Introduction
Glioblastoma (GBM) is the most common primary adult
brain tumor, and it continues to have a dismal median survival of
15 months despite maximal safe surgical resection with radiotherapy
and chemotherapy (1-3). A deeper understanding of the
mechanisms driving the aggressiveness of and high levels of
therapeutic resistance in GBM is needed in order to develop novel
effective therapies. GBM is well characterized as a pathway-driven
disease (4,5) with alterations in receptor tyrosine
kinases (RTKs), activating mutations in PI3CA (p110) and PIK3R1
(p85) or the loss of phosphatase and tensin homolog (PTEN)
occurring in up to 88% of GBM cases (6), which promotes the conversion of
phosphatidylinositol (4,5)-bisphosphate [Ptdlns(4,5)P2] into phosphatidylinositol
(3,4,5)-trisphosphate [Ptdlns(3,4,5)P3]. The accumulation of
Ptdlns(3,4,5)P3 promotes the localization
of proteins containing pleckstrin homology (PH) domains to the
plasma membrane, enhancing downstream signaling, such as the
activation of AKT and mammalian target of rapamycin complex
(mTORC), promoting cancer growth and therapeutic resistance
(7), as well as oncogenic
transformation (8).
Ptdlns(4,5)P2 has important functions in
cell migration (9) and calcium
regulation (10).
Myristoylated alanine-rich C-kinase substrate
(MARCKS) is an intrinsically unstructured protein highly expressed
in the brain that was originally thought to be an 80 kDa protein by
SDS gel electrophoresis due to its unique amino acid composition
(11), but later shown to be 31.75
kDa by cDNA sequencing and mass spectrometry (10-15).
The MARCKS effector domain (ED) is known to electrostatically
sequester Ptdlns(4,5)P2 at the plasma membrane,
blocking its cleavage by phospholipase C (PLC) or phosphorylation
by phosphatidylinositide 3-kinase (PI3K) (13,16)
and crosslink filamentous actin (F-actin) (10,14,17,18).
The ability of MARCKS to sequester Ptdlns(4,5)P2, renders it potentially as
a potent tumor suppressor when membrane-bound due to the frequency
of PI3K hyperactivation and the loss of PTEN in GBM. MARCKS has two
domains that promote plasma membrane binding, an N-terminal
myristoylation moiety and an electrostatically charged ED.
Myristoylation alone has been shown to be insufficient for membrane
binding, instead, requiring contributions from its cationic ED
(13). MARCKS membrane binding is
electrostatically maintained through the attraction of the
positively charged lysine residues of the ED (+13) to negatively
charged phospholipid head groups, such as Ptdlns(4,5)P2 (19), and by the embedding of
phenylalanine residues of the ED into the acyl chain regions of the
phospholipids in the membrane (20). MARCKS plasma membrane and actin
binding are regulated through two major mechanisms: i) The
phosphorylation of up to four serine residues in the ED by PKC
(21) or ROCK kinases (22); and ii) the binding of the ED by
calcium (Ca2+)/calmodulin(CaM) (16,17).
These events, however, are mutually exclusive, allowing competitive
interactions to occur at MARCKS ED, enabling ‘crosstalk’ across
distinct signaling pathways (13).
MARCKS crosslinking of F-actin at the plasma membrane is lost
either with ED phosphorylation or Ca2+/CaM binding
(14,17,18).
In addition to Ptdlns(4,5)P2 sequestration and F-actin
crosslinking, MARCKS ED binds phosphatidylserine (PS) (23,24),
and functions as a nuclear localization sequence (NLS) (25). In cancer, MARCKS expression has
been associated with both tumor-suppressing and tumor-promoting
phenotypes (26-29); however, its inconsistent role in
cancer progression has been attributed to a lack of information on
ED phosphorylation status until more recent studies (10,14,30-32).
