Introduction
Head and neck squamous cell carcinoma (HNSCC) is the
sixth most common type of human cancer worldwide 1). Oral squamous
cell carcinoma (OSCC), which is a subset of HNSCC, is characterized
by a high risk of lymph node metastasis and local invasion.
Although systemic therapeutic strategies, including chemo- and
radiotherapy, have been developed for the treatment of patients
with OSCC, the 5-year overall survival rate remains at ~50%
(2), due to uncontrolled local
adjacent tissue invasion and neck lymph node metastasis (3-5).
Signal transducer and activator of transcription 3
(STAT3) has been identified as a negative prognostic factor in
human cancer, including OSCC (6).
Upon cytokine stimulation, STAT3 translocates into the cell nucleus
where it triggers the transcription of various downstream target
genes, which have important roles in cancer invasion and metastasis
(7,8). Although previous studies have
provided information regarding the oncogenic role of STAT3 in OSCC
(6-8), the detailed regulatory mechanism
underlying the invasion-metastasis cascade remains poorly
understood. Therefore, determining the underlying molecular network
of advanced OSCC is required, in order to identify more efficient
molecular targets with therapeutic potential.
Enhancer of zeste homolog 2 (EZH2) is a component of
polycomb repressive complex 2 (PRC2), which is known to catalyze
trimethylation of lysine 27 in histone 3 (H3K27me3) and
consequently induce transcriptional repression of target genes
(9,10). A previous study reported that EZH2
may mediate the oncogenic role of STAT3 in cancer invasion
(11). Furthermore, phosphorylated
(p)-EZH2 (Ser21) is able to enhance STAT3 activity through
epigenetic modification (12,13).
Members of the microRNA (miR)-200 family, which includes miR-200a,
miR-200b, miR-200c, miR-141 and miR-429, are frequently silenced in
advanced carcinoma (14-16) and serve tumor-suppressive roles in
cell behaviors, such as invasion and metastasis (14,17-20).
Emerging evidence has suggested that miR-200 family members may
regulate epithelial-to-mesenchymal transition (EMT) by targeting
the E-cadherin repressors zinc finger E-box binding homeobox (ZEB)1
and ZEB2 (21,22). For example, overexpression of
miR-429 has been reported to reduce tumor invasion and metastasis
in ovarian cancer via the initiation of mesenchymal-epithelial
transition (MET) (20). Notably, a
previous study demonstrated that miR-200a, miR-200b and miR-429
(miR-200a/b/429) were globally silenced by EZH2 activation through
epigenetic modification in gastric cancer (23).
Based on these previous findings, the present study
took integrated approaches to determine the crosstalk between STAT3
and the EZH2/miR-200a/b/429 axis in OSCC. The results indicated
that targeting STAT3 could significantly inhibit EMT-mediated
invasion in vitro, via the EZH2/miR-200a/b/429 axis.
Furthermore, EZH2 suppression markedly reduced the oncogenic role
of STAT3 in tumor invasion. In addition, animal models suggested
that depletion of STAT3 and EZH2, or elevated miR-200a/b/429, could
globally suppress OSCC invasion and metastasis in vivo.
Materials and methods
Cell culture
The OSCC cell lines SCC25 and SCC15 were purchased
from American Type Culture Collection (ATCC, Manassas, VA, USA).
Cells were cultured in Dulbecco's modified Eagle's medium
(DMEM)/Ham's F-12 supplemented with 10% fetal bovine serum (FBS)
(both from Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
and penicillin (100 U/ml)/streptomycin (100 μg/ml) (HyClone;
GE Healthcare, Logan, UT, USA). Cells were maintained at 37°C in an
atmosphere containing 5% CO2, and were regularly checked
for mycoplasma contamination.
Cell groups
Cells were divided into the following groups: i)
Empty vector (EV) group, cells were infected with lenti-viruses
containing EV (multiplicity of infection, 10) at 37°C for 24 h; ii)
EV + Stattic group, cells were infected with lentiviruses
containing EV (multiplicity of infection, 10) at 37°C for 24 h and
were then treated with Stattic [half maximal inhibitory
concentration (IC50) for each cell line] at 37°C for 24
h; iii) STAT3 group, cells were infected with lentiviruses
containing STAT3 vector (multiplicity of infection, 10) at 37°C for
24 h; iv) negative control (NC) small interfering (si)RNA (si-NC)
group, cells were transfected with si-NC (20 nM) at 37°C for 24 h;
v) si-EZH2 group, cells were transfected with si-EZH2 (20 nM) at
37°C for 24 h; vi) si-NC + EV group, cells were transfected with
si-NC (20 nM) at 37°C for 24 h and were then infected with
lentiviruses containing EV (multiplicity of infection, 10) at 37°C
for 24 h; vii) si-NC + STAT3 group, cells were transfected with
si-NC (20 nM) at 37°C for 24 h and were then infected with
lentiviruses containing STAT3 vector (multiplicity of infection,
10) at 37°C for 24 h; viii) si-EZH2 + STAT3 group, cells were
transfected with si-EZH2 (20nM) at 37°C for 24 h and were then
infected with lentiviruses containing STAT3 vector (multiplicity of
infection, 10) at 37°C for 24 h.
Antibodies and reagents
Dimethyl sulfoxide (DMSO) was purchased from
Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). The STAT3
phosphorylation inhibitor Stattic and the EZH2 inhibitor
3-deazaneplanocin A (DZNeP) were purchased from Selleck Chemicals
(Houston, TX, USA). pLVX-IRES-puro vectors were purchased from
Shanghai GeneChem Co., Ltd. (Shanghai, China) to establish stable
SCC25 or SCC15 clones overexpressing STAT3. The primary antibodies
used in the present study are listed in Table I.
 | Table IPrimary antibodies used in the
present study. |
Table I
Primary antibodies used in the
present study.
| Primary
antibody | Catalog no. | Vendor | Application |
|---|
| STAT3 | ab119352 | Abcam | WB/IHC |
| p-STAT3
(Tyr705) | ab76315 | Abcam | WB/IHC |
| Twist | ab50581 | Abcam | WB |
| EZH2 | 5246 | Cell Signaling
Technology, Inc. | WB/IHC |
| H3K27me3 | 9733 | Cell Signaling
Technology, Inc. | WB/IHC |
| Histone 3 | 4499 | Cell Signaling
Technology, Inc. | WB |
| E-cadherin | 610181 | BD Biosciences | WB/IF/IHC |
| N-cadherin | 610920 | BD Biosciences | WB/IF/IHC |
| Vimentin | 33541 | AbSci | WB/IF/IHC |
| MMP2 | sc-13595 | Santa Cruz
Biotechnology, Inc. | WB |
| MMP9 | sc-21733 | Santa Cruz
Biotechnology, Inc. | WB |
| GAPDH | TA-08 | OriGene
Technologies, Inc. | WB |
MTT assay
SCC25 and SCC15 cell lines (5,000 cells/well) were
seeded into 96-well plates and incubated at 37°C for 24 h to allow
for stabilization, after which, the cells were exposed to Stattic
(0.1, 0.5, 1, 2, 4, 6, 8 or 10 μmol/l) at 37°C for 24 h.
Cell viability was measured using an MTT assay (5 mg/ml;
Sigma-Aldrich; Merck KGaA). The MTT crystals were dissolved in
DMSO, and absorbance was assessed at 490 nm using a microplate
reader (Model 680; Bio-Rad Laboratories, Inc., Hercules, CA, USA).
IC50 was calculated using SPSS software (version 16.0;
SPSS, Inc., Chicago, IL, USA).
Transwell assay
After transfection or drug administration, the
Transwell assays were performed in 24-well culture plates;
Matrigel-coated inserts were used to analyze invasion and uncoated
inserts were used to analyze migration. Briefly, SCC25 or SCC15
cells (50,000 cells/well) were plated into Transwell inserts
(Corning Incorporated, Corning, NY, USA) for invasion or migration
assay, which had been coated with or without Matrigel (BD
Biosciences, Franklin Lakes, NJ, USA), in FBS-free medium. The
lower chamber was filled with complete growth medium containing 20%
FBS. The cells were incubated at 37°C for 16 h, after which the
cells that had passed through the membrane were fixed with 4%
paraformaldehyde and were stained with 0.1% crystal violet (both
from Beijing Solarbio Science & Technology Co., Ltd., Beijing,
China). Cells were counted in four separate fields in three
independent experiments, using an inverted microscope (DMI6000B;
Leica Microsystems, Inc., Buffalo Grove, IL, USA).
