Introduction
Semaphorins are a large family of conserved proteins
originally characterized as directional cues in axonal guidance and
neurite outgrowth in neurogenesis (1–3).
Subsequently, it has been revealed that semaphorins and their
receptors carry out roles beyond neurogenesis and serve interesting
functions in immune regulation, extracellular matrix remodeling,
organogenesis, and angiogenesis (3–7).
Studies have identified the expression of Semaphorin 7A (SEMA7A) in
various tumor types, however few have described functional roles
for SEMA7A in tumor progression (8–11).
Hence, its contribution to tumor progression remains relatively
unknown in comparison to other vertebrate semaphorins.
SEMA7A, or CD108w, is a ~80-kDa GPI-anchored
transmembrane protein expressed by multiple cell types including:
neurons, immune cells, melanocytes, fibroblasts, bone cells, and
tumor cells (12). This protein
can be shed from the cellular membrane by action of ADAM-17(TACE)
(13). Both anchored and soluble
forms of SEMA7A have been shown to bind to Plexin C1 and β-1
integrin (CD29) (14–17). The latter activates the MAPK and
FAK pathways and causes an increase in proinflammatory cytokines
(18). Our group has demonstrated
that DA–3 murine mammary tumor cells exhibit high levels of SEMA7A
and that suppression of tumor-derived SEMA7A resulted in decreased
macrophage-mediated angiogenesis (15).
In this study we further assessed the direct effects
of SEMA7A suppression on the highly malignant 4T1 breast carcinoma
model. Gene silencing of SEMA7A in 4T1 cells yielded a strong
antitumor effect in vivo and SEMA7A shRNA-expressing 4T1
cells showed an impaired ability to proliferate, migrate and
invade. These cells also had a decrease in mesenchymal properties,
with an increase in cell stiffness. Genetic ablation of
host-derived SEMA7A increased the anti-tumor effects of SEMA7A
shRNA. Our study shows a novel functional role for SEMA7A in the
progression of mammary tumors.
Materials and methods
Mice and cell lines
Female BALB/c mice (8–12-week-old) were obtained
from Charles River Laboratories, and SEMA7A−/− mice
generated by Dr A.L. Kolodkin (Johns Hopkins University, Baltimore,
MD, USA), were purchased from Jackson Laboratories. Using a speed
congenic approach (19,20) SEMA7A−/− mice were
backcrossed to a BALB/c background, reaching 99.9% of desired
BALB/c background. Mice were housed and used according to the
National Institutes of Health guidelines, under protocols approved
by Florida Atlantic University Institutional Animal Care and Use
Committee. EpH4 mammary cells were provided by Dr Jenifer Prosperi,
Indiana University School of Medicine (South Bend, IN, USA). EpH4,
67NR, 4T07, 4T1 and 4T1-LUC (Perkin-Elmer, Waltham, MA, USA) cells
were grown in complete DMEM media with 10% FBS. Female BALB/c or
SEMA7A−/− BALB/c mice were inoculated in the mammary fat
pads with 5×105 luciferase transfected 4T1 or 4T1-WT
mammary tumor cells. Bioluminescent imaging of 4T1-LUC tumor
bearers was done up to 3-weeks post-tumor cell implantation. For
4T1 and 4T1-LUC tumor-bearing mice, lungs were collected at 42-days
post-tumor cell implantation.
RNA isolation and real-time reverse
transcriptase-polymerase chain reaction
Total RNA was extracted from murine or human tumor
cells, using the RNeasy Protect Mini kit (Qiagen, Gemantown, MD,
USA) according to the manufacturer's instructions. Briefly, cDNA
was synthesized using Quantitech Reverse Transcription kit (Qiagen)
and gene expression was detected by SYBR Green real-time
quantitative polymerase chain reaction (qPCR) analysis using SYBR
RT2 qPCR primers (Qiagen, proprietary primers, sequence
not disclosed) from SABioscience (Qiagen). The mRNA levels of gene
of interest were normalized to β-actin, GAPDH or HSP90ab mRNA
levels. PCR cycles followed the sequence: 10 min at 95°C of initial
denaturation; 15 sec at 95°C; and 40 cycles of 1 min each at 60°C
for annealing. The samples were amplified using the Stratagene
Mx3005O cycler.
Flow cytometry studies
Ki67 antibodies (Biolegend, San Diego, CA, USA) were
used to determine cellular proliferation by flow cytometry
according to the manufacturer's protocol. Cells (50,000) were
acquired using a FACSCalibur (BD, Franklin Lakes, NJ, USA) flow
cytometer, followed by analysis using FloJo software (Tree Star,
Inc., Ashland, OR, USA).
Silencing of SEMA7A in 4T1 murine mammary
tumor cells
Semaphorin 7A gene silencing in 4T1-LUC mammary
tumor cells was achieved using RNA interference via short hairpin
RNA. To confirm gene knockdown, qPCR was performed using the SEMA7A
specific primers according to the manufacturer's protocol (Qiagen).
A SureSilencing shRNA plasmid (Qiagen) with one of two insert
sequences was used to target SEMA7A in the 4T1-LUC cells, shRNA1
(ccatagcttt gtcttcaatat) or shRNA2 (cctagctgcatcctgttcatt). Cells
were passaged and selected with G418 (800 µg/ml) until at
least a 5-fold decrease in the SEMA7A gene expression was achieved
when compared to the scramble shRNA control. For non-luciferase 4T1
cells, an optimized short hairpin RNA algorithm was used to select
the top three miRE shRNA sequences targeting SEMA7A (21). The miRE shRNA1 targeted the 5′ end
of the SEMA7A mRNA
(tatgatgataagatctatagtgaagccacagatgtatagatcttatcatcataggcttt) and
miRE shRNA2 targeting the 3′ end of the SEMA7A mRNA
(caggagtactagaataatagtgaagccacagatgtattattctagtactcctgggctat)
(Mirimus). As a negative control, we used pooled clones expressing
miRE shRNA against Renilla Firefly Luciferase. Cells were
transfected with shRNA encoding plasmids using Avalanche
transfection reagent (EZ-Biosystems) and selected with puromycin (3
µg/ml). The shRNA vectors also expressed a GFP reporter
protein (21). Gene knockdown was
confirmed by qPCR using SEMA7A specific primers according to the
manufacturer's protocol (Qiagen). The results of gene expression
were then confirmed by determination for the SEMA7A protein.
Atomic force microscopy (AFM) cell
stiffness measurements
Cell stiffness measurements were acquired on living
4T1 cells. The bare AFM tip was lowered onto the cell surface at 4
µm/s (22). The acquired
force-indentation curves of the cells were fit to a model initially
proposed by Hertz to estimate the Young's modulus assuming that the
cell is an isotropic elastic solid and the AFM tip is a rigid cone
(23). The model is as
follows:
where F is the applied force, α the indentation,
K the Young's modulus, θ the angle formed by the
indenter and the plane of the surface (55°) and ν, Poisson
ratio (0.5). Young's modulus was obtained by least square analysis
of the force-indentation curves using Igor Pro software.