In GBM, Micallef et al previously demonstrated that the
epidermal growth factor receptor variant III (EGFR-VIII) invasive
phenotype was driven in part by the phosphorylation of MARCKS ED
(32). Additionally, Jarboe et
al demonstrated that the knockdown of MARCKS in GBM promoted
cell proliferation and radiation resistance through upregulations
in non-homologous end joining (NHEJ) DNA repair mechanisms, and
that patients with a high MARCKS expression, particularly in MGMT
unmethylated GBM tumors, had substantial survival benefits
(33). Since MARCKS itself is not
mutated in GBM (34), it is
suggested that primarily epigenetic, post-transcriptional or
post-translational modifications will overcome the MARCKS
tumor-suppressing effects.
In this study, we further examine the hypothesis
that MARCKS functions as a tumor suppressor in GBM, by
overexpressing MARCKS and investigating its effects on growth
suppression and radiation sensitivity. We hypothesized that the
unphosphorylated ED would have growth-suppressing and
radiation-sensitizing effects, while ED phosphorylation would block
these tumor-suppressing effects.
Materials and methods
Cells and cell culture
U87 and U373 glioblastoma lines were originally
acquired from the University of Uppsala (Uppsala, Sweden), and
293FT cells were acquired from ATCC (Manassas, VA, USA). All cell
lines were cultured as previously described in Dulbecco’s modified
Eagle’s medium with 10% fetal bovine serum and 1%
penicillin-streptomycin at 37°C and 5% CO2 (33). All tetracycline inductions were
accomplished at 2 µg/ml doxycycline in complete DMEM.
MARCKS plasmid production
U87 cells were engineered to overexpress MARCKS or
the MARCKS ED mutants in a tetracycline-dependent manner as
previously described (25). Other
ED mutants were similarly constructed. Concisely, the ViraPower
HiPerform T-REx Gateway Expression System (cat. no. A11141) and the
pENTR221 entry vector containing the wild-type (WT) sequence was
purchased from Thermo Fisher Scientific (Waltham, MA, USA). The
pENTR221-MARCKS vector was cloned into the pLenti6.3/TO/V5-DEST
destination vector. Mutant MARCKS ED constructs were synthesized by
and cloned into the pUC57 vector by GenScript (Piscataway, NJ,
USA). Fragments from these plasmids with the mutations were cloned
into the pLenti6.3/TO/V5-MARCKS-WT using restriction sites and
standard protocols to generate MARCKS mutant lentiviral plasmids
containing blasticidin resistance and a V-5 epitope tag. An empty
vector control plasmid (CTL) was also generated.
Lentiviral particle production
Lentiviral particles were produced as previously
described (33). Concisely,
lentivirus was generated by co-transfection of 293FT cells with an
appropriate amount of MARCKS pLenti6.3/TO/V5 plasmid, pCMV-VSV-G
envelope plasmid (Addgene plasmid 8454) and psPAX2 packaging
plasmid (Addgene plasmid 12260) (both from Addgene, Watertown, MA,
USA) with Lipofectamine 2000 (cat. no. 11668) in Opti-MEM (cat. no.
11058) (both from Thermo Fisher Scientific). The medium was changed
the following morning, and enriched viral medium was collected 24 h
later, filtered through a 0.45-µm filter, aliquoted and
stored at -80°C. Lentivirus was quantified using QuickTiter p24
ELISA (Cell Biolabs, Inc., San Diego, CA, USA).
Stable cell line selection and
validation
U87 cells were first transduced with p24 quantified
tetracycline-repressor (Tet-R) packaged lentiviral particles along
with 8 µg/ml polybrene as previously described (25). A total of 500 µg/ml
Geneticin (G418; Life Technologies/Thermo Fisher Scientific) was
used to select for Tet-R-positive cells. Tet-R-positive cells were
subsequently transduced with similar amounts of p24 quantified CTL,
WT+, NP, PP or ΔED lentiviral particles. Subsequently, 1
µg/ml blasticidin was used to select successfully transduced
cells. Robust tetracycline-dependent MARCKS expression was
validated by western blot analysis following a 72-h induction with
2 µg/ml of doxycycline (Life Technologies/Thermo Fisher
Scientific) or the phosphate-buffered saline vehicle control.
