Introduction
Osteosarcoma is the most common primary malignancy
of the bone in adolescents and is characterized by local invasion
and distant metastasis (1). The
survival rate has improved with the use of multi-agent
chemotherapy, from 11% with surgical resection alone in the 1960s
to 70% with the addition of chemotherapy by the mid-1980s (2). However, long-term outcomes have
reached a plateau over the past 20 years (3). Tumor progression to the invasive and
metastatic states increases morbidity and mortality in cancer and
represents the most formidable obstacle to successful treatment
(4). Overall, ~30% of patients
with a localized form of the disease and 80% of patients with a
metastatic form of the disease at the point of initial diagnosis
will relapse. These patients cannot be described as cured, and they
have <20% chance of long-term survival despite aggressive
therapies. Conventional chemotherapies frequently induce
drug-resistance and cause severe side effects (5,6). For
these reasons, novel therapies are required to treat patients with
osteosarcomas.
Insulin-like growth factor 1 receptor (IGF-1R)
belongs to the large class of tyrosine kinase receptors that are
activated by IGF-1 and by the associated growth factor IGF-2. The
ligand-receptor interaction initiates receptor autophosphorylation
of tyrosine residues and subsequently activates multiple signalling
pathways, including the mitogen-activated protein kinase (MAPK) and
phosphoinositide 3-kinase (PI3K) signaling pathways (7). IGF-1R has been demonstrated to be not
only a key regulator of normal physiological cell processes, but
also to serve important functions in carcinogenesis and tumor
development, including tumor growth, progression, invasion and
metastasis (8–10).
The IGF system is an important regulator of bone
formation and homeostasis (11).
During the adolescent growth spurt, elevated levels of IGF-1
coincide with a high prevalence of osteosarcoma, suggesting that
IGF-1 may contribute to the pathogenesis of osteosarcoma (5,12).
Previous studies have reported that human osteosarcoma cell lines
display functional IGF-1R on their surface (13,14).
In human primary osteosarcoma tissue samples, the levels of IGF-R,
IGF-1, and IGF-2 were increased compared with positive control cell
lines (15,16). In vitro, inhibition of
growth and invasiveness may result from downregulation of IGF-IR
(14,17). Furthermore, IGF-1R blockade
increases radio-sensitivity and induces apoptosis in
multidrug-resistant osteosarcoma cell lines (5,14).
These results suggest that compounds which specifically inhibit the
function of IGF-1R tyrosine kinase in cancer cells may potentially
be an effective treatment strategy. Early studies have revealed
that analogues of and antagonists to growth hormone-releasing
hormone decrease serum IGF-1 levels and inhibit tumor growth in
mice, but failed in their phase 1 clinical trial (18,19).
An alternative approach has been to block the interaction of IGF-1R
with IGF-1 and IGF-2 by targeting IGF-1R, and, most importantly, to
downregulate the receptor by internalization and degradation via
the endosome (20). In the present
study, our group developed a novel quinazoline derivative,
6-fluoro-2-(3-fluorophenyl)-4-(cyanoanilino)quinazoline (HMJ-30;
Fig. 1A), to disrupt IGF-IR
signaling and inhibit tumor invasion and migration in osteosarcoma
cells.
 | Figure 1HMJ-30 selectively targets the IGF-1R
ATP binding site. (A) The chemical structure of HMJ-30. (B)
Following treatment with 0, 20, and 40 μM HMJ-30 for 6 h,
U-2 OS cells were harvested and total proteins were collected.
IGF-1R was evaluated by immune-precipitation and p-IGF-1R
(Tyr1131) and p-tyrosin (4G10) were evaluated by western
blotting. (C) Following treatment with 0, 20 and 40 μM
HMJ-30 or with PPP for 6 h, cells were harvested and total proteins
were collected. IR was evaluated by immune-precipitation, and p-IR
(Tyr1146) was evaluated by western blotting. (D)
p-IGF-1R (Tyr 1131) protein levels were evaluated by ELISA. (E)
Tyrosine kinase activity was evaluated using a tyrosine kinase
assay. (F) In order to predict the major target site of HMJ-30 to
IGF-1R, the docking simulation of HMJ-30 and IGF-1R was performed
using a computational modeling program. The interactions reveal
that HMJ-30 binds readily to the ATP-binding site of IGF-1R with
low potential energy. The superimposed image is of HMJ-30 (yellow
color) and benzimidazole inhibitor A (gray color). The data
represents the mean ± standard deviation of three experiments.
***P<0.001 vs. control. HMJ-30,
6-fluoro-2-(3-fluorophenyl)-4-(cyanoanilino) quinazoline; IGF-1R,
insulin-like growth factor 1 receptor; ATP, adenosine
trisphosphate; p-, phosphorylated; PPP, picropodophyllotoxin; IR,
insulin receptor. |
Epithelial-mesenchymal transition (EMT) is a program
of biological development in which cells lose their epithelial
characteristics and gain mesenchymal properties during
embryogenesis, as well as for the maintenance of homeostasis and
the architecture of epithelial structures during adult life
(21). EMT is controlled by
receptor tyrosine kinase pathways (22). Over the last decade, accumulating
evidence has suggested that EMT regulated by similar pathways is
recapitulated during solid tissue epithelial cancer progression and
invasion, thereby contributing to the formation of metastases
(23). Mesenchymal-epithelial
transition (MET) is the reverse of EMT, and serves an important
function in the formation of somites and in kidney morphogenesis.
This reverse process is characterized by cytoskeletal
reorganization with the reactivation of epithelial cell markers and
the loss of mesenchymal cell markers (24). However, relevant literature is
comparatively rare, particularly concerning cancer.
Quinazoline derivatives have diverse pharmacological
properties, including anti-microbial, anti-malarial, analgesic,
sedative, hypoglycemic, anti-tubercular and anticancer activities
(25). In the past 10 years, a
large number of experimental studies have demonstrated that novel
quinazoline derivatives are able to selectively inhibit important
pathways through blocking growth factor receptors, including
epithelial growth factor receptor, vascular endothelial growth
factor, platelet-derived growth factor, ErbB2 and c-Src (26–30).
One of the most well-characterized derivatives is gefitinib
(Iressa), which is the first selective inhibitor of epidermal
growth factor receptor that binds to the adenosine triphosphate
(ATP)-binding site of the receptor to deter non-small cell lung
cancer (31). There are no reports
on quinazoline derivatives targeting IGF-1R, or concerning the
potential anticancer effects. Our group recently designed and
synthesized quinazoline series compounds, and one of these
compounds, HMJ-30, presented in Fig.
1A, may be a novel anti-IGF-1R agent (32,33).
The present study investigated the novel quinazoline derivative
HMJ-30, which inhibits invasiveness in U-2 OS cells, and elucidated
the potential signaling pathways.
Materials and methods
Chemicals
HMJ-30 was designed and synthesized by Dr Mann-Jen
Hour (School of Pharmacy, China Medical University, Taichung,
Taiwan). Dimethyl sulfoxide (DMSO), potassium phosphate, trypan
blue, propidium iodide (PI), Triton X-100, Tris-HCl, MTT and
ribonuclease-A were obtained from Sigma-Aldrich; Merck KGaA
(Darmstadt, Germany). McCoy's 5a medium, L-glutamine, fetal bovine
serum (FBS), trypsin-EDTA, penicillin G and streptomycin were
obtained from Gibco; Thermo Fisher Scientific (Waltham, MA, USA).
Antibodies against phosphorylated (p-) protein kinase B (AKT) (cat.
no. 4060), AKT (cat. no. 4691), p-extracellular signal-regulated
kinase (ERK) (cat. no. 4370), ERK (cat. no. 4695), p-c-Jun
amino-terminal kinase (JNK) (cat. no. 4668), JNK (cat. no. 9252),
p-p38 (cat. no. 4511), p38 (cat. no. 8690), p-IGF-1R β
(Tyr1131)/insulin receptor β (IRβ; Tyr1146) (cat. no. 3021), and
Ras (cat. no. 3965) were purchased from Cell Signaling Technology,
Inc. (Danvers, MA, USA). Antibodies against β-actin (cat. no.
sc-47778), α-tubulin (cat. no. sc-5286), IGF-1R (cat. no. sc-9038),
IR (cat. no. sc-710), PI3K (p85) (cat. no. sc-1637), IKK (cat. no.
sc-7607), nuclear factor-κB (NF-κB; p65) (cat. no. sc-372), p-IκB
(cat. no. sc-8404), IκB (cat. no. sc-371), matrix metalloproteinase
(MMP)-2 (cat. no. sc-13595), MMP-9 (cat. no. sc-21733), tissue
inhibitor of metalloproteinase (TIMP)-1 (cat. no. sc-5538), TIMP-2
(cat. no. sc-5539), and all goat anti-mouse (cat. no. sc-2005) and
anti-rabbit (cat. no. sc-2004) immunoglobulin (IgG)-horseradish
peroxidase (HRP) secondary antibodies were purchased from Santa
Cruz Biotechnology, Inc. (Dallas, TX, USA). Epithelial cadherin
(E-cadherin) (cat. no. 07-697), β-catenin (cat. no. 05-613),
fibronectin (cat. no. CP70) and vimentin (cat. no. CBL202) primary
antibodies were from EMD Millipore (Billerica, MA, USA). Materials
and chemicals for electrophoresis were obtained from Bio-Rad
Laboratories, Inc. (Hercules, CA, USA).