Migration and invasion assay
4T1-LUC-Scramble-shRNA cells or 4T1-LUC-SEMA7A-shRNA
cells, mammary tumor cells, were cultured under optimal conditions
using DMEM culture media with 10% FBS in 24 wells containing a
500-µM wide culture-insert (Ibidi, Fitchburg, WI, USA) until
~80% confluency was achieved. Subsequently, the 10% FBS DMEM was
replaced with 0.5% FBS DMEM and the culture insert was removed to
expose cell-free gap. Gap width was assessed at 0, 6 and 12-h
post-challenge. For the invasion assay, 10,000 serum-starved
4T1-LUC-Scramble-shRNA cells or 4T1-LUC-SEMA7A-shRNA were seeded
into a 24-well system of 8-µM Transwell inserts coated with
Cultrex Basement Membrane Extract (Corning, Tewksbury, MA, USA) and
cells were allowed to invade for 18 h. Cells that invaded through
insert were dissociated with Tryple Express (Invitrogen, Carlsbad,
CA, USA) and stained with Calcein AM. Fluorescence measurements
were transformed into cell numbers using a pre-determined cell
standard curve and percent invasion was calculated.
Statistical analysis
Results are expressed as means ± standard deviation.
Statistical analyses were performed using GraphPad Prism 6 software
(La Jolla, CA, USA). Statistical comparisons were performed using
an unpaired 2-tailed Student's t-test, with significance at
P<0.05. For multiple comparisons of tumor growth and metastasis,
a two-way ANOVA with a Dunn's post hoc test was performed. For
analyzing the survival of tumor-bearing mice, the Kaplan-Meier
method was used.
Results
SEMA7A is overexpressed in 4T1 mammary
tumor cells and is required for tumor growth in vivo
We evaluated SEMA7A gene expression levels in
non-tumorigenic EpH4 murine mammary cells and three sister murine
mammary tumor cell lines with varying degrees of malignancy (67NR,
4TO7 and 4T1). 67NR can form slow-growing tumors but fail to
disseminate, 4T07 can form tumors that disseminate but cannot
metastasize, and 4T1 can form rapid growing tumors that complete
all the steps required for metastasis (24). Analyses by qPCR showed that EpH4
cells had nearly undetectable levels of SEMA7A and levels were
increased 60-fold in 67NR, 140-fold in 4T07 and 210-fold in 4T1
tumor cells when compared to that of EpH4 cells (Fig. 1A). These results indicate that as
the malignant capabilities of mammary cells increased, so did the
SEMA7A levels. We therefore posed the question: does SEMA7A have a
functional role in mammary tumor cell malignancy? We strategized to
use shRNA to suppress the levels of SEMA7A in the highly aggressive
4T1 cells and assess its effects in vivo. We chose to use
4T1 cells expressing luciferase (4T1-LUC) as they enabled us to
perform non-invasive bioluminescent imaging to monitor tumor
progression. SEMA7A levels were equivalent in 4T1-LUC cells and
wild-type 4T1 cells (data not shown). Experimental control cells
were generated by transfecting 4T1-LUC cells with a plasmid
encoding for a non-targeting scramble shRNA
(4T1-LUC-Scramble-shRNA). 4T1-LUC wild-type and
4T1-LUC-Scramble-shRNA cells had comparable SEMA7A levels (Fig. 1B). 4TI-LUC cells were also
transfected with plasmids encoding for one of four SEMA7A shRNA
sequences. Two of the shRNA strands targeting SEMA7A showed
consistent silencing efficiency, 4T1-LUC-SEMA7A-shRNA1 and
4T1-LUC-SEMA7A-shRNA2 (Fig. 1B).
4T1-LUC-SEMA7A-shRNA1 had an 80% reduction in SEMA7A levels
compared to both 4T1-LUC wild-type cells and 4T1-LUC-Scramble-shRNA
cells, while 4T1-LUC-SEMA7A-shRNA2 had a 55% reduction (Fig. 1B). Given its enhanced silencing
efficiency, we continued our studies using the
4T1-LUC-SEMA7A-shRNA1 clone, which will be referred from here on as
4T1-LUC-SEMA7A-shRNA. The commercially available anti-SEMA7A murine
antibodies at the time of this study were determined to be
unspecific in our hands. The antibodies tested generated false
positives when used for western blotting and immunofluorescence
analysis of SEMA7A-shRNA silenced 4T1 cells and tissue samples from
SEMA7A−/− mice. Hence, we utilized SEMA7A mRNA as an evaluative
readout in this study in order to effectively differentiate its
levels from that of other Semaphorins.
 | Figure 14T1 cells have high levels of SEMA7A
and SEMA7A shRNA gene silencing of 4T1 cells decreases their growth
and metastasis in vivo. (A) Transcript analyses of 67NR,
4T07 and 4T1 tumors cells by qPCR, normalized to SEMA7A levels of
non-tumorigenic EpH4 mammary epithelia cells (n=3, unpaired
two-tailed Student's t-test). (B) qPCR analyses of SEMA7A mRNA
levels in 4T1-LUC-Scramble-shRNA, 4T1-LUC-SEMA7A-shRNA1 and
4T1-LUC-SEMA7A-shRNA2 tumors cells, normalized to SEMA7A levels in
4T1-LUC wild-type (n=3, unpaired two-tailed Student's t-test). (C)
4T1-LUC-Scramble-shRNA or 4T1-LUC-SEMA7A-shRNA tumor cells were
implanted in the mammary fat pads of wild-type female BALB/c, and
non-invasive bioluminescent imaging was done at specified
time-points and (D) reported as normalized photons/sec (n=5 mice,
repeated three times, two-way ANOVA). (E) Kaplan-Meier survival
curve of wild-type BALB/c mice bearing 4T1-LUC-Scramble-shRNA or
4T1-LUC-SEMA7A-shRNA tumor cells (n=5 mice, repeated three times,
log-rank test). (F) On day 42 post-tumor implantation, lungs were
excised from mice bearing 4T1-LUC-Scramble-shRNA or
4T1-LUC-SEMA7A-shRNA and imaged for tumor cell-specific
bioluminescent signals from lung metastasis nodules (n=5 mice,
repeated three times, unpaired two-tailed Student's t-test). (G)
India Black ink staining was done to determine number of
macro-metastasis lesions, which remained unstained (n=5 mice,
repeated three times, unpaired two-tailed Student's t-test). Data
are presented as mean ± SD. ***P≤0.001,
****P≤0.0001. |
We implanted 4T1-LUC-Scramble-shRNA or
4T1-LUC-SEMA7A-shRNA tumor cells into the mammary fat pads of
syngeneic wild-type BALB/c mice and bioluminescent imaging was
performed to monitor tumor growth (Fig. 1C). 4T1-LUC-SEMA7A-shRNA tumor
bearers displayed a significant reduction (P=0.00001) of tumor
burden (Fig. 1D) and a significant
increase (P=0.00001) in survival (Fig.