MARCKS mutations were additionally validated by PCR amplification
using CMV forward primer (5′-CGCAAATGGGCGGTAGGCGTG-3′) and V5
reverse primer (5′-ACCGAGGAGAGGGTTAGGGAT-3′) coupled and sequenced
using Sanger sequencing of an internal MARCKS forward primer
(5′-GAACGGACAGGAGGATGG-3′) and V5 reverse primer
(5′-ACCGAGGAGAGGGTTAGGGAT-3′).
Western blot analysis and antibodies
Western blot analysis was performed as previously
described (35). Briefly, chilled
mammalian protein extraction reagent (MPER) lysis buffer (cat. no.
78501; Pierce/Thermo Fisher Scientific, Rockford, IL, USA) was
supplemented with protease (#P8340) and phosphatase inhibitors
(P0044 and P5726) (both from Sigma-Aldrich, St. Louis, MO, USA)
before lysing the cells for 30 min on ice. The samples were
subsequently centrifuged at 12,000 × g for 10 min at 4°C, and the
supernatant was collected and quantified using the Pierce BCA
protein assay kit. Samples were separated by electrophoresis
through a 10% SDS-polyacrylamide gel (SDS-PAGE) and transferred
onto a PVDF membrane (Immobilon, Emdmilipore, Burlington, MA, USA).
The blots were blocked in 5% BSA for 1 h and probed with the
following antibodies at 4°C overnight with gentle rocking using
manufacturer recommended dilutions: V5-HRP (P/N 46-0708;
Invitrogen/Thermo Fisher Scientific), MARCKS anti-rabbit (ab52616),
MARCKS anti-mouse (ab55451) (both from Abcam, Cambridge, MA, USA),
phosphorylated (p-)histone H2AX S139 (9718S), p-Akt (Ser473; D9E;
#4060), p-Akt (Thr308; C31E5E; #2965), Akt (C67E7; #4691), PKCα
(#2056) (all from Cell Signaling Technology, Danvers, MA, USA),
rabbit IgG control (20304E; Imgenex/Novus Biologicals, Centennial,
CO, USA) and Actin (sc-1616-R), lamin A/C (sc-7292), α-tubulin
(sc-53646) (all from Santa Cruz Biotechnology, Santa Cruz, CA,
USA). The blots were treated with secondary HRP antibody at 1:5,000
for 1 h at room temperature with gentle rocking, and detected with
enhanced chemiluminescence (ECL) using Western Lighting-Plus ECL
substrate (PerkinElmer, Inc., Waltham, MA, USA) and blue X-ray
film. Densitometry was performed using ImageJ software with 8-bit
images and normalized to the loading control.
Isolation of nuclear and cytoplasmic
fractionations
Plate and doxycycline induce for 72 h a sufficient
number of cells to have approximately 10 million cells in a pellet
following collection. The modified nuclear extraction protocol
(#40410; Active Motif, Carlsbad, CA, USA) was as follows: The cells
were collected by removing the media and washing with 1X ice-cold
PBS. This was followed by the addition of 1.5 ml cold PBS and 1 ml
trypsin per plate until the cells lifted. The cells were then
transferred to a 15 ml centrifuge tube and spun for 5 min at 400 ×
g at 4°C. The supernatant discarded, and the cells were then rinsed
once more with 2 ml ice-cold PBS and spun for 5 min at 400 × g at
4°C and the supernatant was removed. The pellet was resuspended in
1 ml of ice-cold 1X hypotonic buffer, with gentle pipetting up and
down and transferring to a chilled 1.5 ml micro-centrifuge tube and
incubation on ice for 30 min. Subsequently, 50 µl detergent
was added and the mixture was vortexed for 10 sec at the highest
setting, and then spun for 2 min at 14,000 × g at 4°C. The
supernatant was then transferred (cytoplasmic fraction) into a new
chilled micro-centrifuge tube and frozen at -20°C for western blot
analysis. The remaining fluid was aspirated until only the pellet
remained. The pellet was then rinsed with 1 ml 1X PBS and spun down
for 2 min at 14,000 × g at 4°C to aspirate off the supernatant.