Cell culture
U-2 OS, a human osteogenic sarcoma cell line, was
obtained from the Food Industry Research and Development Institute
(Hsinchu, Taiwan). U-2 OS cells were cultured in 75 cm2
tissue culture flasks with 90% McCoy's 5a medium containing 1.5 mM
L-glutamine adjusted to 1.5 mg/l sodium bicarbonate and
supplemented with 10% FBS and 1% penicillin-streptomycin (100 U/ml
penicillin G and 100 μg/ml streptomycin) at 37°C under a
humidified 5% CO2 atmosphere, as previously described
(33–36).
Cell viability assay
To evaluate the cytotoxicity of HMJ-30 in
osteocarcinoma U-2 OS cells, an MTT assay was performed. Briefly,
viable cells were counted on a Neubauer chamber with a microscope.
U-2 OS cells were placed in a 96-well cell culture plate at an
initial concentration of 1×105 cells/ml, and treated
with HMJ-30 at different concentrations (0, 5, 10, 20, and 40
μM) or with 0.1% DMSO for 24 h at 37°C. Following a 24 h
incubation period, MTT solution (0.5 mg/ml) was added to each well
for 4 h at 37°C. Subsequently, the culture medium was removed, and
the formazan crystals formed by oxidation of the MTT solution were
dissolved with DMSO in isopropanol and measured
spectrophotometrically at 490 nm. The cell survival ratio was
expressed as a percentage of the control, as previously described
(37–39).
Phase-contrast microscopy of
morphological changes
U-2 OS cells were cultured in 24-well plates at a
density of 2.5×105 cells/well/ml prior to treatment with
20 μM HMJ-30. Following treatment with HMJ-30 for 24 h at
37°C, morphological changes were determined using a phase-contrast
microscope (×200 and ×400) as previously described (6).
Wound healing assay
To determine cell migration, U-2 OS cells were
placed in a 6-well tissue culture plate for 24 h and grown to
80–90% confluence. Individual wells were scratched with a
micropipette tip to create a denuded zone of constant width (1 mm).
Cells were then cultured in serum-free McCoy's 5a medium and
incubated with 0, 5, 10, 20, and 40 μM of HMJ-30 or with
0.1% DMSO for 24 h at 37°C. Cells were photographed under
phase-contrast microscopy (×100) as previously described (35,40,41).
Transwell assay
Cell invasion was measured using a Matrigel-coated
invasion chamber. Initially, a 24-well Transwell insert with 8
μm porosity polycarbonate filters (EMD Millipore) recoated
with 30 μg Englebreth-Holm-Swarm sarcoma tumor matrix (EHS
Matrigel Basement Membrane Matrix) at 25°C for 1 h to form a
genuine reconstituted basement membrane. Cells were then maintained
for 24 h in serum-free McCoy's 5a medium. The serum-starved cells
were then re-suspended in serum-free medium and placed in the upper
chamber of the Transwell insert (5×104 cells/well) and
treated with different concentrations of HMJ-30 (0, 5, 10, 20 and
40 μM) or with 0.1% DMSO for 24 h at 37°C. The culture
medium supplemented with 10% FBS was added to the lower chamber.
The cells were then incubated at 37°C in a humidified atmosphere
with 95% air and 5% CO2 to allow sufficient time for
invasion. Following incubation for 24 h, the cells were fixed with
4% formaldehyde for 15 min in phosphate-buffered-saline (PBS) and
stained with 2% crystal violet for 5 min at room temperature.
Finally, the non-invading cells in the top well were removed with a
cotton swab, and the invading cells which penetrated through the
Matrigel to the bottom wells were counted under a light microscope
(×200) as previously described (6,36,42).
Gelatin zymography
To determine the activity of MMP-2 and MMP-9,
gelatin zymography was used. Briefly, U-2 OS cells were treated
with HMJ-30 at different concentrations (0, 5, 10, 20 and 40
μM) or with 0.1% DMSO for 24 h at 37°C in the absence of
serum. Cells were then collected and separated by dilution in a
zymography sample buffer. Samples were mixed with loading buffer
and electrophoresed on 10% sodium dodecyl sulfate
(SDS)-polyacrylamide gel with 0.1% gelatin. Gels were washed twice
in denaturing buffer (2.5% Triton X-100 in double-distilled
H2O), and incubated with development buffer (50 mM Tris,
pH 7.5, 200 mM NaCl, 5 mM CaCl2, 1 mM ZnCl2,
0.02% Briji-35) at 37°C for 18 h, stained with 0.5% Coomassie blue
G-250 for 1 h, and de-stained with de-staining solution.
Non-staining bands indicating proteolytic activities were digitized
using a scanning digital system and analyzed using NIH image
software. MMP-2 (72-62 kDa) and MMP-9 (92-85 kDa) activity was
expressed as the ratio to MMP-2 standard to avoid differences among
gels (43).
Molecular modeling of the HMJ-30-protein
complex
The crystal structure of the IGF-1R kinase domain
complexed with a benzimidazole (Protein Data Bank code = 2oj9) was
retrieved from the Protein Data Bank (http://www.rcsb.org/pdb). Automated docking was then
performed. LigandFit, within the software package Discovery Studio
2.5 (Accelrys, San Diego, CA, USA) was used to evaluate and predict
the in silico binding free energy of the inhibitors within
the macromolecules. First, the prepare protein protocol was used to
prepare the 2oj9 protein structure, including standardizing atom
names, inserting missing atoms in residues and removing alternate
conformations, inserting missing loop regions based on SEQRES data,
optimizing short and medium size loop regions with Looper
Algorithm, minimizing the remaining loop regions, and calculating
pK and protonate structure. A binding pocket of the native
benzimidazole ligand was selected as the binding site for the
present study. Following typing of the receptor model with the
CHARMm forcefield, the binding site was identified by the LigandFit
flood-filling algorithm. This docking protocol employed total
ligand flexibility, whereby the final ligand conformations were
determined by the Monte Carlo conformation search method set to a
variable number of trial runs. The docked ligands were further
refined using in situ ligand minimization with the Smart
Minimizer algorithm. Each minimization was performed in two steps,
first using the steepest descent minimization for 200 cycles and
then using conjugate gradient minimization, until the average
gradient fell below 0.01 kcal/mol. All atoms within 6.0 Å of the
inhibitor were allowed to relax during the minimization, whereas
those atoms beyond 6.0 Å were held rigid. Finally, the Dock score
was used to estimate the binding free energies of the ligands.
Among the docked conformations, the pose with the highest value of
Dock score was selected for the calculation of binding free energy
(∆Gb) and inhibition constant (Ki).
Tyrosine kinase assay
An assay to determine tyrosine kinase activity was
performed according to the manufacturer's protocols (Tyrosine
kinase assay kit; EMD Millipore). U-2 OS cells were plated in
12-well plates at an initial density of 5.0×106 cells
and incubated with 0, 20 or 40 μM of HMJ-30 or with 10
μM picopodophyllotoxin (PPP) (Sigma-Aldrich; Merck KGaA) for
6 h at 37°C. Cells were harvested and total proteins were collected
under non-denaturing conditions. Samples were incubated with
tyrosine kinase reaction buffer for 30 min at 30°C, followed by the
addition of p-tyrosine (4G10) antibodies at a 1:5,000 dilution for
3 min at room temperature, and then detected using an ELISA reader
(Anthos 2001) (Anthos Labtec Instruments GmbH, Salzburg, Austria)
at a 450 nm wavelength, as previously described (44).
Phosphorylated protein kinase sandwich
ELISA assay
p-IGF-1R β (Tyr1131) (cat. no. 7302), p-p38 MAPK
(Thr180/Tyr182) (cat. no. 7946), p-p44/42 MAPK (Thr202/Tyr204)
(cat. no. 7177), and p-Akt (Thr308) (cat. no. 7252) assays were
performed according to the manufacturer's protocols (PathScan
Sandwich ELISA kits; Cell Signaling Technology, Inc.). In total,
~1×107 U-2 OS cells/ml were plated on 24-well plates and
treated with 0, 20 or 40 μM of HMJ-30 or with specific
protein kinase inhibitors (10 μM PPP, U0126, SB203580 or
wortmannin; Sigma-Aldrich; Merck KGaA) for 6 h at 37°C. These cells
were harvested and total proteins were collected. Samples were
incubated in appropriate antibody-coated microwells for 2 h at 37°C
or overnight at 4°C. To each well, 100 μl of the appropriate
antibody was added for 1 h at 37°C, and HRP-linked secondary
antibody was added for 30 min at 37°C. Absorbance was measured with
an ELISA reader (Anthos 2001) at a wavelength of 450 nm as
previously described (45).