1E). We questioned whether the reduced tumor burden in
4T1-LUC-SEMA7A-shRNA tumor-bearing mice could have changed the
kinetics of metastatic dissemination. We surveyed the lungs of
tumor bearers at day 42 post-tumor implantation given that 4T1
tumor cells are known to metastasis primarily to the lung (25). Detection of the luciferase signals
revealed a significant ~80% decrease (P=0.00001) of tumor
cell-specific bioluminescence in the lungs of 4T1-LUC-SEMA7A-shRNA
tumor bearers (Fig. 1F).
Enumeration of the metastatic nodules, which remain unstained after
India Black staining, revealed >30 metastatic foci in mice
bearing 4T1-LUC-Scramble-shRNA cells, compared to <5 metastatic
foci in lungs of 4T1-LUC-SEMA7A-shRNA mammary tumor bearers
(Fig. 1G). The lung metastasis
nodules that eventually developed in 4T1-LUC-SEMA7A-shRNA
tumor-bearing mice retained SEMA7A expression (data not shown),
indicating they arose from tumor cells in which SEMA7A gene
expression had either never been suppressed or that had lost the
suppression. Taken together, our results indicate that SEMA7A
produced by tumor cells is actively involved in mammary tumor
progression.
SEMA7A shRNA gene silencing decreases the
proliferative, migratory and invasive potential of 4T1 cells
Given that tumor growth was impaired in
4T1-LUC-SEMA7A-shRNA mammary tumor-bearing mice (Fig. 1), we sought to examine the effects
of shRNA-mediated suppression of SEMA7A on 4T1 cells in
vitro. We first observed a reduced growth rate in
4T1-LUC-SEMA7A-shRNA tumor cells (Fig.
2A) that was confirmed by assaying for the expression of the
proliferation marker Ki67 by flow cytometry (Fig. 2B). We found a decrease of nearly
half the expression of Ki67 in 4T1-LUC-SEMA7A-shRNA tumor cells
compared to control (Fig. 2C).
Next, we challenged 4T1-LUC-Scramble-shRNA and 4T1-LUC-SEMA7A-shRNA
cells to populate a 500-µM cell-free gap under serum-low
conditions in order to assess their migratory potential in
vitro (Fig. 2D). At 12-hour
post-challenge, 4T1-LUC-Scramble-shRNA cells succeeded in closing
the gap but 4T1-LUC-SEMA7A-shRNA only populated ~30% of the
original cell-free gap (Fig. 2E).
Given their reduced migratory ability, we surveyed if SEMA7A gene
silencing had also affected the levels of important regulators of
cell migration: RhoA/B/C (26).
When compared to 4T1-LUC-Scramble-shRNA cells, 4T1-LUC-SEMA7A-shRNA
cells exhibited decreased levels of migration/motility promoting
RhoA and RhoC but increased levels of tumor-limiting RhoB (Fig. 2F). We next questioned whether
decreased SEMA7A levels could also affect the invasiveness of 4T1
cells. 4T1-LUC-Scramble-shRNA and 4T1-LUC-SEMA7A-shRNA tumor cells
were labeled with Calcein AM and seeded into the upper chamber of a
Matrigel coated, 8-µM pore Transwell insert. After 12 h, we
quantified the percentage of Calcein AM-positive cells that were
able to invade through the Matrigel-coated insert and into the
lower chamber of the well. We found that 4T1-LUC-SEMA7A-shRNA tumor
cells had a ~60% significant reduction in invasion (P≤0.05) when
compared to 4T1-LUC-Scramble-shRNA tumor cells (Fig. 2G). To further determine the effect
of SEMA7A on invasive abilities, we surveyed for the levels of
various matrix metalloproteinases (MMPs) that have been determined
critical in mediating tumor cell invasion (27). Matrix metalloproteinases MMP-2, -3,
-9, -10 and -13 were significantly (P≤0.001) decreased in
4T1-LUC-SEMA7A-shRNA tumor cells, suggesting a strong linkage
between the gene expression of SEMA7A and levels of MMPs (Fig. 2H). Our results indicate that
suppression of tumor-derived SEMA7A weakens the ability of 4T1
cells not only to proliferate, but also to migrate and invade.
These changes in proliferative, migratory and invasive potentials
may partially account for the decreased tumor progression exhibited
by 4T1-LUC-SEMA7A-shRNA tumor cells in vivo (Fig. 1).
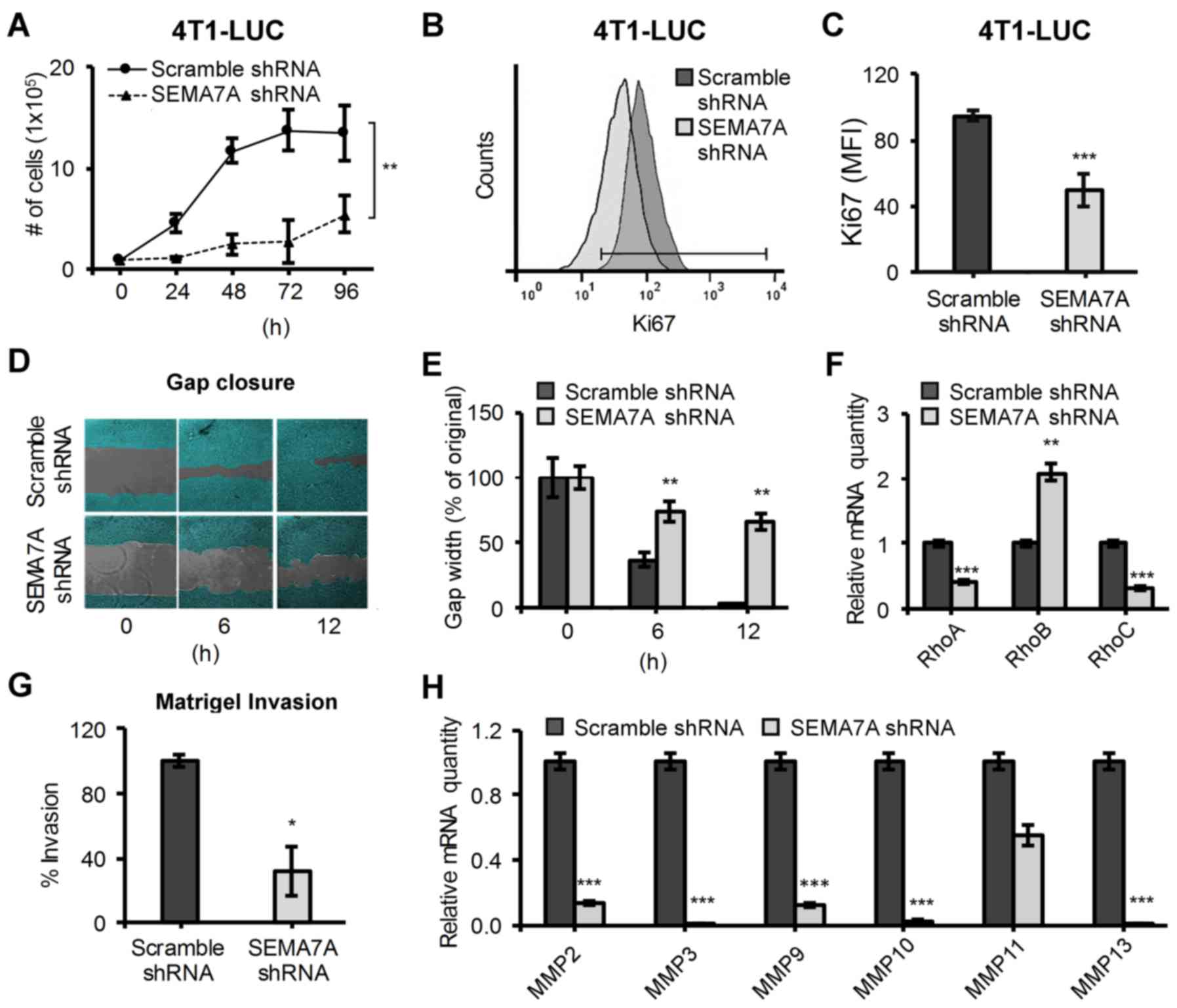 | Figure 2Silencing of SEMA7A gene in 4T1-LUC
cells decreases their proliferation, motility and invasion. (A)
Growth curves of 4T1-LUC-Scramble-shRNA and 4T1-LUC-SEMA7A-shRNA