Nuclear extraction was continued using 50 µl of complete
MPER lysis buffer supplemented with protease (#P8340) and
phosphatase inhibitors (P0044 and P5726) (both from Sigma-Aldrich)
for 30 min on ice. The mixture was then spun for 2 min at 14,000 ×
g at 4°C, and the supernatant was collected and frozen at -20°C for
western blot analysis (nuclear fraction). Protein determination and
probing was performed for western blot analysis using the
above-mentioned protocol.
Cell proliferation assay
A total of 5,000 cells per well (n=12) were counted
using a hemocytometer and plated for each of the validated MARCKS
mutant lines into black 96-well plates containing 100 µl of
DMEM with 10% FBS and 2 µg/ml of the doxycycline
(doxycycline medium). The U87 ATP levels were measured using a
PerkinElmer ATPlite Luminescence Assay System (PerkinElmer,
Waltham, MA, USA) as per the manufacturer’s instructions at 5-7
days after plating. U373 cell viability was determined using same
plating conditions (5,000 cells/well 100 µl), however, using
a CellTiter Glo (Promega, Madison, WI, USA) kit as per the
manufacturer’s protocol (n=4). Luminescence was determined on a
BioTek H1 Hybrid Synergy (BioTek, Winooski, VT, USA).
Colony formation and clonogenic survival
assay
The clonogenic assay was performed as previously
described (33). In brief,
one-step clonogenic fixation and staining solution were made by
combining 750 ml of deionized water with 250 ml of 25%
glutaraldehyde (G6257-1L; Sigma-Aldrich) and 5 g of crystal violet
(C581-100; Thermo Fisher Scientific) in a 1 liter bottle and mixing
at room temperature until the mixture was dissolved. Cells were
then doxycycline-induced for 72 h before counting using a
hemocytometer and diluting to a defined concentration.
Pre-determined cell numbers were plated in 60-mm dishes with a 4 ml
total volume and allowed to attach overnight before irradiating in
a single fraction at indicated doses for clonogenic assay (colony
formation assay does not receive irradiation). Fourteen days after
plating, cells were fixed and stained with the clonogenic staining
solution for 30 min at room temperature, before gently rinsing
plates with cold tap water and drying upside-down overnight.
Colonies were counted at ×45 magnification using a dissecting
microscope (Stereomaster/Thermo Fisher Scientific, Pittsburgh, PA,
USA) and were determined in the presence of >50 cells. The
surviving fraction (SF) was calculated by using the following
equation: (number of colonies formed/number of cells
plated)/(number of colonies from the sham-irradiated group/number
of cells plated). Results are plotted in a semi-logarithmic format
using GraphPad Prism 7.04 and standard error of the mean (SEM)
error bars. Dose enhancement ratio=(dose (Gy) for control
(CTL)/dose for ED mutant) at SF=0.2.
Immunofluorescence staining, and
quantification of fluorescence intensity and localization on the
image cytometer Xcyto10
In total, 50,000 cells were plated in a 24-well
plate containing 12 mm poly-D-Lysine coated round coverslips and
500 µl doxycycline-containing medium. Cells were induced for
72 h before the medium was removed. The cells were then rinsed with
1 ml room temperature PBS and fixed with 500 µl of 4%
paraformaldehyde for 12 min at room temperature. The cells were
then permeabilized with 0.1% Triton X-100 PBS for 20 min and
blocked in 5% BSA, 1% goat serum PBS for 40 min at room
temperature. Subsequently, 250 µl of 1:250 primary antibody
in 0.5% BSA was added per coverslip and incubated overnight at 4°C.