Wound-healing assay
A wound-healing assay was conducted to confirm the
results of the Transwell assay. Briefly, 2×105 SCC25 and
SCC15 cells/well were plated in 6-well plates. Once the cells had
reached 80% confluence, scratches were generated in the cell layer
using a 10 μl pipette tip, thus creating a wound field ~400
mm wide, based on the scale plate in the microscope.
Photomicrographs were captured of live cells at 0 and 48 h, using
an inverted microscope (DMI6000B; Leica Microsystems, Inc.), and
the distance migrated was determined within an appropriate
time.
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated using TRIzol®
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. Equal amounts of RNA were reverse
transcribed into cDNA using the PrimeScript 1st Strand cDNA
Synthesis kit (Nanjing KeyGen BioTech Co., Ltd., Nanjing, China)
according to the manufacturer's protocol. qPCR analysis was
conducted using the SYBR Green PCR Master Mix (Nanjing KeyGen
BioTech Co., Ltd.) according to the manufacturer's protocol
(thermocycling: 95°C initial denaturation for 2 min; followed by 40
cycles of denaturation at 95°C for 15 sec, and annealing and
extension at 60°C for 60 sec), was employed to detect the relative
expression levels of miR-200b, miR-200a and miR-429. U6 was used as
a loading control, and the 2−ΔΔCq method (24) was used to calculate relative gene
abundance. Primers used in the present study are shown in Table II.
| Table IIList of primers and oligonucleotides
used in the present study. |
Table II
List of primers and oligonucleotides
used in the present study.
| Primer | Sequence |
|---|
| miR-200a | |
| RT |
5′-CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGACATCGTT-3′ |
| PCR | F:
5′-ACACTCCAGCTGGGTAACACTGTCTGGTAA-3′ |
| R:
5′-TGGTGTCGTGGAGTCG-3′ |
| miR-200b | |
| RT |
5′-CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGTCATCATT-3′ |
| PCR | F:
5′-ACACTCCAGCTGGGTAATACTGCCTGGTAA-3′ |
| R:
5′-TGGTGTCGTGGAGTCG-3′ |
| miR-429 | |
| RT |
5′-CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGACGGTTTT-3′ |
| PCR | F:
5′-ACACTCCAGCTGGGTAATACTGTCTGGTAA-3′ |
| R:
5′-TGGTGTCGTGGAGTCG-3′ |
| U6 | |
| PCR | F:
5′-CTCGCTTCGGCAGCACA-3′ |
| R:
5′-AACGCTTCACGAATTTGCGT-3′ |
Western blotting
Western blot analysis was employed to assess protein
expression. Cell lysates were prepared in radioimmunoprecipitation
assay buffer supplemented with protease and phosphatase inhibitors
(both from Beijing Solarbio Science & Technology Co., Ltd.).
Protein concentrations were determined using a bicinchoninic acid
(BCA) protein assay kit (Micro BCA Protein Assay kit; Thermo Fisher
Scientific, Inc.). The protein samples (20-30 μg) were
separated by 10% SDS-PAGE and were transferred onto polyvinylidene
difluoride membranes (Merck KGaA). The membranes were blocked with
5% non-fat milk in Tris-buffered saline containing 0.1% Tween-20 at
room temperature for 2 h. Subsequently, the membranes were
incubated with the following antibodies (1:1,000 dilutions)
overnight at 4°C: STAT3, p-STAT3 (Tyr705), Twist (Abcam, Cambridge,
UK), EZH2, H3K27me3 (Cell Signaling Technology, Inc., Danvers, MA,
USA), E-cadherin, N-cadherin (BD Biosciences), Vimentin (AbSci,
Vancouver, WA, USA), matrix metallo-proteinase (MMP)2 and MMP9
(Santa Cruz Biotechnology, Inc., Dallas, TX, USA). GAPDH (OriGene
Technologies, Inc., Beijing, China) and Histone 3 (Cell Signaling
Technology, Inc.) were used as internal controls (Table I). Horseradish
peroxidase-conjugated anti-rabbit (sc-2004) or anti-mouse (sc-2005)
immunoglobulin G antibodies (Santa Cruz Biotechnology, Inc.) were
used as secondary antibodies (1:5,000 dilutions), and blots were
incubated with them at room temperature for 1 h. ImageJ software
(Version 5.2.5; National Institutes of Health, Bethesda, MD, USA)
was used to semi-quantify the relative expression levels of the
target proteins, which were normalized to the respective internal
controls.
Immunofluorescence staining
For immunofluorescence staining, OSCC cells were
grown on 18-mm coverslips for 24 h following treatment.
Immunofluorescence staining was conducted using primary antibodies
against E-cadherin, N-cadherin (1:100 dilutions; BD Biosciences)
and Vimentin (1:100 dilution; AbSci), overnight at 4°C. The cells
were then washed with PBS and incubated with AlexaFluor 488
(anti-rabbit: #4412; anti-mouse: #4408) or AlexaFluor 594
(anti-rabbit: #8889; anti-mouse: #8890) secondary antibodies (1:500
dilutions; Cell Signaling Technology, Inc.) at room temperature for
1 h. For stress fiber formation assessment, cells were stained with
DyLight™ 594-phalloidin (1:500 dilution; #12877; Cell Signaling
Technology, Inc.) at room temperature for 1 h. The nuclei were
stained using DAPI (Thermo Fisher Scientific, Inc.), and each slide
was visualized using an FV-1000 laser scanning confocal microscope
(Olympus Corporation, Tokyo, Japan).
Lentivirus packaging and
transduction
Total RNA from both HNSCC cell lines (SCC15 and
SCC25) was isolated using TRIzol® (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol.
RNA was reverse transcribed into cDNA using the PrimeScript 1st
Strand cDNA Synthesis kit (Nanjing KeyGen BioTech Co., Ltd.)
according to the manufacturer's protocol. Then, STAT3 coding
sequence was amplified by PCR, using the SYBR-Green PCR Master Mix
(Nanjing KeyGen BioTech Co., Ltd.) according to the manufacturer's
instructions, and cloned into the pLVX-IRES-puro vector (Shanghai
GeneChem Co., Ltd.). All constructs were verified by sequencing
(Sanger chain termination method), which was conducted by AuGCT
(Beijing, China). Lentivirus was prepared in 293T cells (ATCC)
using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. For transduction,
cells (30-40% confluence) were infected with lentiviruses
(multiplicity of infection, 10) at 37°C for 24 h. SCC25 or SCC15
clones stably overexpressing STAT3 were labeled as STAT3, and an EV
was used as a negative control.
Transient transfection
SCC25 and SCC15 cell lines were transfected with NC
siRNA or siRNA against EZH2 (20 nM; Guangzhou Ribobio Co., Ltd.,
Guangzhou, China), which were labeled as si-NC
(5′-UUCUCCGAACGUGUCACGU-3′) or si-EZH2 (#1,
5′-GCUGGAAUCAAAGGAUACA-3′; #2, 5′-GTGCCCTTGTGTGATAGCACAA-3′; #3,
5′-GGCACT TTCATTGAAGAACTAA-3′), respectively, using Lipo-fectamine
2000 reagent, according to the manufacturer's protocol. Briefly,
cells (~40% confluence) were transfected with 20 nM siRNAs at 37°C
for 24 h. During transfection, FBS-free Opti-MEM medium was
employed, and after 6 h, the medium was replaced with DMEM-F12 or
DMEM.