tumor cells under optimal conditions (n=6, unpaired two-tailed
Student's t-test). (B) Flow cytometric analyses of proliferation
marker Ki67 in 4T1-LUC-Scramble-shRNA and 4T1-LUC-SEMA7A-shRNA
tumor cells. (C) Quantification of Ki67 expression (n=3, unpaired
two-tailed Student's t-test). (D) 4T1-LUC-Scramble-shRNA and
4T1-LUC-SEMA7A-shRNA cells were grown to confluency in a 24-well
with a culture insert to generate a 500-µM cell-free gap. At
time zero, insert was removed and gap-closure was observed at
specified time-points and (E) reported as percent width of original
gap (n=3, unpaired two-tailed Student's t-test). (F) Gene
expression of RhoA, RhoB and RhoC was assayed by qPCR in
4T1-LUC-SEMA7A-shRNA tumor cells, normalized to
4T1-LUC-Scramble-shRNA (n=3, unpaired two-tailed Student's t-test).
(G) 4T1-LUC-Scramble-shRNA and 4T1-LUC-SEMA7A-shRNA cells' in
vitro invasion through a Matrigel coated 8 µM Transwell,
reported as percent invasion using 4T1-LUC-Scramble-shRNA cells as
100% (n=3, unpaired two-tailed Student's t-test). (H) Gene
expression of specified MMPs was assayed by qPCR in
4T1-LUC-SEMA7A-shRNA tumor cells, normalized to levels of
4T1-LUC-Scramble-shRNA cells (n=3, unpaired two-tailed Student's
t-test). Data are presented as mean ± SD. *P≤0.05,
**P≤0.01, ***P≤0.001. |
Decreased SEMA7A levels in 4T1 mammary
tumor cells promotes an epithelial-like morphology and decreased
levels of mesenchymal-promoting factors
It has been shown that gain of mesenchymal
properties in 4T1 cells may lead to increased invasive behavior
in vivo (28) and that
expression of SEMA7A has been shown to promote a mesenchymal
phenotype in cells (9). We
therefore questioned if SEMA7A gene silencing had altered the
epithelial/mesenchymal properties of 4TI-LUC cells.
4T1-LUC-SEMA7A-shRNA cells displayed an enhanced epithelial-like
morphology with cells growing in tight, rounded colonies (Fig. 3A). This change towards an
epithelial-like morphology led us to probe whether silencing of
SEMA7A in 4T1-LUC-SEMA7A-shRNA cells may have tilted the levels of
mesenchymal-promoting factors. We first assessed TGF-β1 levels as
it has been shown to be a key mediator of mesenchymal programs
(29) and has been tightly linked
to SEMA7A (17). We found that
TGF-β1 levels were significantly decreased (P≤0.001) in
4T1-LUC-SEMA7A-shRNA cells when compared to 4T1-LUC-Scramble-shRNA
cells, but no significant changes were detected in the levels of
TGF-β2 and TGF-β3 (Fig. 3B).
4T1-LUC-SEMA7A-shRNA cells also had a marked decrease in the
expression of known TGF-β1 induced mesenchymal promoting factors:
Snail1, Snail2 and Twist (30)
(Fig. 3C). In addition,
4T1-LUC-SEMA7A-shRNA cells had decreased levels of ZEB1 and ZEB2
compared to 4T1-LUC-Scramble-shRNA (Fig. 3D), both of which are strong
inducers of a mesenchymal phenotype and correlated with enhanced
aggressive behavior in tumor cells (31). Given that Snail1 has been shown to
induce the expression of mesenchymal markers Vimentin (VIM) and
N-cadherin (CDH2) (32), we
assayed for the expression of these markers in 4T1 cells upon
SEMA7A gene silencing. 4T1-LUC-SEMA7A-shRNA cells showed an ~90%
decrease in Vimentin levels and an ~80% decrease in N-cadherin
levels when compared to 4T1-LUC-Scramble-shRNA cells (Fig. 3E). Vimentin and N-cadherin levels
have been shown to be inversely correlated with E-cadherin (CDH1)
(33). We sought to determine if
SEMA7A gene silencing could have affected the E-cadherin (CDH1)
levels, but we found they remained unchanged between
4T1-LUC-Scramble-shRNA and 4T1-LUC-SEMA7A-shRNA cells (Fig. 3F). Like E-cadherin, tight-junction
marker Desmoplakin (DSP) has also been shown to be lost during the
epithelial to mesenchymal transition (32). We found a significant 5-fold
increase (P≤0.01) of Desmoplakin levels in 4T1-LUC-SEMA7A-shRNA
cells compared to 4T1-LUC-Scramble-shRNA cells (Fig. 3F). Our results indicate that shRNA
silencing of the SEMA7A gene in 4T1 cells led to a shift towards a
more epithelial morphology, with decreased levels of mesenchymal
markers that have been linked to enhanced tumor growth and
metastasis.