Immunoprecipitation of IGF-1R and IR
proteins
U-2 OS cells were seeded in a 10-cm dish at an
initial concentration of 1.0×107 cells and treated with
20 or 40 μM HMJ-30 or with 0.1% DMSO for 6 h. Cells were
then harvested and total proteins were collected using PRO-PREP
Protein Extraction Solution (iNtRON Biotechnology, Seongnam,
Korea). Samples were incubated with IGF-1R or IR antibodies
overnight at 4°C, followed by adding A/G-agarose beads with gentle
rocking for 3 h at 4°C. Following centrifugation at 300 x g for 10
min at 4°C, the pellets were washed with 1X cell lysis buffer and
re-suspended in 3X SDS sample buffer. These samples were subjected
to 10% SDS gel electrophoresis followed by incubation with
p-tyrosine (4G10) or p-IGF-1R β (Tyr1131)/IRβ (Tyr1146) antibodies
at a 1:1,000 dilution overnight at 4°C to measure specific protein
levels, as previously described (45).
Immunostaining and confocal laser
microscopy
U-2 OS cells (5×104 cells/well) plated on
a 4-well chamber slide were treated with 20 μM HMJ-30 or
with 0.1% DMSO for 24 h. Cells were then fixed in 4% ice
formaldehyde for 5 min at room temperature and placed in 1% bovine
serum albumin (Sigma-Aldrich; Merck KGaA) containing 0.1% Triton
X-100 at 37°C for 30 min. Subsequently, the cells were washed twice
with PBS for 5 min and incubated with primary antibodies against
E-cadherin (1:250 dilution), β-catenin (1:250 dilution),
fibronectin (1:250 dilution), and vimentin (1:250 dilution)
overnight at 4°C and then exposed to specific secondary antibodies
[fluorescein isothiocyanate-conjugated goat anti-mouse (cat. no.
31569) or anti-rabbit (cat. no. 31635) IgG antibodies; Invitrogen;
Thermo Fisher Scientific] at a 1:500 dilution for 1 h at room
temperature, followed by DNA staining with PI. Photomicrographs
were obtained using a Leica TCS SP2 confocal spectral microscope
(Leica Microsystems GmbH, Wetzlar, Germany) as previously described
(46,47).
Western blotting
To analyze the expression of proteins associated
with invasiveness, western blot analysis was performed. Briefly,
the U-2 OS cells were plated in 10 cm-dish at an initial
concentration of 1.0×107 cells and treated with HMJ-30
at different concentrations (0, 10, 20, and 40 μM) or with
0.1% DMSO for 6 or 24 h at 37°C. Cells were harvested and total
proteins were collected using PRO-PREP Protein Extraction Solution
(iNtRON Biotechnology). Protein samples were quantified using the
Pierce bicinchoninic acid (BCA) Protein assay kit (Pierce; Thermo
Fisher Scientific). Samples (40 μg) were electrophoresed by
10% SDS-polyacrylamide gel electrophoresis and transferred onto
nitrocellulose membranes (Invitrogen; Thermo Fisher Scientific).
The membranes were then incubated in blocking solution (0.1%
Tween-20 in PBS plus 5% powdered non-fat milk) for 1 h at room
temperature, and incubated overnight at 4°C with each of the
indicated primary antibodies [MMP-9, MMP-2, TIMP-1, TIMP-2,
E-cadherin, β-catenin, vimentin, fibronectin, Ras, p-ERK, ERK,
p-JNK, JNK, p-p38, p38, PI3K (p85), p-AKT, AKT, IKK, p-IκB, IκB,
NF-κB (p65), β-actin and α-tubulin] (1:1,000 dilution) diluted in
blocking solution. Subsequently, the membranes were washed with
PBST three times for 10 min and incubated with the appropriate
HRP-conjugated secondary IgG antibodies (1:10,000 dilution;
HRP-conjugated goat anti-rabbit and goat anti-mouse IgG) for 1 h at
room temperature and then washed three times in PBST. Bands were
detected using enhanced chemiluminescence with ECL reagents (GE
Healthcare Life Sciences, Little Chalfont, UK) and exposed to
X-OMAT AR films (Kodak, Rochester, NY, USA). The auto-radiograms
were scanned on a UMAX PowerLook Scanner (UMAX Technologies,
Fremont, CA, USA) with Photoshop CS5 software (Adobe Systems, Inc.,
San Jose, CA, USA), as previously described (43,48).
Analysis of NF-κB binding using an
electrophoretic mobility shift assay (EMSA)
Nuclear proteins were extracted from U-2 OS cells
following treatment with 20 μM HMJ-30 or 10 μM
ammonium pyrrolidinedithiocarbamate (PDTC; NF-κB inhibitor)
(Sigma-Aldrich; Merck KGaA) for 12 h at 37°C using the NE-PER
Nuclear and Cytoplasmic Extraction Reagents kit (Pierce; Thermo
Fisher Scientific, Inc.). The protein concentrations were
determined using the Pierce BCA Protein assay kit (Pierce; Thermo
Fisher Scientific), and biotin end-labeled oligonucleotide
sequences (5′-Biotin-GATCCAGGGGACTTTCCCTAGC-3′) corresponding to
the consensus site of NF-κB were designed. Nuclear extract proteins
(5 μg) were used for EMSA with a LightShift Chemiluminescent
EMSA kit (Pierce; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. Biotin end-labeled duplex DNA was
incubated with a nuclear extract or purified factor and
electrophoresed on a 6% non-denaturing polyacrylamide native gel.
For competition experiments, a 100-fold excess of unlabeled
double-stranded oligonucleotide was added to the reaction. Then,
the DNA was rapidly transferred to a positively-charged
nitrocellulose membrane, UV cross-linked and probed with
streptavidin-HRP conjugate (1:300 dilution) with gentle agitation
for 15 min at room temperature. Bands were detected by enhanced
chemiluminescence with ECL reagents (GE Healthcare Life Sciences)
and exposed to X-OMAT AR films (Kodak) as previously described
(35,49).
Statistical analysis
All data were expressed as the mean ± standard
deviation from three separate experiments. Statistical analysis was
performed using one-way analysis of variance followed by Dunnett's
test, with SPSS software version 16.0 (SPSS, Inc., Chicago, IL,
USA). *P<0.05 was considered to indicate a
statistically significant difference.
Results
HMJ-30 selectively targets insulin-like
growth factor 1 receptor (IGF-1R)
Our group synthesized a series of quinazoline
derivatives targeting tyrosine kinase signaling to inhibit tumor
invasion and migration. One of these compounds, HMJ-30 (Fig. 1A), was proposed to actively target
IGF-1R. This hypothesis was tested using western blotting,
immune-precipitation, kinase activity assays and a computational
modeling program. HMJ-30 reduced protein levels of p-IGF-1R (Tyr
1131) and p-tyrosin (4G10) which are determined by
immune-precipitation and western blotting (Fig. 1B). Several small molecule
inhibitors of IGF-1R tyrosine kinases have been developed and they
are currently undergoing clinical investigations (50). However, this approach may be
unsatisfactory as co-inhibition of IR is expected to cause
hyperglycemia, and this potential effect results from a high
protein sequence similarity between the kinase domains of IGF-1R
and IR (51). In the present
study, p-IR and p-tyrosine (4G10) protein expression levels were
evaluated using immune-precipitation and western blotting, and the
results revealed that there was no statistical significance between
HMJ-30-treated groups and the control (Fig. 1C). The effects of HMJ-30 on
p-IGF-1R (Tyr1131) and tyrosine kinase in U-2 OS cells were further
investigated. p-IGF-1R (Tyr1131) protein expression was
significantly downregulated (Fig.
1D). Tyrosine kinase activity was significantly downregulated,
as measured using a kinase assay (Fig.
1E). PPP, which has been reported to act as a non-competitive,
potent and specific inhibitor of IGF-1R in vitro and in
vivo (52), was used as a
positive control, and was also revealed to reduce kinase
activity.
Molecular modeling of HMJ-30 and IGF-1R
interaction
In order to predict the major target site of HMJ-30,
a docking simulation of HMJ-30 and IGF-1R was performed using the
program Discovery Studio Modeling 2.5 (Accelrys). The
three-dimensional crystalline structure of the IGF-1R kinase domain
in complex with a benzimidazole was downloaded from the RCSB
Protein Data Bank website. The computational modeling of the HMJ-30
and IGF1R interaction indicated that HMJ-30 is able to bind to the
ATP-binding site of IGF-1R. The interaction between HMJ-30 and
IGF-1R is involved in the F-H interaction of 6-fluoro with
Ile1130, the F-H interaction of 2-(3-fluoro) phenyl with
Met1052 and Ala1001, the hydrophobic
interactions of the quinazoline ring with Met1126 and
Thr1127, and the hydrophobic interactions of
2-(3-fluoro)phenyl with Met1049 and Glu1050
(Fig. 1F). These interactions made
HMJ-30 bind readily to IGF-1R, with low potential energy.