Coverslips were rinsed 3 times for 5 min with 500 µl PBS
before a 2-h room temperature incubation in the dark with 1:1,000
AlexaFluor 546 anti-rabbit IgG secondary antibody (A11010) and 1X
phalloidin-FITC (F432) (both from Invitrogen/Thermo Fisher
Scientific) dye in 0.5% BSA PBS. The cells were then rinsed with
500 µl PBS 3 times for 5 min before co-staining with 1:1,000
BlueMask-1 (ChemoMetec, Allerod, Denmark) and 1:250 DAPI (2
µg/ml) at for 30 min at room temperature. Coverslips were
rinsed in 500 µl 1X PBS for 5 min before mounting on Xcyto
2-Sample slides (ChemoMetec) with ProLong Glass Antifade mountant
with DAPI (Thermo Fisher Scientific) overnight in the dark. Slides
were analyzed on the image cytometer Xcyto10 (ChemoMetec) at ×20
magnification with excitation/filter sets AF546 (LED535; 582-636),
AF488 (LED488; 513-555), MASK (LED405; 430-475) DAPI (LED405;
573-613) for high-resolution images and the quantification of
fluorescent intensities and localization. Similarity scores were
calculated using XcytoView (ChemoMetec) and represent the
log-transformed Pearson’s correlation coefficients between two
channels. Similarity scores between MARCKS and DNA were used to
determine MARCKS relative nuclear localization, and similarity
scores between MARCKS and phalloidin were used to compare MARCKS
and F-actin association.
Quantification of γH2AX foci
formation
Cells were adhered to 12 mm poly-D-Lysine-coated
round coverslips (REF354086; Corning, Inc., Corning, NY, USA) in
500 µl medium containing doxycycline for 72 h prior to 8 Gy
irradiation or 0 Gy (Sham). Cells were fixed at indicated
time-points with ice-cold methanol for 12 min at -20°C and blocking
with 5% BSA 1% goat serum for 40 min. 1:400 Histone H2AX S139
(9718S; Cell Signaling Technology) primary and 1:1,000 AlexaFluor
488 (ab150077; Abcam) secondary antibodies were used to stain for
γH2AX and coverslips were subsequently stained with DAPI and
BlueMask-1. Coverslips were mounted slides using with ProLong
diamond anti-fade mountant. Cells were imaged on EVOS FL (Thermo
Fisher Scientific) microscope at ×20 magnification, with 4 images
per time-point collected and were scored by blinded observers.
Positive events were defined as ≥10 foci per cell, and the
percentage of positive cells per field was graphed using Prism
software. The mean nuclear intensity of U373 was acquired using
Xcyto10 at a 20X resolution with fluorescent intensities measured
using XcytoView. The graph and statistics were generated using
GraphPad 7.04 software with error bars in SEM and Log-rank
(Mantel-cox) test used to generate the P-values.
Equipment and settings
The images shown in Figs. 1, S1
and S2 were acquired using a Xcyto10 Image cytometer with a 20X
objective lenses on 2 sample slides as follows: Fig. 1 and S1: MARCKS-AF546 (400 msec, LED535;
582-636), phal-loidin-AF488 (400 msec, LED488; 513-555), DAPI (400
msec, LED405; 573-613); Fig. S2:
MARCKS-AF546 (800 msec, LED535; 582-636), phalloidin-AF488 (800
msec, LED488; 513-555), DAPI (400 msec, LED405; 573-613). Images
were acquired from XcytoView screen captures using linear scale
display properties as follows: Figs.
1 and S1: TMARCKS (Min 142,
Max 2207), phalloidin (Min 1874, Max 89459); DAPI (Min 1142 Max
16188)]; Fig. S2: TMARCKS (Min
44, Max 5441), phalloidin (Min 868, Max 84947); DAPI (Min 1987 Max
1524)]. The colony formation images shown in Fig. 3C were acquired using a 12 MP camera
(Iphone7), and the colony images shown in Fig. 3D were acquired under a dissecting
microscope (×45 magnification) using a 12 MP camera (iPhone X).
Blots were acquired on an Epson Perfection scanner.