SEMA7A gene silencing increases stiffness
of 4T1 cells
Atomic force microscopy (AFM) has recently been
shown to be useful in distinguishing malignant cells from normal
cells (34). Analyses of
biomechanical properties with AFM has shown that aggressive
cancerous cells are less stiff compared to normal cells in 2D
cultures (35,36). AFM measurements were acquired to
determine the relative stiffness of 4T1-LUC-Scramble-shRNA and
4T1-LUC-shRNA-SEMA7A cells. The AFM cantilever was used as a
microindenter, probing the cell <1 µm using applied
forces of <1 nN so as to not damage the cell. Fig. 4A shows representative
force-indentation curves acquired for 4T1-LUC-Scramble-shRNA and
4T1-LUC-shRNA-SEMA7A cells. 4T1-LUC-shRNA-SEMA7A cells indented
less than 4T1-LUC-Scramble-shRNA cells for equivalent applied
forces. One hundred force-indentation curves were acquired for each
cell measured and fitted to the Hertz's model. Histograms in
Fig. 4B reveal the data
distribution of the Young's modulus values for both cell types. The
average Young's modulus value calculated for the 4T1 mammary tumor
cells was 3.7±0.3 kPa (n=35) (Fig.
4C). Following SEMA7A gene knockdown, cell stiffness increased
to 7.5±1 kPa (n=29) in 4T1-LUC-Scramble-shRNA cells. Our AFM data
supports the notion that an increase in cell stiffness in
vitro is inversely related to the malignant behavior of tumor
cells, as we observed that the stiffer 4T1-LUC-shRNA-SEMA7A cells
had reduced malignant potential in vivo (Fig. 1).
Ablation of host-derived SEMA7A impairs
tumor growth and enhances the antitumor effects of shRNA
suppression of tumor-derived SEMA7A
Given that host cells also express SEMA7A (14,17,18,37),
we questioned if genetic ablation of host-derived SEMA7A could
further augment the anti-tumor effects of SEMA7A gene silencing in
4T1 cells. We first determined the effects of solely ablating
host-derived SEMA7A on mammary tumor growth. Mammary pads of
wild-type or SEMA7A−/− female BALB/c mice were
inoculated with wild-type 4T1-LUC cells. Bioluminescent imaging was
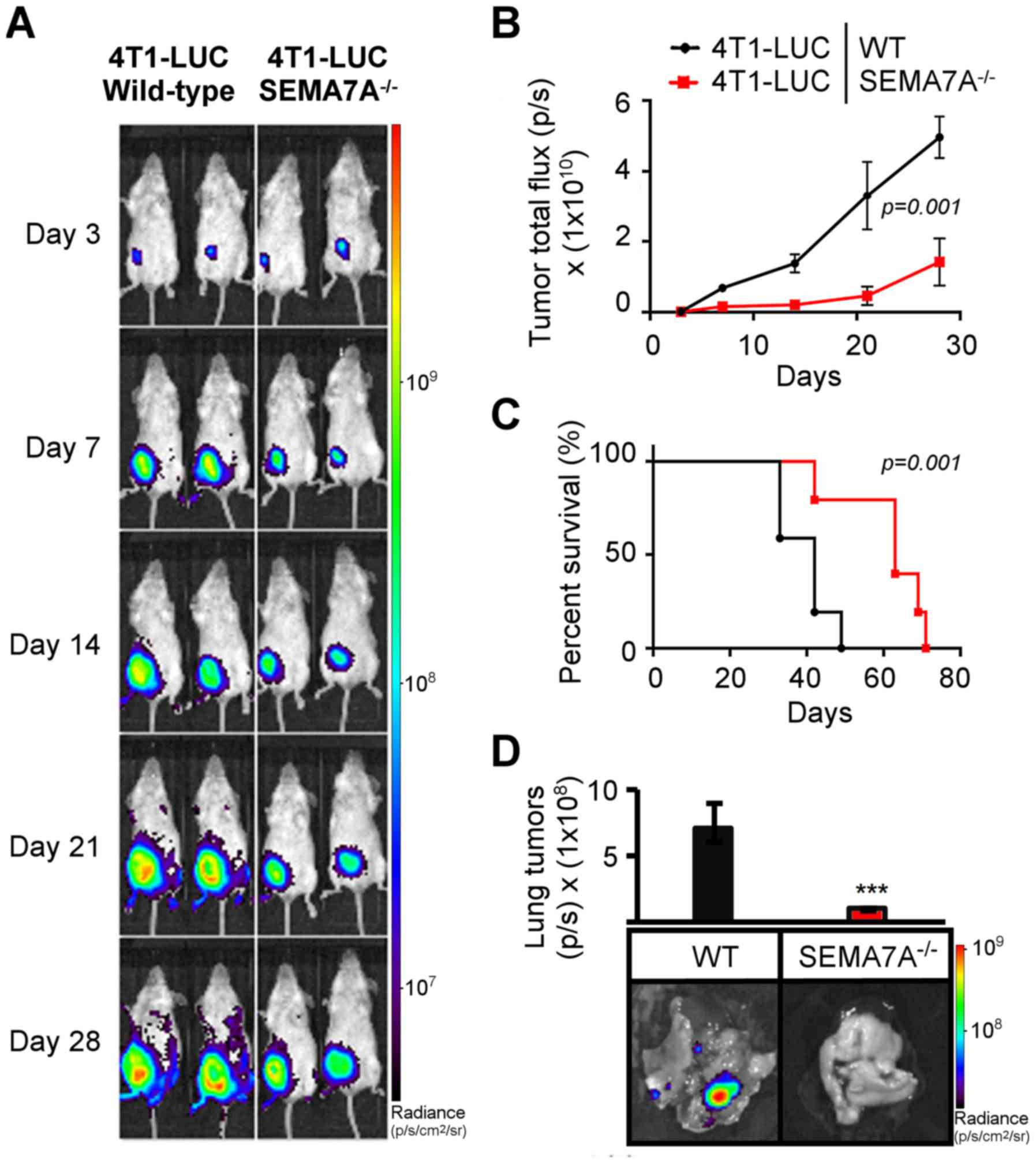
performed for 28 days. SEMA7A−/− tumor-bearing mice
displayed a significant decrease (P=0.001) in tumor growth rate
(Fig. 5A and B), increase in
survival (P=0.001) (Fig. 5C), and
a decrease in metastasis to the lungs (P≤0.001) (Fig. 5D).
We next tested the effects of ablating host-derived
SEMA7A in addition to suppressing tumor-derived SEMA7A. Towards
this, we used an optimized miR-E shRNA system that provided
enhanced stable gene knockdown (21). Wild-type 4T1 were transfected with
a vector encoding for an shRNAmir targeting the 5′ end of the
SEMA7A mRNA (4T1-SEMA7A-shRNA1) or one targeting the 3′ end of the
SEMA7A mRNA (4T1-SEMA7A-shRNA2). 4T1-LUC cells could not be used
with the miR-E system, as puromycin had been used to select for
luciferase expression. To generate an experimental control, 4T1
cells were transfected with a vector encoding for an shRNAmir
targeting Renilla luciferase (4T1-Renilla-shRNA).
4T1-Renilla-shRNA cells had SEMA7A levels equivalent to that
of wild-type 4T1 cells (data not shown). Both SEMA7A shRNAmirs
achieved a >80% decrease in SEMA7A levels when compared to
4T1-Renilla-shRNA cells (Fig.