Furthermore, the structure of HMJ-30 superimposes well onto the
benzimidazole inhibitor
(A)-3-[(5-(1H-imidazol-1-yl)-7-methyl-1H-benzimidazol-2-yl)-4-[(pyridin-2-yl-methyl)
amino]pyridin-2(1H)-one (Fig. 1F,
bottom). Therefore, HMJ-30 may be an IGF-1R inhibitor.
HMJ-30 inhibits invasion in U-2 OS
cells
To determine the effect of HMJ-30 on U-2 OS cell
invasion, wound and Transwell assays were used. HMJ-30 inhibited
cell migration in a concentration-dependent manner (Fig. 2A and B). HMJ-30 also inhibited cell
invasion in a concentration-dependent manner (Fig. 2C and D). Tumor cell viability was
inhibited, however, only following treatment with 40 μM
HMJ-30 for 24 h (Fig. 2E). These
data suggested that a concentration of HMJ-30 <40 μM
primarily inhibited tumor migration and invasion, but toxicity in
U-2 OS cells may require a higher concentration and an increased
incubation time.
 | Figure 2HMJ-30 suppresses invasion and
migration in human osteogenic sarcoma U-2 OS cells. (A) Following
incubation with 0, 5, 10, 20, and 40 μM HMJ-30 for 24 h in
serum-free medium, cells were photographed, and (B) migrated cells
were quantified. (C) The invasion of U-2 OS cells was measured
using Matrigel-coated invasion chambers. Following treatment with
the indicated concentrations of HMJ-30 and incubation for 24 h, the
invading cells which penetrated through the Matrigel to the bottom
wells were counted under a light microscope (×200), and (D) cell
invasion was quantified. (E) Following treatment with 0, 5, 10, 20,
and 40 μM HMJ-30 for 24 h, the cell viability of U-2 OS
cells was evaluated. The data represents the mean ± standard
deviation of three experiments. *P<0.05 and
***P<0.001 vs. 0 μM. HMJ-30,
6-fluoro-2-(3-fluorophenyl)-4-(cyanoanilino)quinazoline. |
HMJ-30 inhibits MMP-2 and MMP-9 protein
activity and expression levels in U-2 OS cells
Previous studies have reported that tumor invasion
requires the degradation of basement membranes and proteolysis of
the extracellular matrix (ECM) (4). Multiple proteolytic enzymes,
including MMPs and serine proteinases, are involved in tumor-host
interactions to transmigrate limiting basement membranes and the
ECM (53). MMP-2 and MMP-9 serve
an important function in cancer cell invasion, and degrade type IV
collagen and gelatin which are primarily found in the basal lamina
(54). To further evaluate the
invasive activity, protein levels of MMP-2 and MMP-9 were
determined by gelatin zymography, sandwich ELISA assay and western
blotting. HMJ-30 significantly inhibited MMP-2 and MMP-9 activity
(Fig. 3A and B) and protein levels
(Fig. 3C and D).
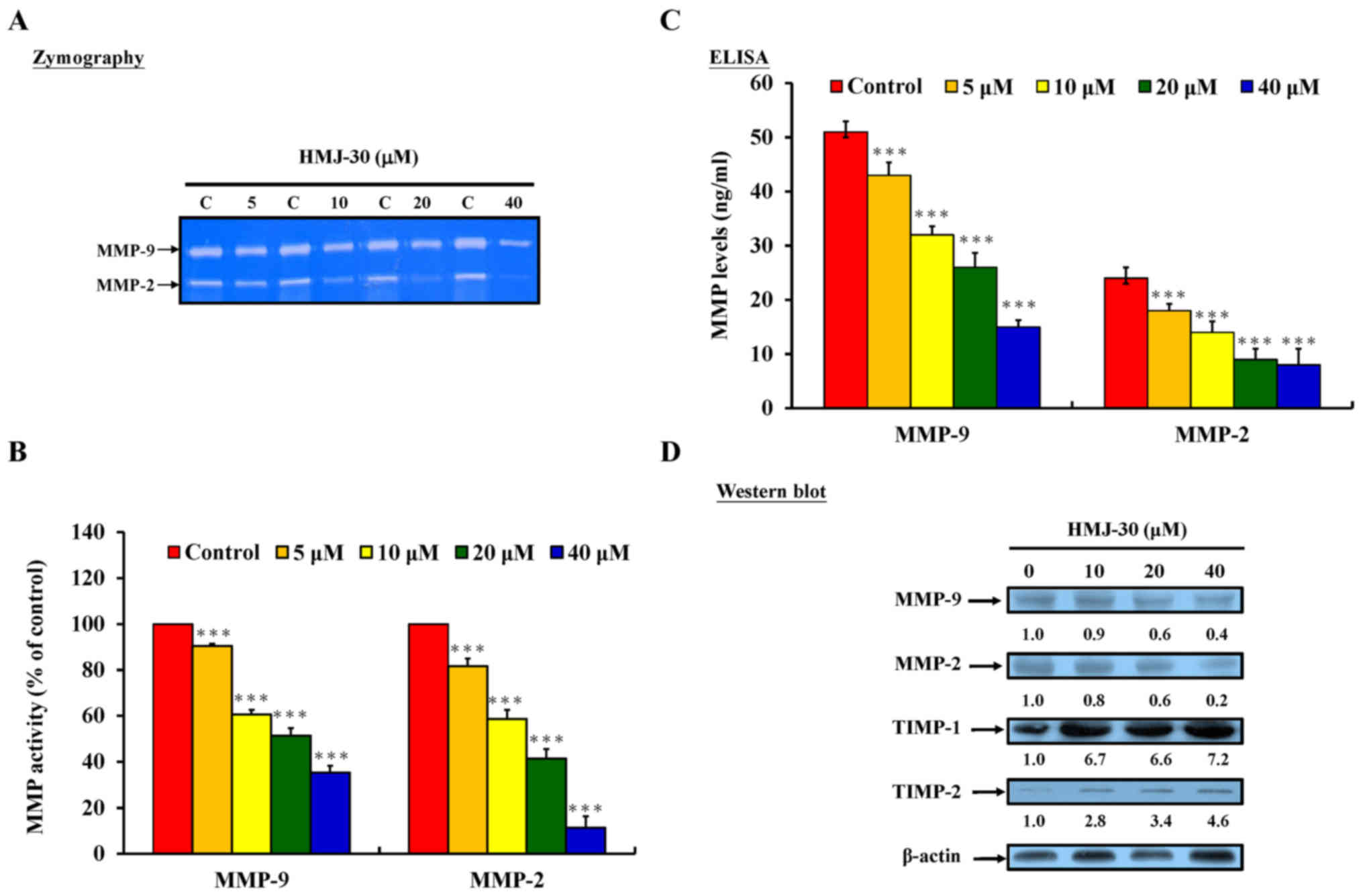 | Figure 3HMJ-30 inhibits MMP-2 and MMP-9
activity and expression levels in human osteogenic sarcoma U-2 OS
cells. (A) Gelatin zymography was used to measure the activity of
MMP-2 and MMP-9, and (B) MMP-9 and MMP-2 levels were quantified.
(C) Following treatment with 0, 5, 10, 20 and 40 μM HMJ-30
for 24 h, MMP-9 and MMP-2 protein levels were determined by
sandwich ELISA. (D) Protein levels of MMP-2, MMP-9, TIMP-1, and
TIMP-2 were evaluated by western blotting. The data represents the
mean ± standard deviation of three experiments.
***P<0.001 vs. control. HMJ-30,
6-fluoro-2-(3-fluorophenyl)-4-(cyanoanilino)quinazoline; MMP,
matrix metalloproteinase; TIMP, tissue inhibitor of
metalloproteinase. |
The tissue inhibitors of metalloproteinase (TIMP)
family, which includes TIMP-1 to TIMP-4, is a group of endogenous
MMP inhibitors that serve a central post-translational regulatory
role in the majority of known MMPs (55). TIMP-1, the inducible form, has been
identified as an inhibitor of the majority of metalloproteinases.
TIMP-2, a primarily constitutive protein, has been observed
interacting with MMP-1, -2, -9 and -14 (55,56).
The present study evaluated the protein levels of TIMP-1 and
TIMP-2, which were revealed to be significantly increased in
HMJ-30-treated U-2 OS cells (Fig.
3D). These results suggested that HMJ-30 may also be involved
in the regulation of MMP-2 and MMP-9 activity.
HMJ-30 induces morphological changes and
inhibits EMT in U-2 OS cells
During the progression and invasion of solid tissue
epithelial cancers, tumor cells will lose their epithelial
characteristics and gain mesenchymal properties, thus undergoing
EMT (23). In contrast, previous
studies have reported that the reactivation of epithelial
characteristics and the loss of mesenchymal cell markers causes
cells to undergo a reverse EMT process, which is also known as MET.
