, Xiong S, Fang W, Leong DT and Loo JS: The role of the
tumor suppressor p53 pathway in the cellular DNA damage response to
zinc oxide nanoparticles. Biomaterials. 32:8218–8225. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lee YS, Yoon S, Park MS, Kim JH, Lee JH
and Song CW: Influence of p53 expression on sensitivity of cancer
cells to bleomycin. J Biochem Mol Toxicol. 24:260–269. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schmittgen TD, Lee EJ, Jiang J, Sarkar A,
Yang L, Elton TS and Chen C: Real-time PCR quantification of
precursor and mature microRNA. Methods. 44:31–38. 2008. View Article : Google Scholar
|
|
17
|
Agarwal V, Bell GW, Nam JW and Bartel DP:
Predicting effective microRNA target sites in mammalian mRNAs.
eLife. 4:e050052015. View Article : Google Scholar :
|
|
18
|
Ma Z, Sun X, Xu D, Xiong Y and Zuo B:
MicroRNA, miR-374b, directly targets Myf6 and negatively regulates
C2C12 myoblasts differentiation. Biochem Biophys Res Commun.
467:670–675. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Suzuki HI and Miyazono K: Emerging
complexity of microRNA generation cascades. J Biochem. 149:15–25.
2011. View Article : Google Scholar
|
|
20
|
Guil S and Esteller M: DNA methylomes,
histone codes and miRNAs: Tying it all together. Int J Biochem Cell
Biol. 41:87–95. 2009. View Article : Google Scholar
|
|
21
|
Marsico A, Huska MR, Lasserre J, Hu H,
Vucicevic D, Musahl A, Orom U and Vingron M: PROmiRNA: A new miRNA
promoter recognition method uncovers the complex regulation of
intronic miRNAs. Genome Biol. 14:R842013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chang CJ, Chao CH, Xia W, Yang JY, Xiong
Y, Li CW, Yu WH, Rehman SK, Hsu JL, Lee HH, et al: p53 regulates
epithelial-mesenchymal transition and stem cell properties through
modulating miRNAs. Nat Cell Biol. 13:317–323. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Muller PA and Vousden KH: p53 mutations in
cancer. Nat Cell Biol. 15:2–8. 2013. View
Article : Google Scholar
|
|
24
|
Jiang F-Z, He Y-Y, Wang H-H, Zhang HL,
Zhang J, Yan XF, Wang XJ, Che Q, Ke JQ, Chen Z, et al: Mutant p53
induces EZH2 expression and promotes epithelial-mesenchymal
transition by disrupting p68-Drosha complex assembly and
attenuating miR-26a processing. Oncotarget. 6:44660–44674.
2015.PubMed/NCBI
|
|
25
|
Gurtner A, Falcone E, Garibaldi F and
Piaggio G: Dysregulation of microRNA biogenesis in cancer: The
impact of mutant p53 on Drosha complex activity. J Exp Clin Cancer
Res. 35:452016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Engels BM and Hutvagner G: Principles and
effects of microRNA-mediated post-transcriptional gene regulation.
Oncogene. 25:6163–6169. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Qian D, Chen K, Deng H, Rao H, Huang H,
Liao Y, Sun X, Lu S, Yuan Z, Xie D, et al: MicroRNA-374b suppresses
proliferation and promotes apoptosis in T-cell lymphoblastic
lymphoma by repressing AKT1 and Wnt-16. Clin Cancer Res.
21:4881–4891. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bates S and Vousden KH: Mechanisms of
p53-mediated apoptosis. Cell Mol Life Sci. 55:28–37. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kuribayashi K, Finnberg N, Jeffers JR,
Zambetti GP and El-Deiry WS: The relative contribution of
pro-apoptotic p53-target genes in the triggering of apoptosis
following DNA damage in vitro and in vivo. Cell Cycle.
10:2380–2389. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dihlmann S, Kloor M, Fallsehr C and von
Knebel Doeberitz M: Regulation of AKT1 expression by
beta-catenin/Tcf/Lef signaling in colorectal cancer cells.
Carcinogenesis. 26:1503–1512. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Green BD, Jabbour AM, Sandow JJ, Riffkin
CD, Masouras D, Daunt CP, Salmanidis M, Brumatti G, Hemmings BA,
Guthridge MA, et al: Akt1 is the principal Akt isoform regulating
apoptosis in limiting cytokine concentrations. Cell Death Differ.
20:1341–1349. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tucka J, Yu H, Gray K, Figg N, Maguire J,
Lam B, Bennett M and Littlewood T: Akt1 regulates vascular smooth
muscle cell apoptosis through FoxO3a and Apaf1 and protects against
arterial remodeling and atherosclerosis. Arterioscler Thromb Vasc
Biol. 34:2421–2428. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jackson JG, Pant V, Li Q, Chang LL,
Quintás-Cardama A, Garza D, Tavana O, Yang P, Manshouri T, Li Y, et
al: p53-mediated senescence impairs the apoptotic response to
chemotherapy and clinical outcome in breast cancer. Cancer Cell.
21:793–806. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen GX, Zheng LH, Liu SY and He XH:
rAd-p53 enhances the sensitivity of human gastric cancer cells to
chemotherapy. World J Gastroenterol. 17:4289–4297. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gu J, Tang Y, Liu Y, Guo H, Wang Y, Cai L,
Li Y and Wang B: Murine double minute 2 siRNA and wild-type p53
gene therapy enhances sensitivity of the SKOV3/DDP ovarian cancer
cell line to cisplatin chemotherapy in vitro and in vivo. Cancer
Lett. 343:200–209. 2014. View Article : Google Scholar
|
|
36
|
Guo L, Wu H, Zhu J, Zhang C, Ma J, Lan J
and Xie X: Genetic variations in the PI3K/AKT pathway predict
platinum-based neoadjuvant chemotherapeutic sensitivity in squamous
cervical cancer. Life Sci. 143:217–224. 2015. View Article : Google Scholar : PubMed/NCBI
|