6A). 4T1-Renilla-shRNA cells, 4T1-SEMA7A-shRNA1 or
4T1-SEMA7A-shRNA2 cells were implanted into the mammary fat pads of
either wild-type or SEMA7A−/− female BALB/c mice. Gene
silencing of SEMA7A in 4T1 cells resulted in a significant
reduction (P≤0.0001) in tumor burden, but ablation of host-derived
SEMA7A further decreased tumor growth by an additional ~15%
(Fig. 6B). At day 42, lungs were
excised from tumor-bearing mice and assessed for metastatic lesions
(Fig. 6C). Ablating host-derived
SEMA7A yielded an additional ~30% significant reduction (P≤0.0001)
in the number of metastatic lung lesions compared to suppressing
tumor-derived SEMA7A alone (Fig.
6D). Our results show that ablation of host-derived SEMA7A and
tumor-derived SEMA7A can significantly improve outcomes in our
breast carcinoma model.
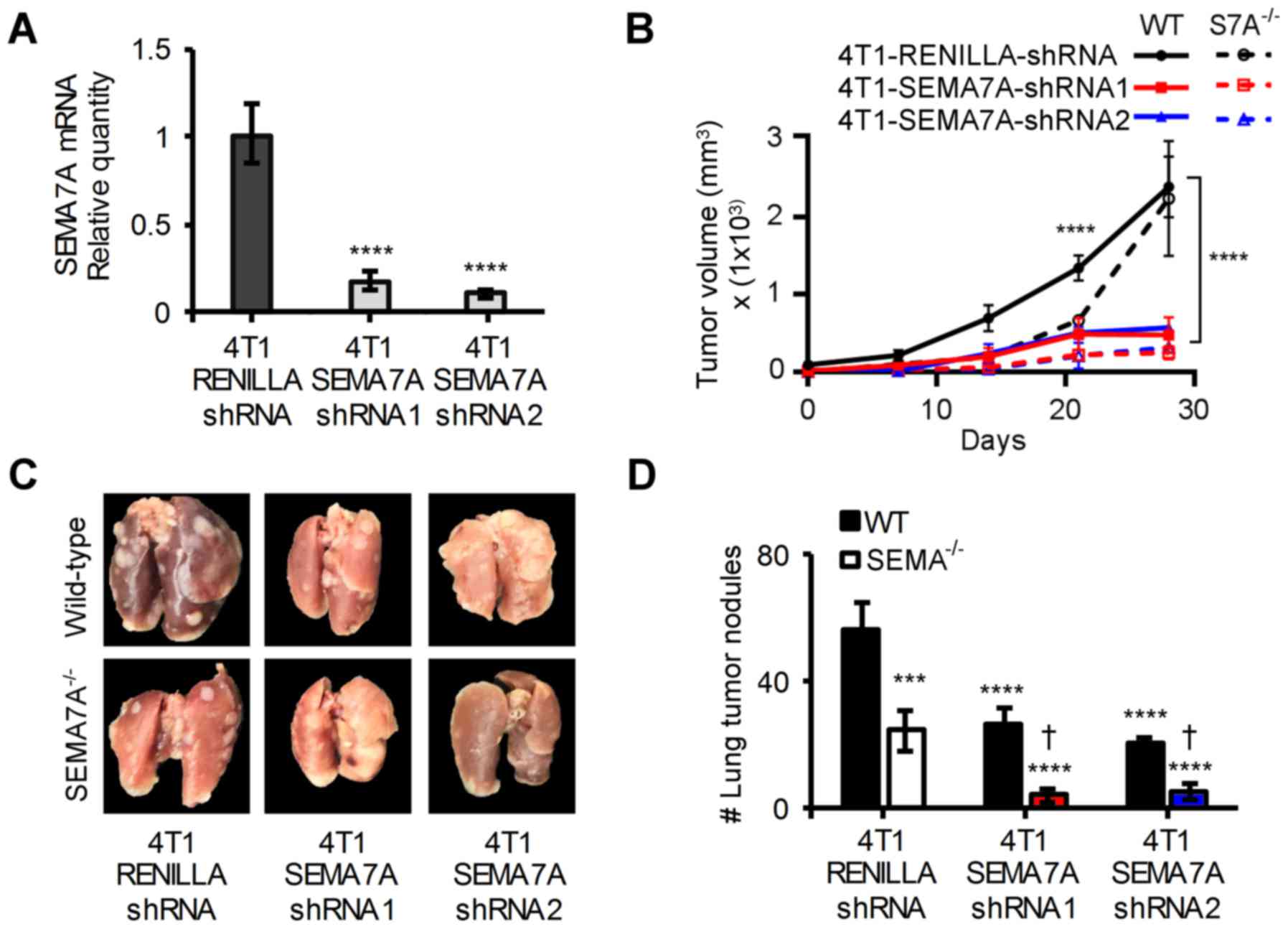 | Figure 6Inhibition of host-derived and
tumor-derived SEMA7A decreases rate of tumor growth and metastasis
in 4T1 tumor-bearing mice. (A) qPCR analyses of SEMA7A mRNA levels
in 4T1-SEMA7A-shRNA1 (targeting 5′ end of SEMA7A mRNA) and
4T1-SEMA7A-shRNA2 (targeting 3′ end of SEMA7A mRNA) tumors cells,
normalized to SEMA7A levels of 4T1-Renilla-shRNA (non-target
control) (n=3, unpaired two-tailed Student's t-test). (B)
4T1-LUC-Scramble-shRNA, 4T1-LUC-SEMA7A-shRNA1 or shRNA2 tumor cells
were implanted in the mammary fat pads of wild-type BALB/c or
SEMA7A−/− BALB/c mice and tumor volume was measured at
specified time-points (n=5 mice, repeated three times, two-way
ANOVA. (C) Photographic images of excised and 4% PFA fixed lungs at
day 42 post-tumor implantation, and (D) quantification of
macro-metastatic lesions in the lungs at day 42 post-tumor
implantation (n=5 mice, repeated three times, two-way ANOVA). Data
are presented as mean ± SD. To represent statistical significance,
asterisks denote differential significance of values compared to
that of 4T1-Renilla-shRNA/WT-BALB/c and a dagger denotes
differential significance compared to
4T1-Renilla-shRNA/SEMA7A−/−.
***P≤0.001, ****P≤0.0001,
†P≤0.0001. |
Discussion
The objective of this study was to elucidate the
role of SEMA7A in breast cancer. To do so, we utilized the 4T1
murine model of advanced breast carcinoma. We found that shRNA
suppression of SEMA7A in 4T1 mammary tumor cells significantly
inhibited tumor growth, which in turn deferred metastasis and
increased survival. When we implanted wild-type 4T1 cells in
SEMA7A-deficient mice, we found that lack of host-derived SEMA7A
further decreased tumor growth. However, shRNA inhibition of
tumor-derived SEMA7A resulted in a greater decrease of tumor burden
than genetic ablation of host-derived SEMA7A. When we combined both
approaches, the antitumor effects of SEMA7A shRNA were augmented.