In vivo orthotopic tumor model
To establish orthotopic models, 5×106
SCC15 cells, which were transduced with luciferase lentivirus
(Promega Corporation, Madison, WI, USA), were injected into the
base of the oral cavity of 4-week-old, male BALB/c-nu mice (n=40;
weight, ~15 g) (4,25). The animals were obtained from the
Animal Center at the Cancer Institute, Chinese Academy of Medical
Science (Beijing, China). Mice were maintained under the following
conditions: Temperature, 18-22°C; humidity, 50-60%; 12-h light/dark
cycle; food and water access, ad libitum. To verify tumor
establishment, fluorescence images were captured 7 days after
injection using an IVIS Lumina Imaging system (PerkinElmer, Inc.,
Waltham, MA, USA). Mice were randomly assigned to five groups
(n=8/group): DMSO (20 μl dissolved in 100 μl PBS),
Stattic (3.75 mg/kg) (26), DZNeP
(1 mg/kg) (27), NC mimics
(5′-UUUGUACUACACAAAAGUACUG-3′, 10 nM/mice) or miR-429 mimics
(5′-UAAUACUGUCUGGUAAAACCGU-3′, 10 nM/mice; Guangzhou Ribobio Co.,
Ltd.); treatments were administered by intraperitoneal injection
every 3 days for 21 days. Bioluminescence imaging was conducted to
measure tumor volume each week, and body weight was measured daily.
Finally, the animals were sacrificed by cervical dislocation and
the orthotopic tumors were collected for further examination. Tumor
volume was finally measured using a caliper (Volume = long diameter
x short diameter2/2). Tumor specimens were used for IHC
and RT-qPCR. The present study was approved by the Institutional
Animal Care and Use Committee of Tianjin Medical University Cancer
Institute and Hospital (Tianjin, China).
Immunohistochemistry (IHC) and
hematoxylin and eosin (H&E) staining
For IHC, tumor samples were fixed with 40% formalin
in methanol for 2 days at room temperature and were embedded in
paraffin. Subsequently, formalin-fixed, paraffin-embedded
SCC15-derived OSCC orthotopic tumor samples were deparaffinized,
rehydrated and incubated with primary antibodies (1:100 dilutions)
overnight at 4°C. Subsequently, tissue sections were incubated with
a biotin-labeled secondary antibody (PV-9000; OriGene Technologies,
Inc.) for 40 min at 37°C. Cells were visualized using
ABC-peroxidase and diaminobenzidine (Vector Laboratories, Inc.,
Burlingame, CA, USA) at room temperature for 30 sec, counterstained
with hematoxylin and visualized using light microscopy. The primary
antibodies used in this investigation are listed in Table I. In addition, H&E staining was
performed as previously described (28).
Database analysis
The online software cBioPortal (www.cbio-portal.org) and Gene Expression Profiling
Interactive Analysis (GEPIA; gepia.cancer-pku.cn) were used to
analyze TCGA (The Cancer Genome Atlas) database.
Statistical analysis
SPSS 16.0 (SPSS, Inc.) was used for all statistical
analyses. Statistical comparisons between two groups were made
using Student's t-test. Differences between more than two groups
were determined by two-way analysis of variance followed by
Dunnett's test. All data are presented as the means ± standard
deviation, and represent the average of at least three experiments
performed in triplicate. P<0.05 was considered to indicate a
statistically significant difference.
Results
Inhibition of STAT3 suppresses OSCC tumor
invasion and migration in vitro
According to previous studies, Stattic, a
STAT3-selective phosphorylation inhibitor, may inhibit the
expression levels of p-STAT3 (Tyr705), without any effect on total
STAT3 (26,29). As shown in Fig. 1A, western blot analysis
demonstrated that treatment with Stattic (24 h IC50 for
SCC15: 1.979 μM; IC50 for SCC25: 1.388 μM;
Fig. 1B) markedly suppressed MMP2
and MMP9 expression in SCC25 and SCC15 cell lines, which may be
induced by inhibition of p-STAT3 (Tyr705). Furthermore, STAT3
silencing significantly impaired the invasive and migratory
capabilities of OSCC cells, according to the results of a Transwell
assay and scratch test analysis in vitro (Fig. 1C and D). Meanwhile, stable OSCC
clones overexpressing STAT3 were generated, in order to verify the
function of STAT3 in OSCC cells. Stable SCC25 or SCC15 clones
overexpressing STAT3 were labeled STAT3, and cells infected with EV
were used as a negative control. Compared with in the EV group,
cells overexpressing STAT3 possessed markedly increased MMP2 and
MMP9 protein expression, leading to a significant elevation in
tumor cell migration and invasion (Fig. 1A, C and D).
 | Figure 1STAT3 contributes to invasion and
migration of OSCC cells. (A) Western blot detection of STAT3,
p-STAT3 (Tyr705), MMP2 and MMP9 expression. GAPDH was used as a
loading control. (B) MTT analysis was used to determine the
IC50 values for both OSCC cell lines. (C) Targeting
STAT3 significantly inhibited SCC25 and SCC15 cell invasion and
migration ability, as determined by Transwell assay (magnification,
×40). (D) Cell migration ability was measured by a wound-healing
assay (magnification, ×40). ***P<0.001 vs. EV group;
###P<0.001 EV + Stattic group vs. STAT3 group. EV,
empty vector; IC50, half maximal inhibitory
concentration; MMP, matrix metalloproteinase; OSCC, oral squamous
cell carcinoma; p-STAT3, phosphorylated-STAT3; STAT3, signal
transducer and activator of transcription 3. |
Regulatory role of the
EZH2/miR-200/a/b/429 axis in EMT of OSCC
Previous studies have indicated that downregulation
of EZH2 may increase miR-200b/a/429 expression in human cancers
(23,30). In order to explore the regulatory
role in OSCC, two EZH2 siRNAs (si#1 and si#2) were used to inhibit
the expression of EZH2. Subsequently si#2 was selected for further
analysis (Fig. 2A). In
siRNA-transfected OSCC cells, EZH2 expression was markedly
inhibited, as was H3K27me3 (Fig.
2B). p-EZH2 (Ser21) has previously been reported to
significantly enhance STAT3 activity through epigenetic
modification (12,13). In the present study, p-STAT3
expression was suppressed by EZH2 attenuation. Furthermore, qPCR
was used to detect miR-200-a/b/429 expression in both cell lines.
Compared with in the untreated SCC15 and SCC25 cells, EZH2-depleted
cells exhibited significantly increased miR-200-b/a/429 expression
(Fig. 2C).
 | Figure 2EZH2/miR-200/b/a/429 axis regulates
the invasiveness of OSCC cells in vitro. (A) Following
transfection with EZH2 siRNAs (si#1 and si#2) for 3 days, the
expression levels of EZH2 were detected using western blotting. (B)
OSCC cells underwent western blot analysis using antibodies
specific to STAT3, p-STAT3 (Tyr705), EZH2, H3K27me3, H3 and GAPDH.
EZH2 siRNA markedly inhibited EZH2 and H3K27me3 expression. In
addition, p-STAT3 (Tyr705), but not STAT3, was suppressed by EZH2
depletion. (C) EZH2 siRNA induced miR-200b/a/429 expression. (D)
Results of a Transwell assay demonstrated that EZH2 knockdown
markedly attenuated migration (without Matrigel) and invasion (with
Matrigel) of SCC25 and SCC15 cells (scale bar, 100 μm;
magnification, ×40). (E) Scratch assay demonstrated that EZH2
silencing markedly delayed wound healing in SCC25 and SCC15 cells
(magnification, ×40). *P<0.05 and ***P<0.001 vs.
si-NC group. EZH2, enhancer of zeste homolog 3; H3, histone 3;
H3K27me3, trimethylation of lysine 27 in H3; miR-200b/a/429,
microRNA-200b, -200a and -429; NC, negative control; OSCC, oral
squamous cell carcinoma; p-STAT3, phosphorylated-STAT3; siRNA/si,
small interfering RNA; STAT3, signal transducer and activator of
transcription 3. |
The present study also aimed to determine whether
cell motility was governed by the EZH2/miR-200-b/a/429 regulatory
network. The results of Transwell and wound healing assays
indicated that EZH2 silencing markedly inhibited the invasion and
migration of OSCC cell lines (Fig. 2D
and E). To confirm these results, immunofluorescence staining
was conducted to directly visualize the effects of EZH2 abrogation
on E-cadherin expression, localization and cell morphology. As
shown in Fig. 3,
si-EZH2-transfected SCC15 and SCC25 cells possessed an epithelial
phenotype, as indicated by an increase in E-cadherin expression on
the cell membrane and rearrangement of F-actin to a relatively
cortical pattern, which is a hallmark of the epithelial
phenotype.