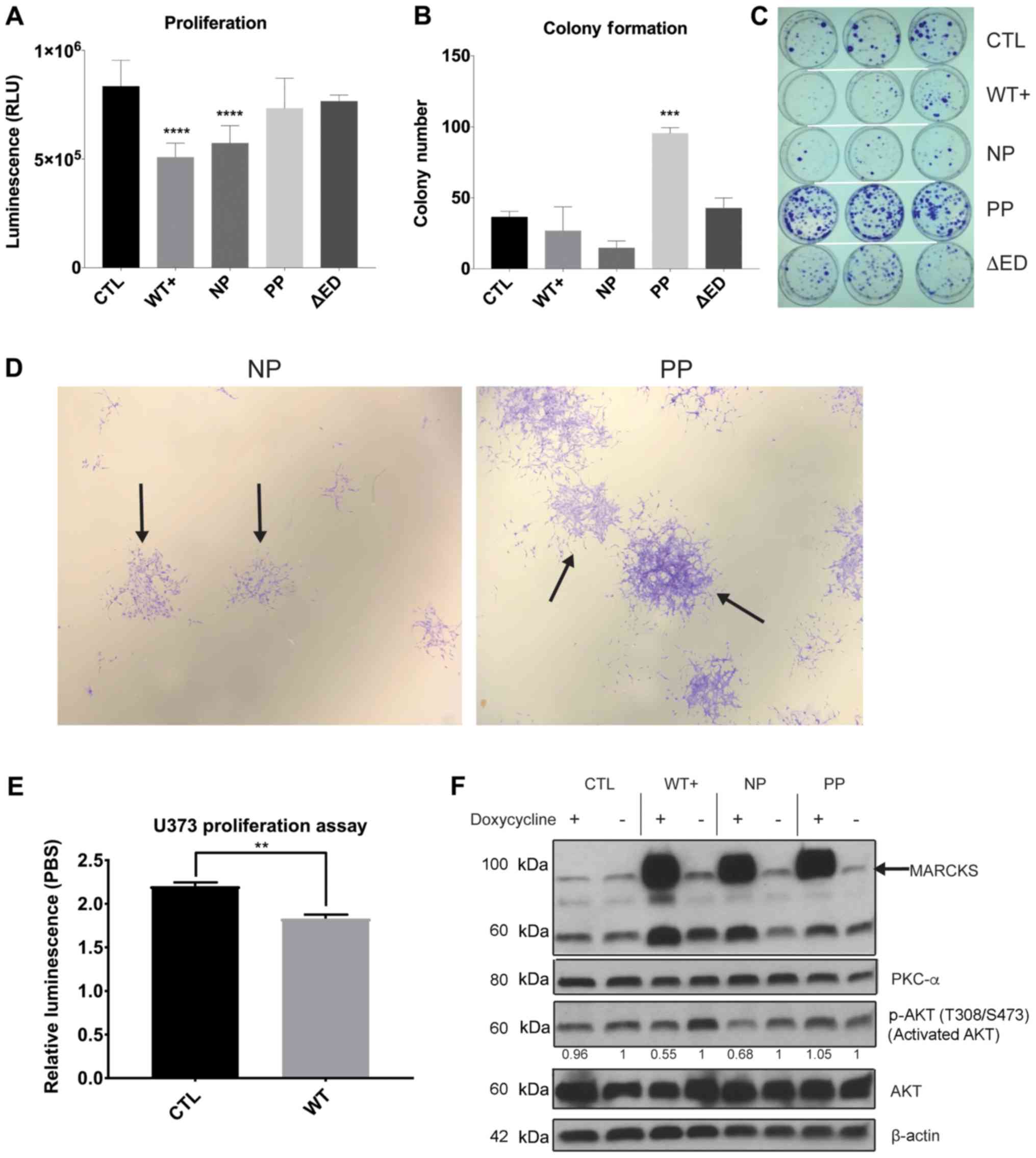 | Figure 3MARCKS ED phosphorylation overcomes
the growth suppressive effects of MARCKS. (A) A total of 5,000
cells were plated in a black-walled 96 well plate with doxycycline
and assessed for cell viability at day 7 using ATPlite assay
(n=12). Comparison of mutants PBS vs. doxycycline induction is
shown in Fig. S1. (B) Quantification of colonies at day 14 day
containing >50 cells after plating 2,000 cells per plate. (C)
Image of colony formation at day 14. (D) Magnified view of NP and
PP colonies from colony formation 14 days after plating (×10
magnification). The solid arrows indicate example colonies. (E) A
total of 5,000 cells were plated in a black-walled 96 well plate
with doxycycline and assessed using CellTiter glow cell viability
assay at 5 days in U373 (n=4). (F) Western blot analysis
demonstrating the inducible nature of MARCKS expression 7 days
after treating with 2 µg/ml doxycycline, and the effects on
activated AKT (T308 and S473), AKT and PKCα expression. All probing
done on same membrane with stripping. Cropped boundaries indicated
by black line. Full-length blots and probing order are available in
Fig. S5. Adjusted relative densitometry of p-AKT between
Dox-induced and PBS un-induced (=1) calculated in ImageJ
(CTL=0.962, WT+=0.549, NP=0.678, PP=1.05). Statistical analysis was
carried out in GraphPad Prism using (A and B) ordinary one-way
ANOVA with Dunnett’s multiple comparison tests to CTL, or (E)
two-tailed t-test. Data are the means ± SEM.