The reverse process has been revealed to be associated with the
inhibition of invasion and migration in cancer cells (57–59).
To evaluate whether or not HMJ-30 induces MET in U-2 OS cells,
morphological changes and mesenchymal/epithelial cell markers were
investigated. HMJ-30-treated U-2 OS cells acquired a flattened,
less elongated and cobblestone-like shape, which is typical of
epithelial cell appearance (Fig.
4A). Furthermore, the cells were more aggregated and formed
tighter clusters compared with the control group. Expression of
vimentin and fibronectin (mesenchymal markers) and of E-cadherin
and β-catenin (epithelial markers) were evaluated by
immunofluorescent staining and western blotting. HMJ-30-treated U-2
OS cells gained E-cadherin and β-catenin (epithelial markers) and
lost vimentin and fibronectin protein (mesenchymal markers)
(Fig. 4B and C). These results
indicated that MET acts as an important anti-invasive function in
HMJ-30-treated U-2 OS cells.
 | Figure 4HMJ-30 alters cell morphology and
inhibits epithelial-mesenchymal transition in human osteogenic
sarcoma U-2 OS cells. (A) Following treatment with 0 and 20
μM HMJ-30 for 24 h, cells were photographed using
phase-contrast microscopy. U-2 OS cells were scattered; however,
HMJ-30-treated cells were more aggregated and formed tight clusters
(top, ×200). The U-2 OS cells had spindle-like morphology; however,
HMJ-30-treated cells gained flattened, less elongated and
cobblestone-like shapes (bottom, ×400). (B) Epithelial markers
(E-cadherin and β-catenin; top) and mesenchymal markers (vimentin
and fibronectin; bottom) were evaluated by immunofluorescent
staining. (C) The protein levels of E-cadherin, β-catenin, vimentin
and fibronectin were evaluated by western blotting. HMJ-30,
6-fluoro-2-(3-fluorophenyl)-4-(cyanoanilino)quinazoline;
E-cadherin, epithelial cadherin. |
HMJ-30 inhibits invasiveness through
IGF-1R-mediated Ras/MAPK, PI3K/AKT and NF-κB signaling pathways in
U-2 OS cells
Activated IGF-IR initiates signaling through two
primary cascades, the PI3K/AKT and Ras/MAPK pathways (7). These cascades serve critical
functions in diverse cellular processes (60). MAPKs are part of a phosphorylation
regulatory system composed of three sequentially activated kinases,
ERK1/2, p38 kinase and c-Jun amino-terminal kinases (JNKs)
(60). To elucidate the potential
molecular signaling pathways in HMJ-30-treated U-2 OS cells, the
levels of associated proteins in the PI3K/AKT and Ras/MAPK
signaling pathways were assessed using western blotting. Key
phosphorylated proteins in these signaling pathways were
reconfirmed via sandwich ELISA assay. HMJ-30 caused a decrease in
the protein levels of Ras, p-ERK1/2, p-p38, and p-JNK in U-2 OS
cells (Fig. 5A). Inhibitors of MEK
1 and MEK 2 (U0126) and p38 (SB203580) also reduced the protein
levels of p-p44/22 MAPK (ERK) and p-p38, respectively (Fig. 5B and C). HMJ-30 caused a decrease
in the protein levels of PI3K (p85) and p-AKT in U-2 OS cells
(Fig. 5D). Inhibitors of PI3K
(wortmannin) also reduced the protein expression of p-AKT (Fig. 5E).
 | Figure 5HMJ-30 inhibits the Ras/MAPK,
PI3K/AKT, and NF-κB signaling pathways in human osteogenic sarcoma
U-2 OS cells. (A) Protein levels of Ras, p-ERK, p-JNK and p-p38
were downregulated, as determined by western blotting. Expression
of the key proteins in MAPK signaling, (B) p-ERK (p-p44/42) and (C)
p-p38, were reconfirmed using ELISA assays. (D) The protein levels
of PI3K (p85) and p-AKT were downregulated, as determined by
western blot-ting. (E) The key protein in p-AKT was reconfirmed by
ELISA assay. (F) Protein levels of IKK, p-IκB and NF-κB (p65) were
downregulated, as determined by western blotting. (G) NF-κB binding
activity was determined using an electrophoretic mobility shift
assay. Inhibitors of p44/22 MAPK (U0126), p38 (SB203580), PI3K
(wortmannin) and NF-κB (PDTC), as positive controls, reduced
protein kinase levels. The data represents the mean ± standard
deviation of three experiments. ***P<0.001 vs.
control. HMJ-30,
6-fluoro-2-(3-fluorophenyl)-4-(cyanoanilino)quinazoline; MAPK,
mitogen-activated protein kinase; PI3K, phosphoinositide 3-kinase;
AKT, protein kinase B; NF-κB, nuclear factor-κB; p-,
phosphorylated; ERK, extracelullar signal-regulated kinase; JNK,
c-Jun amino-terminal kinase; PDTC, ammonium
pyrrolidinedithiocarbamate. |
The PI3K and AKT signaling pathway allows the
translocation of NF-κB to the nucleus to regulate gene
transcription. The accumulation of IκB, resulting from the decrease
of the phosphorylation of IκB by IKKs, prevents the translocation
of NF-κB and the activation of transcription by retaining NF-κB in
the cytoplasm (61). NF-κB serves
an important function in the modulation of the expression of
oncogenes, including MMP-2/9, cyclin D1, cell survival proteins and
vascular endothelial growth factor; the activities of which are
associated with the growth as well as the invasive and metastatic
propertoes of cancer cells (61,62).
HMJ-30 caused a decrease in protein levels of IKK and NF-κB (p65)
and an increase in the protein level of IκB, as confirmed by
western blotting (Fig. 5F). HMJ-30
caused a decrease in the protein levels of IKK and NF-κB (p65) and
an increase in the protein levels of IκB (Fig. 5F). Furthermore, HMJ-30 caused a
decrease in NF-κB activity, and the inhibitor of NF-κB, PDTC, also
reduced the activity of NF-κB, as determined by EMSA (Fig. 5G). These results suggested that
HMJ-30 inhibited PI3K/AKT, Ras/MAPK, and IκB/NF-κB signaling
pathways in U-2 OS cells.
Discussion
Several quinazoline derivatives have been developed
to act as potent and selective inhibitors of the growth factor
receptor tyrosine kinase (25).
The quinazoline ring has been reported to occupy the adenine
binding site of the tyrosine kinase receptor, and to inhibit its
phosphorylation. Anilinoquinazolines have also been reported to act
as potent therapeutic agents in invasive cancers to prevent
metastasis (27). Our group has
designed and synthesized several anilinoquinazolines in order to
inhibit tumor metastasis. One of these compounds, HMJ-30 (Fig. 1A), selectively targeted the ATP
binding site of IGF-1R and downregulated MAPKs, as well as PI3K/AKT
signaling. In order to gain further insights into HMJ-30 inhibiting
the IGF-1R signaling pathway in human osteosarcoma, the antitumor
effects in HMJ-30-treated U-2 OS cells were evaluated. The U-2 OS
cell line, with its spindle-like morphology (Fig. 4A), was established by Ponten and
Saksela from the osteosarcoma of a 15-year old Caucasian female
(63). In vitro studies
have demonstrated that U-2 OS cells grow via autocrine stimulation
by expressing IGF-IR and insulin-like growth
factor-II/mannose-6-phosphate (IGF-II/M6P) receptors, and
synthesizing and secreting IGF-like peptides (13). Initially, migration and invasion
were evaluated in HMJ-30-treated U-2 OS cells. HMJ-30 inhibited
cell migration and invasion in a concentration-dependent manner
(Fig. 2). These results are in
agreement with a previous study in vitro which revealed that
the downregulation of endogenous IGF-1R expression by
lentivirus-mediated RNAi reduces tumor invasion in osteosarcoma
cells (14). The present study
revealed that <40 μM HMJ-30 inhibited tumor migration and
invasion; whereas a higher concentration and an increased
incubation time induced toxicity in U-2 OS cells.
The degradation of basement membranes and the
proteolysis of the ECM has been reported to enable tumors to
migrate into adjacent tissues or transmigrate, limiting basement
membranes and extracellular matrices (4). MMPs are zinc-dependent endopeptidases
and serve a major function in ECM degradation and remodeling.
Previous studies have demonstrated that overexpression of MMPs,
particularly the gelatinases MMP-2 and MMP-9, is associated with
the invasive properties of several tumor cell lines (54,64).
The results of the present study demonstrated that HMJ-30 decreased
the protein activities and levels of MMP-2 and MMP-9 in U-2 OS
cells (Fig. 3).
Regulation of MMPs consists of three levels: i) gene
expression; ii) activation of the latent pro-enzymes; and iii)
inhibition by tissue inhibitor of metalloproteinases (TIMPs)
(4,54,60,66).