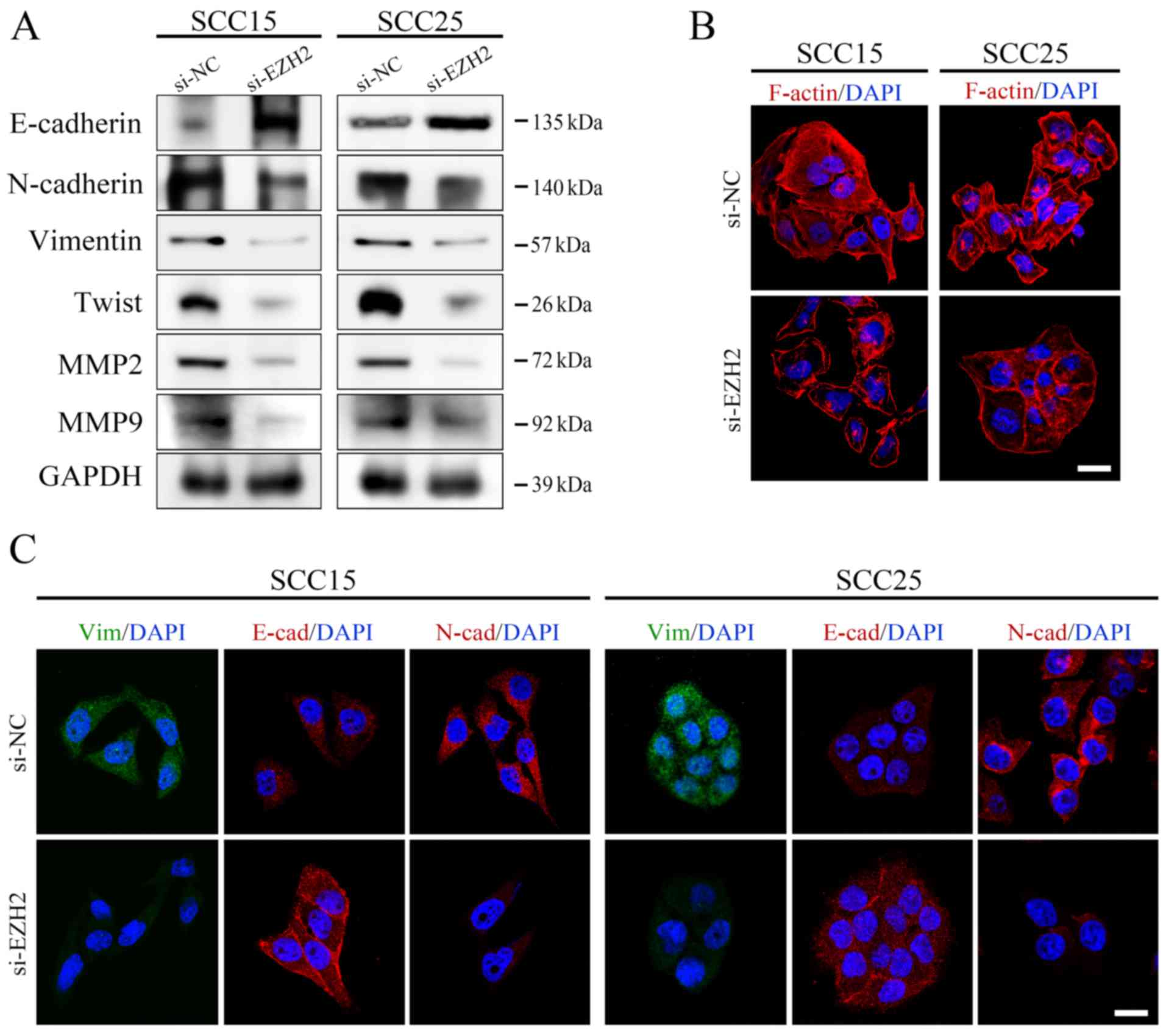 | Figure 3EZH2 silencing affects the EMT
process in OSCC cells. (A) Protein expression levels of
EMT-associated markers were analyzed by western blotting and were
normalized to GAPDH. (B) F-actin staining exhibited a stress-fiber
pattern in NC siRNA-transfected cells, whereas a cortical pattern
was observed in EZH2 siRNA-transfected cells, as assessed by
immunofluorescence (scale bar, 20 μm). (C) E-cadherin,
N-cadherin and Vimentin staining of OSCC cells transfected with
si-EZH2 or si-NC. Cell nuclei were stained with DAPI (scale bar, 20
μm). EMT, epithelial-mesenchymal transition; EZH2, enhancer
of zeste homolog 3; MMP, matrix metalloproteinase; NC, negative
control; OSCC, oral squamous cell carcinoma; siRNA/si, small
interfering RNA. |
Targeting STAT3 signaling regulates EZH2
and miR-200b/a/429, thus affecting EMT in OSCC in vitro
A previous study verified that the
EZH2/miR-200b/a/429 axis is a critical regulatory network
associated with tumor motility. Notably, STAT3 has been reported to
transcriptionally regulate EZH2 in human colorectal cancer
(11). Based on these findings,
the present study aimed to determine whether STAT3 could trigger
OSCC invasion via the EZH2/miR-200b/a/429 axis.
Analysis of TCGA demonstrated that EZH2 expression
was positively correlated with STAT3 expression in human HNSCC
specimens (Fig. 4A), which was in
line with the outcome of the OSCC dataset in TCGA analyzed by
GEPIA. Moreover, western blot analysis demonstrated that treatment
with Stattic markedly suppressed p-STAT3 (Tyr705, Fig. 1A) and EZH2 (Fig. 4B), and inhibited EZH2-induced
H3K27me3 in both OSCC cell lines (Fig.
4B). Notably, STAT3 stable clones exhibited increased EZH2
expression (Fig. 4B).
Subsequently, the present study examined whether targeting STAT3
could regulate the expression levels of miR-200b/a/429. The
expression levels of miR-200b/a/429 were detected prior to and
following treatment with Stattic using qPCR. The results of qPCR
revealed that Stattic-induced STAT3 inhibition markedly enhanced
mir-200b/a/429 expression (Fig.
4C). Conversely, overexpression of STAT3 globally silenced the
tumor suppressor miRNAs, miR-200b/a/429, compared with in the EV
group (Fig. 4C). These data
indicated that STAT3 may inhibit the expression levels of
mir-200b/a/429 in OSCC cells via EZH2-induced epigenetic
silencing.
 | Figure 4EZH2/miR-200/b/a/429 axis mediates
the prometastatic role of STAT3 in an EMT-dependent manner. (A) The
Cancer Genome Atlas data were used to determine the correlation
between STAT3 and EZH2 in head and neck squamous cell carcinoma
(P<0.05). (B) Western blot analysis of EZH2, H3K27me3 and EMT
markers following Stattic treatment or STAT3 transduction. (C)
Stattic upregulated miR-200b/a/429 expression, whereas ectopic
STAT3 overexpression significantly attenuated mir-200b/a/429
expression. (D) Expression of the epithelial marker E-cadherin, and
the mesenchymal markers N-cadherin and Vimentin, were detected by
immunofluorescence staining (scale bar, 20 μm).
***P<0.001 vs. EV group; ###P<0.001 EV
+ Stattic group vs. STAT3 group. EMT, epithelial-mesenchymal
transition; EV, empty vector; EZH2, enhancer of zeste homolog 3;
H3, histone 3; H3K27me3, trimethylation of lysine 27 in H3;
miR-200b/a/429, microRNA-200b, -200a and -429; STAT3, signal
transducer and activator of transcription 3. |
SCC25 and SCC15 cells treated with EV, Stattic or a
STAT3 plasmid were examined by immunofluorescence; Stattic
treatment resulted in less regularly shaped cells compared with in
the control group (Fig. 4D).