We will further delineate the contributions of tumor-derived SEMA7A
versus that of host-derived SEMA7A. In addition, determining if
there are any inherent variations between tumor-specific and
host-derived SEMA7A could be useful when designing inhibitory
strategies. To date, no tumor-enhancing mutations or variations of
SEMA7A have been described.
In addition to breast cancer, SEMA7A has also been
shown to be expressed in melanoma, glioblastoma and oral squamous
cell carcinoma (8,11,15,38).
Multiple studies (9,10,15,38)
suggest a strong linkage between SEMA7A and the potential of tumor
cells to proliferate, migrate and invade. In terms of invasive
potential, 4T1-LUC-shRNA-SEMA7A cells had decreased levels of
MMP-2,-3,-9,-10 and -13. Encouragingly, oral squamous cell
carcinoma cell lines shRNA gene silenced for SEMA7A displayed
decreased MMP-2, -9 and MT1-MMP (38). Hence, these overlapping findings
suggest a conserved role for SEMA7A in mediating pro-migration and
pro-metastatic MMPs among different cancers.
Further characterization of 4T1-LUC-shRNA-SEMA7A
cells in our study revealed decreased levels of mesenchymal
promoting factors: Snail, Twist, ZEB1 and ZEB2. Additional studies
will determine the specific pathway(s) by which SEMA7A affects the
levels of these factors. We speculate that SEMA7A induction of
TGF-β may play a critical role in promoting a mesenchymal
phenotype, as TGF-β has been shown to directly induce EMT-promoting
transcription factors (32).
Supporting the role of SEMA7A in promoting mesenchymal phenotypes,
it has been shown that SEMA7A can serve as a differentiation marker
for mesenchymal stem cells (39).
It is proposed that gain of mesenchymal properties
causes a decrease in cell stiffness (40). A decrease in stiffness allows cells
to spread more easily on a substrate and thus in turn could
facilitate migration and invasion. Transformation of non-metastatic
human breast cancer cells into metastatic also caused a decrease in
cell stiffness in 2D cultures (41). Our results support the notion that
cell stiffness and malignant behavior can be inversely related.
Microindentation with an AFM probe showed that 4T1-LUC-shRNA-SEMA7A
cells have increased cell stiffness compared to SEMA7A-expressing
4T1 cells. Both in vitro and in vivo, these stiffer
cells showed lessened invasion potential. A recent study used AFM
to show that SEMA7A decreases the adhesion strength of dendritic
cells to the extracellular matrix (42). It would be interesting to know if
inhibition of SEMA7A in tumor cells affects their ability to adhere
to the extracellular matrix. A potential increase in adhesion to
the ECM, coupled with the decreased levels of MMPs we observed,
could potentially hinder the ability of tumor cells to migrate and
disseminate.
We and others now show that shRNA inhibition of
SEMA7A in tumor cells lessens their malignant potential (10,15,38).
Although useful in delineating the function of SEMA7A in tumor
progression, shRNA has limited therapeutic potential. In order to
translate these preclinical findings into therapies, the
development of SEMA7A inhibitors will be critical. To date, there
are no known agents that specifically target SEMA7A. Black et
al corroborated that inhibiting SEMA7A in breast cancer would
be beneficial as SEMA7A levels correlated with poor prognosis in
breast cancer patients (10).
Overall, our collective results support our
hypothesis that SEMA7A promotes breast cancer progression. Our
findings postulate a novel role for SEMA7A in breast cancer that
may lead to further findings of therapeutic value.
Acknowledgments
This study was supported by the National Institutes
of Health grants NIH R15 CA135513-01 and R15 CA135513-01-OS1, and
by the Boca Raton Regional Hospital Foundation. We remember and
thank the late Kathy Tabor-McEwan, M.D., for her instrumental
efforts in establishing the FAU-BRRH alliance to support this study
and her commitment to further breast cancer research.
References
|
1
|
Pasterkamp RJ and Kolodkin AL: Semaphorin
junction: Making tracks toward neural connectivity. Curr Opin
Neurobiol. 13:79–89. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhou Y, Gunput RA and Pasterkamp RJ:
Semaphorin signaling: Progress made and promises ahead. Trends
Biochem Sci. 33:161–170. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Műzes G and Sipos F: Relation of immune
semaphorin/plexin signaling to carcinogenesis. Eur J Cancer Prev.
23:469–476. 2014. View Article : Google Scholar
|
|
4
|
Epstein JA, Aghajanian H and Singh MK:
Semaphorin signaling in cardiovascular development. Cell Metab.
21:163–173. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Garcia-Areas R, Libreros S and
Iragavarapu-Charyulu V: Semaphorin7A: Branching beyond axonal
guidance and into immunity. Immunol Res. 57:81–85. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ito D, Nojima S and Kumanogoh A: The role
of Semaphorin family in immune systems. Nihon Rinsho Meneki Gakkai
Kaishi. 37:1–10. 2014.In Japanese. View Article : Google Scholar
|
|
7
|
Morihana T and Kumanogoh A: Immune
semaphorins and allergic diseases. Arerugi. 62:155–162. 2013.In
Japanese. PubMed/NCBI
|
|
8
|
Ma B, Herzog EL, Lee CG, Peng X, Lee CM,
Chen X, Rockwell S, Koo JS, Kluger H, Herbst RS, et al: Role of
chitinase 3-like-1 and Semaphorin 7a in pulmonary melanoma
metastasis. Cancer Res. 75:487–496. 2015. View Article : Google Scholar :
|
|
9
|
Allegra M, Zaragkoulias A, Vorgia E,
Ioannou M, Litos G, Beug H and Mavrothalassitis G: Semaphorin-7a
reverses the ERF-induced inhibition of EMT in Ras-dependent mouse
mammary epithelial cells. Mol Biol Cell. 23:3873–3881. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Black SA, Nelson AC, Gurule NJ, Futscher
BW and Lyons TR: Semaphorin 7a exerts pleiotropic effects to
promote breast tumor progression. Oncogene. 35:5170–5178. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Formolo CA, Williams R, Gordish-Dressman
H, MacDonald TJ, Lee NH and Hathout Y: Secretome signature of
invasive glioblastoma multiforme. J Proteome Res. 10:3149–3159.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jongbloets BC, Ramakers GM and Pasterkamp
RJ: Semaphorin7A and its receptors: Pleiotropic regulators of
immune cell function, bone homeostasis, and neural development.
Semin Cell Dev Biol. 24:129–138. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fong KP, Barry C, Tran AN, Traxler EA,
Wannemacher KM, Tang HY, Speicher KD, Blair IA, Speicher DW,
Grosser T, et al: Deciphering the human platelet sheddome. Blood.