MMP gene expression at the transcriptional and post-transcriptional
levels is regulated through multiple signaling pathways, and is
directed by the protein kinases (60). Highly selective inhibitors of PI3K
(wortmannin), MEK (U0126), p38 MAPK (SB203580) and JNK (SP600125)
confirm the regulation of MMP gene expression by the PI3K/AKT and
Ras/MAPK pathways (60). The
transcription factor, NF-κB, has binding sites in the promoter of
MMPs genes, including MMP-2 and -9. PDTC, an inhibitor of NF-κB,
determined the modulation of MMP production (60,62).
In a recent study, IGF-1R signaling was reported to act as a
positive and negative regulator of MMP expression and function
(64). Based on previous studies
and present results, it would appear that HMJ-30 suppressed MMP-2
and MMP-9 expression through IGF-1R-mediated PI3K/AKT and Ras/MAPK
signaling pathways in U-2 OS cells. On the other hand, the
upregulation of TIMP-1 and TIMP-2 suggests that HMJ-30 may be
involved in post-translational regulation of MMP activity.
Normally, the expression of the majority of MMPs is low in tissue;
however, as it is required for remodeling ECM, it is induced by
several extracellular stimuli (60). MMPs in human cancers have functions
beyond ECM degradation. They induce cancer cells to produce
numerous growth factors, cytokines and chemokines which contribute
to the expression of MMPs by stromal cells, creating a favorable
environment for metastasis (53).
The present study indicated that the upregulation of TIMP-1 and
TIMP-2 by HMJ-30 may not only inhibit MMPs produced by U-2 OS
cells, but may also reduce the activity of stromal-derived
MMPs.
Epithelial cells form layers kept together by
specialized membrane structures which limit the movement of
epithelial cells to the two-dimensional space of the epithelial
plane. In contrast, mesenchymal cells only have focal contacts with
their neighbors without forming organized cell layers, which are
elongated and motile in a three-dimensional space, enriching MMPs
(66). During cancer progression,
EMT occurs in which epithelial cells lose their epithelial
characteristics and gain mesenchymal properties (23). These differentiated mesenchymal
cells easily spread into tissues surrounding the original tumor, as
well as travelling to other locations. Finally, the disseminated
tumor cells undergo a reverse EMT process, MET, recapitulating the
corresponding primary tumors at the site of metastasis to establish
a secondary tumor (23). Previous
studies have reported that specific inhibition of tumor invasion
and migration reactivates epithelial characteristics and lose
mesenchymal cell markers, resulting in MET in malignancies
(57–59). Pax3 has also been reported to
induce the formation of multi-layered condensed cell aggregates
with epithelial characteristics and regulated phenotypic
mesenchymal-epithelial interconversion in osteogenic Saos-2 cells
(67,68). However, few further discussions
concerning the association between metastasis and MET in
osteosarcoma cells have occurred. HMJ-30-treated U-2 OS cells
acquired a flattened, less elongated and cobblestone-like shape,
which is typical of an epithelial appearance, and underwent MET as
determined by an increase of epithelial markers and a reduction of
mesenchymal markers (Fig. 4).
E-cadherin is a key component of intercellular
junctions, and spans membranes to hook into E-cadherin molecules on
adjacent cells. β-catenin, an intracellular attachment protein,
binds to the intracellular domain of E-cadherin and links
E-cadherin to the intracellular cytoskeletons. The
E-cadherin/β-catenin complex therefore provides the major strength
underlying cell-cell associations, establishing cell polarity and
supporting the cellular structure. Loss of E-cadherin and junction
complex has been implicated in the gain of invasive potential by
cancer cells (69). In the present
study, HMJ-30 upregulated protein levels of E-cadherin and
β-catenin around the cytoplasm and membrane (Fig. 4B and C). One interpretation of
these results is that HMJ-30 caused tumor cell aggregation and
formed tight clusters through increasing cell-cell junctions to
limit the invasion and migration of these differentiated epithelial
cells from the primary site.
Several transcription factors, including Snail,
Slug, Twist, and zinc finger E-box binding homeobox 1 (Zeb1), are
crucial to EMT. NF-κB was recently demonstrated to be essential for
EMT and metastasis via binding to the Snail promoter and increasing
its activity (70). Furthermore,
inhibition of NF-κB prevented EMT in Ras-transformed epithelial
cells and caused a reversal of EMT in mesenchymal cells, suggesting
that the induction and maintenance of EMT critically depend on
NF-κB activity (71). Activation
of IGF-1R kinase was also reported to cause EMT and promote
metastasis in prostate and breast epithelial tumors through
PI3K/AKT and Ras/MAPK signaling, increasing the transcriptional
actives of Snail and Zeb1 (70,72).
In the present study, blockage of IGF-1R by HMJ-30 reduced NF-κB
activity and downregulated PI3K/AKT and Ras/MAPK pathway signaling,
resulting in MET in U-2 OS cells.
In conclusion, the present study has demonstrated
that HMJ-30 inhibits tumor invasiveness and induces
mesenchymal-epithelial transition via selective blockage of IGF-1R
signaling, involving PI3K/AKT and Ras/MAPK pathways in human
osteosarcoma U-2 OS cells. Furthermore, this occurred without
targeting IR, which indicated that HMJ-30 causes fewer toxic
side-effects compared with the conventional chemotherapy.
Downregulated protein levels and MMP-2 and MMP-9 activity, and
upregulated TIMP-1 and TIMP-2 protein levels indicated that HMJ-30
not only inhibited MMP gene expression but also acted through
post-translational regulation. HMJ-30 induced dramatic morphology
changes in U-2 OS cells and increased the expression of epithelial
markers and reduced mesenchymal markers, suggesting that HMJ-30
treatment inhibited invasiveness in tumor cells. The proposed
signal pathways underlying HMJ-30-induced IGF-1R blockage are
presented in Fig. 6. These results
provide important information regarding the potential molecular
mechanisms of the effect of HMJ-30 on osteosarcoma, and confirm
that HMJ-30 may be a candidate osteosarcoma drug in the future.
Acknowledgments
Not applicable.
Notes
[1]
Funding
The present study was supported by the Ministry of
Science and Technology, Taiwan (grant no. MOST
104-2320-B-039-004-MY3), and China Medical University Hospital
(grant no. DMR-106-122).
[2] Availability
of data and materials
The data sets generated during the study are
available from the corresponding author on reasonable request.
[3] Authors'
contributions
YJC, MJH and JSY conceived and designed the
experiments. YAJ, CCL and TLC performed the experiments. FJT, HM
and YNJ analyzed the data. YJC, MJH and JSY wrote and modified the
paper. All authors read and approved the final manuscript.
[4] Ethics
approval and consent to participate
Not applicable.
[5] Consent for
publication
Not applicable.
[6] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Arndt CA and Crist WM: Common
musculoskeletal tumors of childhood and adolescence. N Engl J Med.
341:342–352. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chou AJ and Gorlick R: Chemotherapy
resistance in osteosarcoma: Current challenges and future
directions. Expert Rev Anticancer Ther. 6:1075–1085. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
O'Day K and Gorlick R: Novel therapeutic
agents for osteosarcoma. Expert Rev Anticancer Ther. 9:511–523.
2009. View Article : Google Scholar
|
|
4
|
Wells A: Tumor invasion: Role of growth
factor-induced cell motility. Adv Cancer Res. 78:31–101. 2000.
View Article : Google Scholar
|
|
5
|
Duan Z, Choy E, Harmon D, Yang C, Ryu K,
Schwab J, Mankin H and Hornicek FJ: Insulin-like growth factor-I
receptor tyrosine kinase inhibitor cyclolignan picropodophyllin
inhibits proliferation and induces apoptosis in multidrug resistant
osteosarcoma cell lines. Mol Cancer Ther. 8:2122–2130. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liao CL, Lai KC, Huang AC, Yang JS, Lin
JJ, Wu SH, Gibson Wood W, Lin JG and Chung JG: Gallic acid inhibits
migration and invasion in human osteosarcoma U-2 OS cells through
suppressing the matrix metalloproteinase-2/-9, protein kinase B
(PKB) and PKC signaling pathways. Food Chem Toxicol. 50:1734–1740.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ullrich A, Gray A, Tam AW, Yang-Feng T,
Tsubokawa M, Collins C, Henzel W, Le Bon T, Kathuria S, Chen E, et
al: Insulin-like growth factor I receptor primary structure:
Comparison with insulin receptor suggests structural determinants
that define functional specificity. EMBO J. 5:2503–2512.