Furthermore, STAT3-depleted cells exhibited reduced formation of
robust actin stress fibers, suggesting that features of mesenchymal
cells and mobility features were decreased (Fig. 4D). In addition, in STAT3-depleted
cells E-cadherin expression was increased, whereas N-cadherin and
Vimentin expression were decreased (Fig. 4D). Conversely, STAT3 enhancement
markedly induced the mesenchymal phenotype, which was characterized
by an absence of E-cadherin on the cell membrane and rearrangement
of F-actin from a cortical to stress-fiber pattern, thus indicating
the presence of robust mesenchymal features. Subsequent western
blot analysis verified the immunofluorescence results of EMT
markers (Fig. 4B).
EZH2 silencing counteracts STAT3
activation in OSCC cells in vitro
To verify the underlying mechanism by which STAT3
governs OSCC invasion and migration, the role of EZH2 was
determined in STAT3-mediated cell biology. When OSCC cells
overexpressing STAT3 were transfected with si-EZH2 for 72 h, the
effects of STAT3 on H3K27me3 and miR-200b/a/429 were significantly
attenuated in vitro (Fig. 5A
and B). Furthermore, invasion assays demonstrated that EZH2
knockdown markedly reversed the oncogenic effects of STAT3 on tumor
invasion and migration (Fig. 5C and
D).
 | Figure 5EZH2 silencing counteracts
STAT3-induced invasion by targeting miR-200b/a/429. (A) STAT3 and
EZH2 expression levels were evaluated by western blotting. (B) EZH2
depletion markedly reduced the inhibitory effects of STAT3 on
miR-200b/a/429 expression. (C and D) EZH2 knockdown reduced the
invasion and migration of oral squamous cell carcinoma cells
overexpressing STAT3 (magnification, ×40). *P<0.05
and ***P<0.001 vs. si-NC + EV group;
###P<0.001 si-NC + STAT3 group vs. si-EZH2 + STAT3
group. EV, empty vector; EZH2, enhancer of zeste homolog 3; H3,
histone 3; H3K27me3, tri-methylation of lysine 27 in H3;
miR-200b/a/429, microRNA-200b, -200a and -429; p-STAT3,
phosphorylated-STAT3; siRNA/si, small interfering RNA; STAT3,
signal transducer and activator of transcription 3. |
Western blot analysis was used to determine whether
EZH2/miR-200/b/a/429 mediated the prometastatic effects of STAT3 on
the expression of EMT markers. In both cell lines, EZH2 knockdown
reduced the oncogenic effects of STAT3, leading to increased
E-cadherin and decreased N-cadherin expression (Fig. 6A). To further confirm these
results, immunofluorescence staining was performed to directly
visualize EMT markers and cell morphology. As shown in Fig. 6B, si-EZH2-transfected OSCCs
possessed epithelial cell features, as characterized by a typical
cobblestone structure and membrane-localized E-cadherin.
Conversely, cells transfected with si-NC possessed a mesenchymal
phenotype following ectopic overexpression of STAT3. These results
suggested that the EZH2/miR-200b/a/429 axis may contribute to the
STAT3-directed OSCC invasion and migration.
Targeting STAT3 and EZH2 reduces OSCC
tumor invasion in orthotopic mouse models
The results of the present study suggested that the
EZH2/miR-200/b/a/429 axis may trigger the oncogenic role of STAT3
in OSCC. To further validate the therapeutic potential of targeting
STAT3 and EZH2, a proof-of-concept experiment was employed using an
orthotopic OSCC tumor model, derived from the SCC15 cell line
(Fig. 7). Bioluminescence assay
results (Fig. 7A) and tumor
volumes (Fig. 7D) indicated that
Stattic-treated (3.75 mg/kg) and DZNeP-treated (1 mg/kg) mice
exhibited significantly reduced growth stasis compared with the
DMSO group (Table III;
P<0.05). None of the mice developed numerous tumors. No
significant alteration in body weight (Fig. 7B) was observed in the mice treated
with the inhibitors (initial weight: DMSO, 17.6 g; Stattic, 17.3 g;
DZNeP, 18.3 g; weight at sacrifice: DMSO, 19.5g; Stattic, 19.4 g;
DZNeP, 18.6 g). To evaluate the therapeutic effects on tumor
invasion, the tumors were resected from each group. As shown in
Fig. 7C, Stattic-treated and
DZNeP-treated mice exhibited reduced local invasion compared with
in the control group.
 | Figure 7Targeting STAT3 and EZH2 inhibits
tumorigenesis and invasiveness in an orthotopic OSCC model. (A)
Representative bioluminescence images of mice implanted with
orthotopic tumors, which were intraperitoneally treated with 3.75
mg/kg Stattic, 1 mg/kg DZNeP or DMSO every 3 days. (B) Based on
body weight, no detectable toxicity was observed at the tested
dose. (C) Representative photographs of the tumor resection
procedure. Stattic and DZNeP treatment inhibited local invasion of
orthotopic OSCC tumors. (D) Tumor diameter and volume were
measured. (E) Quantitative polymerase chain reaction was used to
detect miR-200b/a/429 expression in OSCC tumor sections. (F) Tumor
samples from three distinct groups underwent immunohistochemistry
for STAT3, p-STAT3 (Tyr705), EZH2, H3K27me3, MMP2, MMP6,
E-cadherin, N-cadherin and Vimentin expression (scale bar, 100
μm; magnification, ×200). ***P<0.001 vs. DMSO
group. DMSO, dimethyl sulfoxide; DZNeP, 3-deazaneplanocin A; EZH2,
enhancer of zeste homolog 3; H3K27me3, trimethylation of lysine 27
in histone 3; H&E, hematoxylin and eosin; miR, microRNA; MMP,
matrix metalloproteinase; p-STAT3, phosphorylated-STAT3; STAT3,
signal transducer and activator of transcription 3. |
| Table IIIBioluminescence data obtained from
the animal study (107 p/sec/cm2/sr). |
Table III
Bioluminescence data obtained from
the animal study (107 p/sec/cm2/sr).
| Group | Mouse
|
|---|
| #1 | #2 | #3 | #4 | #5 | #6 | #7 | #8 |
|---|
| DMSO | | | | | | | | |
| D0 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| D7 | 0.980 | 1.040 | 0.450 | 1.010 | 1.050 | 0.660 | 1.122 | 0.990 |
| D14 | 2.110 | 2.770 | 3.310 | 2.090 | 1.980 | 3.150 | 2.220 | 3.010 |
| D21 | 5.310 | 5.010 | 6.500 | 5.420 | 5.120 | 5.990 | 4.500 | 4.620 |
| D28 | 9.870 | 8.340 | 7.990 | 8.550 | 7.040 | 9.010 | 9.550 | 8.010 |
| D35 | 14.110 | 15.550 | 14.770 | 14.010 | 15.310 | 14.980 | 14.410 | 15.010 |
| D42 | 18.110 | 19.100 | 22.100 | 19.020 | 18.960 | 20.810 | 18.410 | 18.760 |
| Stattic | | | | | | | | |
| D0 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| D7 | 0.410 | 0.350 | 0.220 | 0.390 | 0.370 | 0.200 | 0.430 | 0.340 |
| D14 | 0.660 | 0.870 | 0.410 | 0.600 | 0.940 | 0.360 | 0.700 | 0.880 |
| D21 | 1.710 | 2.240 | 0.880 | 1.820 | 2.020 | 1.980 | 1.410 | 2.010 |
| D28 | 3.110 | 2.330 | 3.010 | 3.010 | 2.330 | 3.320 | 2.880 | 2.130 |
| D35 | 5.120 | 6.110 | 5.110 | 5.010 | 6.410 | 5.020 | 5.310 | 6.010 |
| D42 | 9.010 | 11.210 | 9.010 | 10.100 | 10.310 | 8.510 | 8.670 | 10.010 |
| DZNeP | | | | | | | | |
| D0 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| D7 | 0.340 | 0.120 | 0.410 | 0.300 | 0.160 | 0.330 | 0.440 | 0.090 |
| D14 | 0.440 | 0.560 | 0.320 | 0.410 | 0.590 | 0.300 | 0.450 | 0.580 |
| D21 | 1.200 | 1.110 | 0.990 | 1.310 | 0.990 | 1.010 | 1.220 | 1.040 |
| D28 | 1.400 | 1.890 | 1.550 | 3.010 | 1.120 | 2.780 | 1.250 | 1.110 |
| D35 | 4.110 | 3.030 | 7.010 | 4.010 | 3.710 | 7.310 | 3.910 | 5.120 |
| D42 | 9.010 | 5.020 | 6.990 | 7.040 | 5.010 | 9.210 | 10.910 | 5.210 |
| Negative control
mimic | | | | | | | | |
| D0 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| D7 | 0.980 | 1.040 | 0.980 | 0.870 | 1.120 | 1.010 | 0.990 | 1.120 |
| D14 | 2.110 | 2.770 | 3.450 | 2.230 | 2.540 | 2.780 | 2.450 | 3.010 |
| D21 | 5.310 | 5.010 | 4.000 | 6.010 | 4.980 | 3.780 | 5.870 | 4.120 |
| D28 | 10.120 | 8.000 | 7.000 | 10.120 | 8.120 | 7.000 | 10.120 | 8.240 |
| D35 | 14.110 | 15.120 | 16.000 | 13.990 | 15.610 | 16.870 | 14.210 | 14.980 |
| D42 | 22.000 | 19.100 | 21.000 | 23.120 | 18.120 | 22.340 | 21.980 | 19.870 |
| microRNA-429 | | | | | | | | |
| D0 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| D7 | 0.410 | 0.350 | 0.410 | 0.430 | 0.400 | 0.360 | 0.340 | 0.380 |
| D14 | 0.660 | 0.870 | 0.660 | 0.570 | 0.890 | 0.760 | 0.550 | 0.910 |
| D21 | 2.110 | 1.880 | 1.980 | 2.010 | 1.780 | 1.990 | 1.750 | 2.070 |
| D28 | 4.000 | 2.010 | 3.110 | 4.010 | 2.450 | 3.410 | 3.780 | 2.310 |
| D35 | 5.000 | 7.010 | 7.000 | 4.560 | 7.010 | 6.310 | 4.780 | 6.010 |
| D42 | 8.000 | 8.000 | 11.000 | 7.670 | 8.120 | 12.010 | 7.890 | 10.110 |
Notably, IHC demonstrated that p-STAT3 (Tyr705),
EZH2 and H3K27me3 expression in Stattic- and DZNeP-treated groups
were markedly suppressed (Fig.