117:e15–e26. 2011. View Article : Google Scholar :
|
|
14
|
Holmes S, Downs AM, Fosberry A, Hayes PD,
Michalovich D, Murdoch P, Moores K, Fox J, Deen K, Pettman G, et
al: Sema7A is a potent monocyte stimulator. Scand J Immunol.
56:270–275. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Garcia-Areas R, Libreros S, Amat S,
Keating P, Carrio R, Robinson P, Blieden C and Iragavarapu-Charyulu
V: Semaphorin7A promotes tumor growth and exerts a pro-angiogenic
effect in macrophages of mammary tumor-bearing mice. Front Physiol.
5:172014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu H, Juo ZS, Shim AH, Focia PJ, Chen X,
Garcia KC and He X: Structural basis of semaphorin-plexin
recognition and viral mimicry from Sema7A and A39R complexes with
PlexinC1. Cell. 142:749–761. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kang HR, Lee CG, Homer RJ and Elias JA:
Semaphorin 7A plays a critical role in TGF-beta1-induced pulmonary
fibrosis. J Exp Med. 204:1083–1093. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Suzuki K, Okuno T, Yamamoto M, Pasterkamp
RJ, Takegahara N, Takamatsu H, Kitao T, Takagi J, Rennert PD,
Kolodkin AL, et al: Semaphorin 7A initiates T-cell-mediated
inflammatory responses through alpha1beta1 integrin. Nature.
446:680–684. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Markel P, Shu P, Ebeling C, Carlson GA,
Nagle DL, Smutko JS and Moore KJ: Theoretical and empirical issues
for marker-assisted breeding of congenic mouse strains. Nat Genet.
17:280–284. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wakeland E, Morel L, Achey K, Yui M and
Longmate J: Speed congenics: A classic technique in the fast lane
(relatively speaking). Immunol Today. 18:472–477. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fellmann C, Hoffmann T, Sridhar V,
Hopfgartner B, Muhar M, Roth M, Lai DY, Barbosa IA, Kwon JS, Guan
Y, et al: An optimized microRNA backbone for effective single-copy
RNAi. Cell Rep. 5:1704–1713. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wojcikiewicz EP, Zhang X, Chen A and Moy
VT: Contributions of molecular binding events and cellular
compliance to the modulation of leukocyte adhesion. J Cell Sci.
116:2531–2539. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hoh JH and Schoenenberger CA: Surface
morphology and mechanical properties of MDCK monolayers by atomic
force microscopy. J Cell Sci. 107:1105–1114. 1994.PubMed/NCBI
|
|
24
|
Aslakson CJ and Miller FR: Selective
events in the metastatic process defined by analysis of the
sequential dissemination of subpopulations of a mouse mammary
tumor. Cancer Res. 52:1399–1405. 1992.PubMed/NCBI
|
|
25
|
Tao K, Fang M, Alroy J and Sahagian GG:
Imagable 4T1 model for the study of late stage breast cancer. BMC
Cancer. 8:2282008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ridley AJ: RhoA, RhoB and RhoC have
different roles in cancer cell migration. J Microsc. 251:242–249.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Brown GT and Murray GI: Current
mechanistic insights into the roles of matrix metalloproteinases in
tumour invasion and metastasis. J Pathol. 237:273–281. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xiang X, Zhuang X, Ju S, Zhang S, Jiang H,
Mu J, Zhang L, Miller D, Grizzle W and Zhang HG: miR-155 promotes
macroscopic tumor formation yet inhibits tumor dissemination from
mammary fat pads to the lung by preventing EMT. Oncogene.
30:3440–3453. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pickup M, Novitskiy S and Moses HL: The
roles of TGFβ in the tumour microenvironment. Nat Rev Cancer.
13:788–799. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Padua D and Massagué J: Roles of TGFbeta
in metastasis. Cell Res. 19:89–102. 2009. View Article : Google Scholar
|
|
31
|
Sánchez-Tilló E, Siles L, de Barrios O,
Cuatrecasas M, Vaquero EC, Castells A and Postigo A: Expanding
roles of ZEB factors in tumorigenesis and tumor progression. Am J
Cancer Res. 1:897–912. 2011.PubMed/NCBI
|
|
32
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Serrano-Gomez SJ, Maziveyi M and Alahari
SK: Regulation of epithelial-mesenchymal transition through
epigenetic and post-translational modifications. Mol Cancer.
15:182016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lekka M: Discrimination between normal and
cancerous cells using AFM. Bionanoscience. 6:65–80. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lekka M, Laidler P, Gil D, Lekki J,
Stachura Z and Hrynkiewicz AZ: Elasticity of normal and cancerous
human bladder cells studied by scanning force microscopy. Eur
Biophys J. 28:312–316. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cross SE, Jin YS, Rao J and Gimzewski JK:
Nanomechanical analysis of cells from cancer patients. Nat
Nanotechnol. 2:780–783. 2007. View Article : Google Scholar
|
|
37
|
De Minicis S, Rychlicki C, Agostinelli L,
Saccomanno S, Trozzi L, Candelaresi C, Bataller R, Millán C,
Brenner DA, Vivarelli M, et al: Semaphorin 7A contributes to
TGF-β-mediated liver fibrogenesis. Am J Pathol. 183:820–830. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Saito T, Kasamatsu A, Ogawara K, Miyamoto
I, Saito K, Iyoda M, Suzuki T, Endo-Sakamoto Y, Shiiba M, Tanzawa
H, et al: Semaphorin7A promotion of tumoral growth and metastasis
in human oral cancer by regulation of G1 cell cycle and matrix
metalloproteases: Possible contribution to tumoral angiogenesis.
PLoS One. 10:e01379232015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wetzig A, Alaiya A, Al-Alwan M, Pradez CB,
Pulicat MS, Al-Mazrou A, Shinwari Z, Sleiman GM, Ghebeh H,
Al-Humaidan H, et al: Differential marker expression by cultures
rich in mesenchymal stem cells. BMC Cell Biol. 14:542013.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bongiorno T, Kazlow J, Mezencev R,
Griffiths S, Olivares-Navarrete R, McDonald JF, Schwartz Z, Boyan
BD, McDevitt TC and Sulchek T: Mechanical stiffness as an improved
single-cell indicator of osteoblastic human mesenchymal stem cell
differentiation. J Biomech. 47:2197–2204. 2014. View Article : Google Scholar :
|
|
41
|
Guck J, Schinkinger S, Lincoln B, Wottawah
F, Ebert S, Romeyke M, Lenz D, Erickson HM, Ananthakrishnan R,
Mitchell D, et al: Optical deformability as an inherent cell marker
for testing malignant transformation and metastatic competence.
Biophys J. 88:3689–3698. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
van Rijn A, Paulis L, te Riet J, Vasaturo
A, Reinieren-Beeren I, van der Schaaf A, Kuipers AJ, Schulte LP,
Jongbloets BC, Pasterkamp RJ, et al: Semaphorin 7A promotes
chemokine-driven dendritic cell migration. J Immunol. 196:459–468.
2016. View Article : Google Scholar
|