1986.PubMed/NCBI
|
|
8
|
Zwick E, Bange J and Ullrich A: Receptor
tyrosine kinase signalling as a target for cancer intervention
strategies. Endocr Relat Cancer. 8:161–173. 2001. View Article : Google Scholar
|
|
9
|
Rowlands MA, Gunnell D, Harris R, Vatten
LJ, Holly JM and Martin RM: Circulating insulin-like growth factor
peptides and prostate cancer risk: A systematic review and
meta-analysis. Int J Cancer. 124:2416–2429. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kurmasheva RT and Houghton PJ: IGF-I
mediated survival pathways in normal and malignant cells. Biochim
Biophys Acta. 1766:1–22. 2006.PubMed/NCBI
|
|
11
|
Conover CA: Insulin-like growth
factor-binding proteins and bone metabolism. Am J Physiol
Endocrinol Metab. 294:E10–E14. 2008. View Article : Google Scholar
|
|
12
|
Johnson LC: A general theory of bone
tumors. Bull N Y Acad Med. 29:164–171. 1953.PubMed/NCBI
|
|
13
|
Raile K, Höflich A, Kessler U, Yang Y,
Pfuender M, Blum WF, Kolb H, Schwarz HP and Kiess W: Human
osteosarcoma (U-2 OS) cells express both insulin-like growth
factor-I (IGF-I) receptors and insulin-like growth
factor-II/mannose-6-phosphate (IGF-II/M6P) receptors and synthesize
IGF-II: Autocrine growth stimulation by IGF-II via the IGF-I
receptor. J Cell Physiol. 159:531–541. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang YH, Wang ZX, Qiu Y, Xiong J, Chen YX,
Miao DS and De W: Lentivirus-mediated RNAi knockdown of
insulin-like growth factor-1 receptor inhibits growth, reduces
invasion, and enhances radiosensitivity in human osteosarcoma
cells. Mol Cell Biochem. 327:257–266. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Burrow S, Andrulis IL, Pollak M and Bell
RS: Expression of insulin-like growth factor receptor, IGF-1, and
IGF-2 in primary and metastatic osteosarcoma. J Surg Oncol.
69:21–27. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Benini S, Baldini N, Manara MC, Chano T,
Serra M, Rizzi S, Lollini PL, Picci P and Scotlandi K: Redundancy
of autocrine loops in human osteosarcoma cells. Int J Cancer.
80:581–588. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kappel CC, Velez-Yanguas MC, Hirschfeld S
and Helman LJ: Human osteosarcoma cell lines are dependent on
insulin-like growth factor I for in vitro growth. Cancer Res.
54:2803–2807. 1994.PubMed/NCBI
|
|
18
|
Pinski J, Schally AV, Halmos G, Szepeshazi
K and Groot K: Somatostatin analog RC-160 inhibits the growth of
human osteosarcomas in nude mice. Int J Cancer. 65:870–874. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mansky PJ, Liewehr DJ, Steinberg SM,
Chrousos GP, Avila NA, Long L, Bernstein D, Mackall CL, Hawkins DS
and Helman LJ: Treatment of metastatic osteosarcoma with the
somatostatin analog OncoLar: Significant reduction of insulin-like
growth factor-1 serum levels. J Pediatr Hematol Oncol. 24:440–446.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yee D: Targeting insulin-like growth
factor pathways. Br J Cancer. 94:465–468. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Boyer B, Vallés AM and Edme N: Induction
and regulation of epithelial-mesenchymal transitions. Biochem
Pharmacol. 60:1091–1099. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Thiery JP and Sleeman JP: Complex networks
orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell
Biol. 7:131–142. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hugo H, Ackland ML, Blick T, Lawrence MG,
Clements JA, Williams ED and Thompson EW: Epithelial - mesenchymal
and mesenchymal - epithelial transitions in carcinoma progression.
J Cell Physiol. 213:374–383. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nakaya Y, Kuroda S, Katagiri YT, Kaibuchi
K and Takahashi Y: Mesenchymal-epithelial transition during somitic
segmentation is regulated by differential roles of Cdc42 and Rac1.
Dev Cell. 7:425–438. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Michael JP: Quinoline, quinazoline and
acridone alkaloids. Nat Prod Rep. 25:166–187. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hwang SH, Rait A, Pirollo KF, Zhou Q,
Yenugonda VM, Chinigo GM, Brown ML and Chang EH: Tumor-targeting
nanodelivery enhances the anticancer activity of a novel
quinazolinone analogue. Mol Cancer Ther. 7:559–568. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Plé PA, Green TP, Hennequin LF, Curwen J,
Fennell M, Allen J, Lambert-Van Der, Brempt C and Costello G:
Discovery of a new class of anilinoquinazoline inhibitors with high
affinity and specificity for the tyrosine kinase domain of c-Src. J
Med Chem. 47:871–887. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hennequin LF, Stokes ES, Thomas AP,
Johnstone C, Plé PA, Ogilvie DJ, Dukes M, Wedge SR, Kendrew J and
Curwen JO: Novel 4-anilinoquinazolines with C-7 basic side chains:
Design and structure activity relationship of a series of potent,
orally active, VEGF receptor tyrosine kinase inhibitors. J Med
Chem. 45:1300–1312. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhou Y, Li S, Hu YP, Wang J, Hauser J,
Conway AN, Vinci MA, Humphrey L, Zborowska E, Willson JK, et al:
Blockade of EGFR and ErbB2 by the novel dual EGFR and ErbB2
tyrosine kinase inhibitor GW572016 sensitizes human colon carcinoma
GEO cells to apoptosis. Cancer Res. 66:404–411. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Al-Obaid AM, Abdel-Hamide SG, El-Kashef
HA, Abdel-Aziz AA, El-Azab AS, Al-Khamees HA and El-Subbagh HI:
Substituted quinazolines, part 3. Synthesis, in vitro antitumor
activity and molecular modeling study of certain
2-thieno-4(3H)-quinazolinone analogs. Eur J Med Chem. 44:2379–2391.
2009. View Article : Google Scholar
|
|
31
|
Ciardiello F, Caputo R, Bianco R, Damiano
V, Pomatico G, De Placido S, Bianco AR and Tortora G: Antitumor
effect and potentiation of cytotoxic drugs activity in human cancer
cells by ZD-1839 (Iressa), an epidermal growth factor
receptor-selective tyrosine kinase inhibitor. Clin Cancer Res.
6:2053–2063. 2000.PubMed/NCBI
|
|
32
|
Lu CC, Chen HP, Chiang JH, Jin YA, Kuo SC,
Wu TS, Hour MJ, Yang JS and Chiu YJ: Quinazoline analog HMJ-30
inhibits angiogenesis: Involvement of endothelial cell apoptosis
through ROS-JNK-mediated death receptor 5 signaling. Oncol Rep.
32:597–606. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chiu YJ, Hour MJ, Lu CC, Chung JG, Kuo SC,
Huang WW, Chen HJ, Jin YA and Yang JS: Novel quinazoline HMJ-30
induces U-2 OS human osteogenic sarcoma cell apoptosis through
induction of oxidative stress and up-regulation of ATM/p53
signaling pathway. J Orthop Res. 29:1448–1456. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tsai YF, Huang CW, Chiang JH, Tsai FJ, Hsu
YM, Lu CC, Hsiao CY and Yang JS: Gadolinium chloride elicits
apoptosis in human osteosarcoma U-2 OS cells through extrinsic
signaling, intrinsic pathway and endoplasmic reticulum stress.
Oncol Rep. 36:3421–3426. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen HJ, Lin CM, Lee CY, Shih NC, Peng SF,
Tsuzuki M, Amagaya S, Huang WW and Yang JS: Kaempferol suppresses
cell metastasis via inhibition of the ERK-38-JNK and AP-1 signaling
pathways in U-2 OS human osteosarcoma cells. Oncol Rep. 30:925–932.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chueh FS, Chen YY, Huang AC, Ho HC, Liao
CL, Yang JS, Kuo CL and Chung JG: Bufalin-inhibited migration and
invasion in human osteosarcoma U-2 OS cells is carried out by
suppression of the matrix metalloproteinase-2, ERK, and JNK
signaling pathways. Environ Toxicol. 29:21–29. 2014. View Article : Google Scholar
|
|
37
|
Ma YS, Weng SW, Lin MW, Lu CC, Chiang JH,
Yang JS, Lai KC, Lin JP, Tang NY, Lin JG, et al: Antitumor effects
of emodin on LS1034 human colon cancer cells in vitro and in vivo:
Roles of apoptotic cell death and LS1034 tumor xenografts model.
Food Chem Toxicol. 50:1271–1278. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wu SH, Hang LW, Yang JS, Chen HY, Lin HY,
Chiang JH, Lu CC, Yang JL, Lai TY, Ko YC, et al: Curcumin induces
apoptosis in human non-small cell lung cancer NCI-H460 cells
through ER stress and caspase cascade- and mitochondria-dependent
pathways. Anticancer Res. 30:2125–2133. 2010.PubMed/NCBI
|
|
39
|
Lee MR, Lin C, Lu CC, Kuo SC, Tsao JW,
Juan YN, Chiu HY, Lee FY, Yang JS and Tsai FJ: YC-1 induces
G0/G1phase arrest and mitochondria-dependent apoptosis in
cisplatin-resistant human oral cancer CAR cells. Biomedicine
(Taipei). 7:122017. View Article : Google Scholar
|
|
40
|
Yang JS, Lin CA, Lu CC, Wen YF, Tsai FJ
and Tsai SC: Carboxamide analog ITR-284 evokes apoptosis and
inhibits migration ability in human lung adenocarcinoma A549 cells.