7F). In addition, the expression levels of mir-200b/a/429 were
elevated in both groups (Fig. 7E).
Notably, compared with in the control tumors, Stattic and DZNeP
treatment was sufficient to attenuate the cell invasion proteins
MMP2 and 9, and the EMT-associated biomarkers N-cadherin and
Vimentin in vivo, alongside an increase in E-cadherin
expression (Fig. 7F).
Systematic delivery of miR-200b/a/429
markedly suppresses N-cadherin and induces E-cadherin expression in
vivo
The present study indicated that the
EZH2/miR-200b/a/429 axis may mediate the oncogenic role of STAT3 in
tumor invasion via EMT interaction. To further confirm these
results, another in vivo experiment was conducted to verify
the inhibitory role of miR-200b/a/429 in tumor invasion. Since
miR-429 is the most sensitive miRNA, according to upstream
stimulation in vitro in the present study, as assessed by
RT-qPCR (Figs. 2C, 4C and 5B), miR-429 was selected for further
analysis.
A total of 7 days after tumor implantation,
miR-control (miR-Ctrl) and miR-429 were intraperitoneally injected
every 3 days (Fig. 8). Body weight
was assessed daily, and tumor volume was measured each week using
bioluminescence imaging. As shown in Fig. 8A and D, delivery of miR-429
markedly reduced tumor volume compared with in the miR-Ctrl group
(Table III). None of the mice
developed numerous tumors. No significant alteration in body weight
was observed during treatment (Fig.
8B) (initial weight: miR-Ctrl, 18.1 g; miR-429, 17.9 g; weight
at sacrifice: miR-Ctrl, 19.32 g; miR-429, 18.7 g). Furthermore,
miR-429 overexpression markedly reduced tumor invasion (Fig. 8C).
 | Figure 8miR-429 inhibits tumor progression in
the orthotopic mouse model of OSCC. (A) Representative
bioluminescence images of mice implanted with orthotopic tumors and
treated intraperitoneally with miR-Ctrl (10 nM/mice) or miR-429 (10
nM/mice) every 3 days. (B) Quantification of body weight in control
and miR-429-treated mice. (C) miR-429 inhibits OSCC local invasion
in an orthotopic mouse model. (D) Tumor diameter and volume were
measured. (E) Quantitative polymerase chain reaction was used to
detect the expression levels of mir-429 in OSCC tumor sections. (F)
Tumor samples from control and miR-429-treated mice underwent
immunohistochemistry for MMP2, MMP9, E-cadherin, N-cadherin and
Vimentin (scale bar, 100 μm magnification, ×200).
***P<0.001 vs. miR-Ctrl group. Ctrl, control;
H&E, hematoxylin and eosin; miR, microRNA; MMP, matrix
metalloproteinase; NC, negative control. |
RT-qPCR was used to compare the expression levels of
mir-429 between the two groups (Fig.
8E). In addition, IHC was conducted to determine whether
systemic delivery of miR-429 affected the expression levels of EMT
markers. As shown in Fig. 8F,
miR-429 treatment markedly inhibited N-cadherin and Vimentin
expression, and increased E-cadherin expression, compared with in
the miR-Ctrl group. Furthermore, MMP2 and MMP9 were attenuated by
miR-429 administration. These results indicated that elevating
miR-200b/a/429 expression may be considered a potential therapeutic
strategy for the treatment of patients with OSCC.
Discussion
Increasing attention has recently been devoted to
the aberrant activation of STAT3, due to its critical role in the
progression of numerous human malignancies, including lung cancer
(31), pancreatic cancer (32,33),
colorectal cancer (34) and OSCC
(35-37). Extensive functional studies have
characterized STAT3 as a potential therapeutic target in OSCC. The
present study demonstrated that targeting STAT3 could inhibit OSCC
invasion and metastasis in vitro and in vivo via the
EZH2/miR-200b/a/429 axis.
STAT3 signaling is well known to contribute to
cancer progression, invasion and metastasis. It has previously been
suggested that targeting STAT3 may be considered a promising
therapeutic strategy for the treatment of solid tumors. In the
present study, inhibition of STAT3 decreased MMP2 and MMP9
expression, which was accompanied by reduced invasion and migration
of OSCC cells. In addition, targeting STAT3 could inhibit tumor
local invasion in vivo. Conversely, cells overexpressing
STAT3 exhibited a robust malignant phenotype. These findings
suggested that STAT3 may have a contributing role in OSCC invasion
and migration.
It has been reported that EZH2 induces
transcriptional repression of numerous target genes through the
formation of a PRC2 complex with EED and SUZ12 proteins, and is
highly relevant to cancer invasion and metastasis (38). In particular, the present
integrated analysis of EZH2 highlighted its important role in OSCC.
In the present study, EZH2 depletion markedly suppressed OSCC cell
motility in vitro and in animal models. Subsequent western
blotting and immunofluorescence staining revealed that EZH2
silencing significantly hindered EMT by retaining E-cadherin
expression in the cell membrane. IHC staining also detected MET
progression following DZNeP exposure in vivo.
Recent efforts have focused on the regulatory
network between STAT3 and EZH2. In experimental models, EZH2 may
significantly enhance STAT3 accumulation in tumor cells via
methylation, thus promoting tumorigenicity in humans (12,13,39).
In addition, STAT3 has been globally validated to transcriptionally
regulate EZH2 in gastric cancer (40) and colorectal cancer (11). Therefore, a feed-forward loop
between STAT3 and EZH2 has been successfully established and
verified in human cancer. Therefore, the present study focused on
the regulatory relationship between STAT3 and EZH2, in order to
explore whether this crosstalk contributes to OSCC invasion and
metastasis. TCGA network analysis confirmed the positive
correlation between STAT3 and EZH2 among ~500 OSCC cases on the
basis of transcriptome data. In the present study, treatment with
Stattic significantly attenuated EZH2 expression via targeting
STAT3. Furthermore, the present mechanistic investigation revealed
that knockdown of EZH2 may partially reduce the oncogenic role of
STAT3 in OSCC migration and invasion. In addition, STAT3-induced
EMT was markedly suppressed by EZH2 knockdown. Knockdown of EZH2
also inhibited STAT3 phosphorylation, whereas inhibition or
activation of STAT3 phosphorylation affected EZH2 levels
accordingly, establishing a feed-forward loop between these two
proteins. Consequently, the present study established a regulatory
network between STAT3 and EZH2 by in vitro experiments and
animal models. This finding may provide a rationale for the
treatment of OSCC.
miR-200b/a/429 genes have been reported to serve as
tumor suppressors in numerous types of human cancer (41,42)
and have been demonstrated to be major regulators of EMT (14,19,43).