Oncol Rep. 37:1786–1792. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tsai SC, Tsai MH, Chiu CF, Lu CC, Kuo SC,
Chang NW and Yang JS: AMPK-dependent signaling modulates the
suppression of invasion and migration by fenofibrate in CAL 27 oral
cancer cells through NF-κB pathway. Environ Toxicol. 31:866–876.
2016. View Article : Google Scholar
|
|
42
|
Hsu SC, Yang JS, Kuo CL, Lo C, Lin JP,
Hsia TC, Lin JJ, Lai KC, Kuo HM, Huang LJ, et al: Novel quinolone
CHM-1 induces apoptosis and inhibits metastasis in a human
osterogenic sarcoma cell line. J Orthop Res. 27:1637–1644. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lai KC, Huang AC, Hsu SC, Kuo CL, Yang JS,
Wu SH and Chung JG: Benzyl isothiocyanate (BITC) inhibits migration
and invasion of human colon cancer HT29 cells by inhibiting matrix
metalloproteinase-2/-9 and urokinase plasminogen (uPA) through PKC
and MAPK signaling pathway. J Agric Food Chem. 58:2935–2942. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chen HJ, Jiang YL, Lin CM, Tsai SC, Peng
SF, Fushiya S, Hour MJ and Yang JS: Dual inhibition of EGFR and
c-Met kinase activation by MJ-56 reduces metastasis of HT29 human
colorectal cancer cells. Int J Oncol. 43:141–150. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Han J, Lee JD, Bibbs L and Ulevitch RJ: A
MAP kinase targeted by endotoxin and hyperosmolarity in mammalian
cells. Science. 265:808–811. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lai WW, Hsu SC, Chueh FS, Chen YY, Yang
JS, Lin JP, Lien JC, Tsai CH and Chung JG: Quercetin inhibits
migration and invasion of SAS human oral cancer cells through
inhibition of NF-κB and matrix metalloproteinase-2/-9 signaling
pathways. Anticancer Res. 33:1941–1950. 2013.PubMed/NCBI
|
|
47
|
Yu CS, Huang AC, Yang JS, Yu CC, Lin CC,
Chung HK, Huang YP, Chueh FS and Chung JG: Safrole induces G0/G1
phase arrest via inhibition of cyclin E and provokes apoptosis
through endoplasmic reticulum stress and mitochondrion-dependent
pathways in human leukemia HL-60 cells. Anticancer Res.
32:1671–1679. 2012.PubMed/NCBI
|
|
48
|
Huang WW, Chiu YJ, Fan MJ, Lu HF, Yeh HF,
Li KH, Chen PY, Chung JG and Yang JS: Kaempferol induced apoptosis
via endoplasmic reticulum stress and mitochondria-dependent pathway
in human osteosarcoma U-2 OS cells. Mol Nutr Food Res.
54:1585–1595. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hour MJ, Tsai SC, Wu HC, Lin MW, Chung JG,
Wu JB, Chiang JH, Tsuzuki M and Yang JS: Antitumor effects of the
novel quinazolinone MJ-33: Inhibition of metastasis through the
MAPK, AKT, NF-κB and AP-1 signaling pathways in DU145 human
prostate cancer cells. Int J Oncol. 41:1513–1519. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Weroha SJ and Haluska P: IGF-1 receptor
inhibitors in clinical trials - early lessons. J Mammary Gland Biol
Neoplasia. 13:471–483. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hofmann F and García-Echeverría C:
Blocking the insulin-like growth factor-I receptor as a strategy
for targeting cancer. Drug Discov Today. 10:1041–1047. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Girnita A, Girnita L, del Prete F,
Bartolazzi A, Larsson O and Axelson M: Cyclolignans as inhibitors
of the insulin-like growth factor-1 receptor and malignant cell
growth. Cancer Res. 64:236–242. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sengupta N and MacDonald TT: The role of
matrix metalloproteinases in stromal/epithelial interactions in the
gut. Physiology (Bethesda). 22:401–409. 2007.
|
|
54
|
Björklund M and Koivunen E:
Gelatinase-mediated migration and invasion of cancer cells. Biochim
Biophys Acta. 1755:37–69. 2005.PubMed/NCBI
|
|
55
|
Brew K, Dinakarpandian D and Nagase H:
Tissue inhibitors of metalloproteinases: Evolution, structure and
function. Biochim Biophys Acta. 1477:267–283. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wojtowicz-Praga SM, Dickson RB and Hawkins
MJ: Matrix metalloproteinase inhibitors. Invest New Drugs.
15:61–75. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Hong KO, Kim JH, Hong JS, Yoon HJ, Lee JI,
Hong SP and Hong SD: Inhibition of Akt activity induces the
mesenchymal-to-epithelial reverting transition with restoring
E-cadherin expression in KB and KOSCC-25B oral squamous cell
carcinoma cells. J Exp Clin Cancer Res. 28:282009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Jo M, Lester RD, Montel V, Eastman B,
Takimoto S and Gonias SL: Reversibility of epithelial-mesenchymal
transition (EMT) induced in breast cancer cells by activation of
urokinase receptor-dependent cell signaling. J Biol Chem.
284:22825–22833. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Funasaka T, Hu H, Yanagawa T, Hogan V and
Raz A: Down-regulation of phosphoglucose isomerase/autocrine
motility factor results in mesenchymal-to-epithelial transition of
human lung fibrosarcoma cells. Cancer Res. 67:4236–4243. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Reuben PM and Cheung HS: Regulation of
matrix metal-loproteinase (MMP) gene expression by protein kinases.
Front Biosci. 11:1199–1215. 2006. View
Article : Google Scholar
|
|
61
|
Sliva D: Signaling pathways responsible
for cancer cell invasion as targets for cancer therapy. Curr Cancer
Drug Targets. 4:327–336. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Clark IM, Swingler TE, Sampieri CL and
Edwards DR: The regulation of matrix metalloproteinases and their
inhibitors. Int J Biochem Cell Biol. 40:1362–1378. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Pontén J and Saksela E: Two established in
vitro cell lines from human mesenchymal tumours. Int J Cancer.
2:434–447. 1967. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Li S, Zhang D, Yang L, Burnier JV, Wang N,
Lin R, Lee ER, Glazer RI and Brodt P: The IGF-I receptor can alter
the matrix metalloproteinase repertoire of tumor cells through
transcriptional regulation of PKC-{alpha}. Mol Endocrinol.
23:2013–2025. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Yan C and Boyd DD: Regulation of matrix
metalloproteinase gene expression. J Cell Physiol. 211:19–26. 2007.
View Article : Google Scholar
|
|
66
|
Berx G, Raspé E, Christofori G, Thiery JP
and Sleeman JP: Pre-EMTing metastasis? Recapitulation of
morphogenetic processes in cancer. Clin Exp Metastasis. 24:587–597.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Wiggan O, Fadel MP and Hamel PA: Pax3
induces cell aggregation and regulates phenotypic
mesenchymal-epithelial interconversion. J Cell Sci. 115:517–529.
2002.PubMed/NCBI
|
|
68
|
Wiggan O, Shaw AE and Bamburg JR:
Essential requirement for Rho family GTPase signaling in Pax3
induced mesenchymal-epithelial transition. Cell Signal.
18:1501–1514. 2006. View Article : Google Scholar
|
|
69
|
Hsu YM, Chen YF, Chou CY, Tang MJ, Chen
JH, Wilkins RJ, Ellory JC and Shen MR: KCl cotransporter-3
down-regulates E-cadherin/beta-catenin complex to promote
epithelial-mesenchymal transition. Cancer Res. 67:11064–11073.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Kim HJ, Litzenburger BC, Cui X, Delgado
DA, Grabiner BC, Lin X, Lewis MT, Gottardis MM, Wong TW, Attar RM,
et al: Constitutively active type I insulin-like growth factor
receptor causes transformation and xenograft growth of immortalized
mammary epithelial cells and is accompanied by an
epithelial-to-mesenchymal transition mediated by NF-kappaB and
snail. Mol Cell Biol. 27:3165–3175. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Huber MA, Azoitei N, Baumann B, Grünert S,
Sommer A, Pehamberger H, Kraut N, Beug H and Wirth T: NF-kappaB is
essential for epithelial-mesenchymal transition and metastasis in a
model of breast cancer progression. J Clin Invest. 114:569–581.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Graham TR, Zhau HE, Odero-Marah VA,
Osunkoya AO, Kimbro KS, Tighiouart M, Liu T, Simons JW and O'Regan
RM: Insulin-like growth factor-I-dependent up-regulation of ZEB1
drives epithelial-to-mesenchymal transition in human prostate
cancer cells. Cancer Res. 68:2479–2488. 2008. View Article : Google Scholar : PubMed/NCBI
|