Silencing miR-200 genes by methylation has been reported to promote
tumor growth and invasion via EMT induction (44,45).
The present results demonstrated that systemic delivery of miR-429
induced a marked decrease in OSCC cell growth and local invasion.
Notably, EZH2 was validated to abrogate miR-200b/a/429 expression
levels through epigenetic regulation. The EZH2/miR-200b/a/429 axis
has previously been identified as a critical therapeutic target in
cancers of epithelial origin (23). In the present study, EZH2 depletion
significantly increased miR-200b/a/429 expression in vitro
and in vivo. Together with the confirmed role of EZH2 in
EMT, the EZH2/miR-200b/a/429 axis may serve a critical role in
tumor invasion and EMT-directed metastasis. Furthermore, STAT3 was
revealed to regulate miR-200b/a/429 in an EZH2-dependent manner.
These data provide evidence to suggest that the EZH2/miR-200b/a/429
axis may mediate the oncogenic role of STAT3 in vitro and
in vivo.
On the basis of increasing integrated research and
experimental evidence, STAT3 has been revealed to serve a central
role in cancer progression, and its overexpression is associated
with tumor cell proliferation, invasiveness and metastasis in human
cancers, including OSCC. Accordingly, the present study revealed
the therapeutic effect of targeting STAT3, and the mechanism by
which STAT3 exerts its oncogenic role in OSCC. The results
indicated that STAT3 or EZH2 inhibition, as well as miR-429
systemic delivery, may markedly attenuate local tumor invasion and
outgrowth, which may have potential significance for the
identification of targeted therapies for patients with OSCC.
In conclusion, the present study is the first, to
the best of our knowledge, to identify the mechanism by which STAT3
governs OSCC invasion and metastasis, and revealed a master
regulatory network between STAT3 and the EZH2/miR-200b/a/429 axis
in OSCC. Although STAT3-based therapeutics are in their infancy,
these findings are still encouraging and suggest that STAT3 is a
potential target; therefore, these findings may be translated
clinically for the treatment of OSCC.
Glossary
Abbreviations
Abbreviations:
|
EMT
|
epithelial-mesenchymal transition
|
|
HNSCC
|
head and neck squamous cell
carcinoma
|
|
OSCC
|
oral squamous cell carcinoma
|
|
STAT3
|
signal transducer and activator of
transcription 3
|
|
EZH2
|
enhancer of zeste homolog 2
|
|
PRC2
|
polycomb repressive complex 2
|
|
H3K27me3
|
trimethylation of lysine 27 in histone
3
|
|
DMSO
|
dimethyl sulfoxide
|
|
IHC
|
immunohistochemistry
|
Acknowledgments
The authors would like to thank Dr Jie Zhao at
Tianjin Hospital (Tianjin, China) for his kind assistance.
Notes
[1]
Funding
This study was supported by the National Natural
Science Foundation of China (grant no. 81572492) and the CSCO-Merck
Serono Oncology Research Fund, SCORE (grant no. Y-MT2015-017).
[2] Availability
of data and materials
All data generated or analyzed during this study are
included in this published article.
[3] Authors'
contributions
YWa, YWu, XZ, MZ, XQ and LK made substantial
contributions to the conception and design of the study. ZL, CZ,
LZ, JD, RJ, YQ and WG made substantial contributions to data
acquisition. ZL, CJ, YS, JC, XW and YR made substantial
contributions to data analysis and interpretation. MZ, LZ, SS, CW
and JD were also involved in drafting the manuscript. LK, SS, CW,
YQ, RJ and XQ were involved in revising the manuscript critically
for important intellectual content. XZ, SS and CW also gave final
approval of the version to be published. All authors read and
approved the final manuscript.
[4] Ethics
approval and consent to participate
The present study was approved by the Institutional
Animal Care and Use Committee of Tianjin Medical University Cancer
Institute and Hospital (Tianjin, China).
[5] Consent for
publication
Not applicable.
[6] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Pulte D and Brenner H: Changes in survival
in head and neck cancers in the late 20th and early 21st century: A
period analysis. Oncologist. 15:994–1001. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lam L, Logan RM and Luke C:
Epidemiological analysis of tongue cancer in South Australia for
the 24-year period, 1977–2001. Aust Dent J. 51:16–22. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Beasley NJ, Prevo R, Banerji S, Leek RD,
Moore J, van Trappen P, Cox G, Harris AL and Jackson DG:
Intratumoral lymphangiogenesis and lymph node metastasis in head
and neck cancer. Cancer Res. 62:1315–1320. 2002.PubMed/NCBI
|
|
4
|
Patel V, Marsh CA, Dorsam RT, Mikelis CM,
Masedunskas A, Amornphimoltham P, Nathan CA, Singh B, Weigert R,
Molinolo AA, et al: Decreased lymphangiogenesis and lymph node
metastasis by mTOR inhibition in head and neck cancer. Cancer Res.
71:7103–7112. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Roepman P, Kemmeren P, Wessels LF,
Slootweg PJ and Holstege FC: Multiple robust signatures for
detecting lymph node metastasis in head and neck cancer. Cancer
Res. 66:2361–2366. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Duan Z, Foster R, Bell DA, Mahoney J,
Wolak K, Vaidya A, Hampel C, Lee H and Seiden MV: Signal
transducers and activators of transcription 3 pathway activation in
drug-resistant ovarian cancer. Clin Cancer Res. 12:5055–5063. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Giraud S, Bienvenu F, Avril S, Gascan H,
Heery DM and Coqueret O: Functional interaction of STAT3
transcription factor with the coactivator NcoA/SRC1a. J Biol Chem.
277:8004–8011. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nakashima K, Yanagisawa M, Arakawa H,
Kimura N, Hisatsune T, Kawabata M, Miyazono K and Taga T:
Synergistic signaling in fetal brain by STAT3-Smad1 complex bridged
by p300. Science. 284:479–482. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Margueron R and Reinberg D: The Polycomb
complex PRC2 and its mark in life. Nature. 469:343–349. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ciferri C, Lander GC, Maiolica A, Herzog
F, Aebersold R and Nogales E: Molecular architecture of human
polycomb repressive complex 2. Elife. 1:e000052012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lin YW, Ren LL, Xiong H, Du W, Yu YN, Sun
TT, Weng YR, Wang ZH, Wang JL, Wang YC, et al: Role of STAT3 and
vitamin D receptor in EZH2-mediated invasion of human colorectal
cancer. J Pathol. 230:277–290. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim E, Kim M, Woo DH, Shin Y, Shin J,
Chang N, Oh YT, Kim H, Rheey J, Nakano I, et al: Phosphorylation of
EZH2 activates STAT3 signaling via STAT3 methylation and promotes
tumorigenicity of glioblastoma stem-like cells. Cancer Cell.
23:839–852. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Methylation by EZH2 activates STAT3 in
glioblastoma. Cancer Discov. 3:OF212013.PubMed/NCBI
|
|
14
|
Xue X, Zhang Y, Zhi Q, Tu M, Xu Y, Sun J,
Wei J, Lu Z, Miao Y and Gao W: MiR200-upregulated Vasohibin 2
promotes the malignant transformation of tumors by inducing
epithelial-mesenchymal transition in hepatocellular carcinoma. Cell
Commun Signal. 12:622014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sossey-Alaoui K, Bialkowska K and Plow EF:
The miR200 family of microRNAs regulates WAVE3-dependent cancer
cell invasion. J Biol Chem. 284:33019–33029. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sun N, Zhang Q, Xu C, Zhao Q, Ma Y, Lu X,
Wang L and Li W: Molecular regulation of ovarian cancer cell
invasion. Tumour Biol. 35:11359–11366. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Holzner S, Senfter D, Stadler S,
Staribacher A, Nguyen CH, Gaggl A, Geleff S, Huttary N, Krieger S,
Jäger W, et al: Colorectal cancer cell-derived microRNA200
modulates the resistance of adjacent blood endothelial barriers in
vitro. Oncol Rep. 36:3065–3071. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xiao P, Liu W and Zhou H: miR-200b
inhibits migration and invasion in non-small cell lung cancer cells
via targeting FSCN1. Mol Med Rep. 14:1835–1840. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yuan D, Xia H, Zhang Y, Chen L, Leng W,
Chen T, Chen Q, Tang Q, Mo X, Liu M, et al: P-Akt/miR 200 signaling
regulates epithelial-mesenchymal transition, migration and invasion
in circulating gastric tumor cells. Int J Oncol. 45:2430–2438.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen J, Wang L, Matyunina LV, Hill CG and
McDonald JF: Overexpression of miR-429 induces
mesenchymal-to-epithelial transition (MET) in metastatic ovarian
cancer cells. Gynecol Oncol. 121:200–205. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gregory PA, Bert AG, Paterson EL, Barry
SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y and Goodall GJ:
The miR-200 family and miR-205 regulate epithelial to mesenchymal
transition by targeting ZEB1 and SIP1. Nat Cell Biol. 10:593–601.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Park SM, Gaur AB, Lengyel E and Peter ME:
The miR-200 family determines the epithelial phenotype of cancer
cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes
Dev. 22:894–907. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ning X, Shi Z, Liu X, Zhang A, Han L,
Jiang K, Kang C and Zhang Q: DNMT1 and EZH2 mediated methylation
silences the microRNA-200b/a/429 gene and promotes tumor
progression. Cancer Lett. 359:198–205. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
25
|
Lee TK, Poon RT, Wo JY, Ma S, Guan XY,
Myers JN, Altevogt P and Yuen AP: Lupeol suppresses
cisplatin-induced nuclear factor-kappaB activation in head and neck
squamous cell carcinoma and inhibits local invasion and nodal
metastasis in an orthotopic nude mouse model. Cancer Res.
67:8800–8809. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Scuto A, Kujawski M, Kowolik C, Krymskaya
L, Wang L, Weiss LM, Digiusto D, Yu H, Forman S and Jove R: STAT3
inhibition is a therapeutic strategy for ABC-like diffuse large
B-cell lymphoma. Cancer Res. 71:3182–3188. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fiskus W, Wang Y, Sreekumar A, Buckley KM,
Shi H, Jillella A, Ustun C, Rao R, Fernandez P, Chen J, et al:
Combined epigenetic therapy with the histone methyltransferase EZH2
inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor
panobinostat against human AML cells. Blood. 114:2733–2743. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Feldman AT and Wolfe D: Tissue processing
and hematoxylin and eosin staining. Methods Mol Biol. 1180:31–43.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang H, Yamazaki T, Pietrocola F, Zhou H,
Zitvogel L, Ma Y and Kroemer G: STAT3 inhibition enhances the
therapeutic efficacy of immunogenic chemotherapy by stimulating
type 1 interferon production by cancer cells. Cancer Res.
75:3812–3822. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sharma V, Purkait S, Takkar S, Malgulwar
PB, Kumar A, Pathak P, Suri V, Sharma MC, Suri A, Kale SS, et al:
Analysis of EZH2: micro-RNA network in low and high grade
astrocytic tumors. Brain Tumor Pathol. 33:117–128. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen J, Lan T, Zhang W, Dong L, Kang N,
Zhang S, Fu M, Liu B, Liu K and Zhan Q: Feed-forward reciprocal
activation of PAFR and STAT3 regulates epithelial-mesenchymal
transition in non-small cell lung cancer. Cancer Res. 75:4198–4210.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Loncle C, Bonjoch L, Folch-Puy E,
Lopez-Millan MB, Lac S, Molejon MI, Chuluyan E, Cordelier P, Dubus
P, Lomberk G, et al: IL17 Functions through the Novel
REG3β-JAK2-STAT3 inflammatory pathway to promote the transition
from chronic pancreatitis to pancreatic cancer. Cancer Res.
75:4852–4862. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Baumgart S, Chen NM, Siveke JT, König A,
Zhang JS, Singh SK, Wolf E, Bartkuhn M, Esposito I, Heßmann E, et
al: Inflammation-induced NFATc1-STAT3 transcription complex
promotes pancreatic cancer initiation by KrasG12D. Cancer Discov.
4:688–701. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kesselring R, Glaesner J, Hiergeist A,
Naschberger E, Neumann H, Brunner SM, Wege AK, Seebauer C, Köhl G,
Merkl S, et al: IRAK-M expression in tumor cells supports
colorectal cancer progression through reduction of antimicrobial
defense and stabilization of STAT3. Cancer Cell. 29:684–696. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fan TF, Bu LL, Wang WM, Ma SR, Liu JF,
Deng WW, Mao L, Yu GT, Huang CF, Liu B, et al: Tumor growth
suppression by inhibiting both autophagy and STAT3 signaling in
HNSCC. Oncotarget. 6:43581–43593. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhou X, Ren Y, Liu A, Han L, Zhang K, Li
S, Li P, Li P, Kang C, Wang X, et al: STAT3 inhibitor WP1066
attenuates miRNA-21 to suppress human oral squamous cell carcinoma
growth in vitro and in vivo. Oncol Rep. 31:2173–2180. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhou X, Ren Y, Liu A, Jin R, Jiang Q,
Huang Y, Kong L, Wang X and Zhang L: WP1066 sensitizes oral
squamous cell carcinoma cells to cisplatin by targeting
STAT3/miR-21 axis. Sci Rep. 4:74612014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhou X, Ren Y, Kong L, Cai G, Sun S, Song
W, Wang Y, Jin R, Qi L, Mei M, et al: Targeting EZH2 regulates
tumor growth and apoptosis through modulating mitochondria
dependent cell-death pathway in HNSCC. Oncotarget. 6:33720–33732.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dasgupta M, Dermawan JK, Willard B and
Stark GR: STAT3-driven transcription depends upon the dimethylation
of K49 by EZH2. Proc Natl Acad Sci USA. 112:3985–3990. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Pan YM, Wang CG, Zhu M, Xing R, Cui JT, Li
WM, Yu DD, Wang SB, Zhu W, Ye YJ, et al: STAT3 signaling drives
EZH2 transcriptional activation and mediates poor prognosis in
gastric cancer. Mol Cancer. 15:792016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wiklund ED, Bramsen JB, Hulf T, Dyrskjøt
L, Ramanathan R, Hansen TB, Villadsen SB, Gao S, Ostenfeld MS,
Borre M, et al: Coordinated epigenetic repression of the miR-200
family and miR-205 in invasive bladder cancer. Int J Cancer.
128:1327–1334. 2011. View Article : Google Scholar
|
|
42
|
Manavalan TT, Teng Y, Litchfield LM,
Muluhngwi P, Al-Rayyan N and Klinge CM: Reduced expression of
miR-200 family members contributes to antiestrogen resistance in
LY2 human breast cancer cells. PLoS One. 8:e623342013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Izumchenko E, Chang X, Michailidi C,
Kagohara L, Ravi R, Paz K, Brait M, Hoque MO, Ling S, Bedi A, et
al: The TGFβ-miR200-MIG6 pathway orchestrates the EMT-associated
kinase switch that induces resistance to EGFR inhibitors. Cancer
Res. 74:3995–4005. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Davalos V, Moutinho C, Villanueva A, Boque
R, Silva P, Carneiro F and Esteller M: Dynamic epigenetic
regulation of the microRNA-200 family mediates epithelial and
mesenchymal transitions in human tumorigenesis. Oncogene.
31:2062–2074. 2012. View Article : Google Scholar :
|
|
45
|
Enkhbaatar Z, Terashima M, Oktyabri D,
Tange S, Ishimura A, Yano S and Suzuki T: KDM5B histone demethylase
controls epithelial-mesenchymal transition of cancer cells by
regulating the expression of the microRNA-200 family. Cell Cycle.
12:2100–2112. 2013. View Article : Google Scholar : PubMed/NCBI
|