**P<0.01, ***P<0.001 and
****P<0.0001, compared to CTL. MARCKS, myristoylated
alanine-rich C-kinase substrate; ED, effector domain; WT+,
V5-tagged MARCKS vector; CTL, control; NP, non-phosphorylatable ED
mutant; PP, pseudo-phosphorylated ED mutant; ΔED, deleted effector
domain mutant. |
Comet assay
A total of 100,000 cells were plated in 60-mm dishes
and induced with doxycycline for 72 h. At 24 h prior to the assay,
the medium was exchanged for fresh doxycycline-containing media to
remove any floating cells. Cells were irradiated with 16 Gy and
collected at the following time-points [immediately
post-irradiation (T0), 30 min, 1 h and 4 h] by gently lifting them
off the plate into the medium using a rubber policeman. T0 cells
were irradiated on ice and scraped immediately following
irradiation. The Trevigen Comet assay (Trevigen, Gaithersburg, MD,
USA) was used under neutral conditions and manufacturer-provided
materials and protocol. Tail moments from 200 cells per spot (3X
replicates) were determined using a Zeiss AX10 observer A1
microscope and Comet Assay IV version 4.3 software. The graph was
generated using tail moment values in GraphPad 7.04 software.
Immunoprecipitation coupled with mass
spectrometry
U87 WT+ cells were cultured to 90% confluence in a
75 cm2 flask with standard doxycycline medium for 72 h,
prior to trypsin disassociation, rinsing with PBS and
flash-freezing in liquid nitrogen. The cells were later lysed in
chilled MPER lysis buffer supplemented with protease (#P8340) and
phosphatase inhibitors (P0044 and P5726) (both from Sigma-Aldrich)
for 30 min on ice. The lysate was centrifuged at 12,000 × g for 10
min at 4°C and protein was quantified by BCA assay. Catch and
Release version 2.0 (EMD Millipore, Temecula, CA, USA) was used for
immunoprecipitation with the following modifications to the
manufacturer’s protocol. 500 µg protein was loaded into a
1.5 ml centrifuge tube, along with a 1:200 dilution of MARCKS
antibody (cat. no. 20661-1-AP; Proteintech, Rosemont, IL, USA) or
normal rabbit IgG antibody (sc-2027; Santa Cruz Biotechnology). The
lysate antibody mixture was rotated at 4°C for 15 min. The lysate
antibody mixture was then added to the spin column along with 10
µl affinity ligand and 1X wash buffer for 500 µl
total volume. The spin column was rotated overnight at 4°C before
proceeding with standard protocol eluting with 70 µl of
provided non-denaturing elution buffer. Flow through, washes and
elutions were collected for evaluation by western blot analysis
before submitting a sample to UAB mass spectrometry core for
analysis. The sample was run on 10% Bistris gel and stained using
the colloidal blue staining kit (LC60225; Thermo Fisher Scientific)
following the manufacturer’s protocol, with fixation for 10 min at
room temperature using 50% methanol, 10% acetic acid and staining
for 12 h at room temperature. In total, 6 separate fractions were
isolated using in-gel digestion with trypsin and analyzed using an
nLC LTQ Velos Pro Orbitrap mass spectrometer (Thermo Fisher
Scientific) for analysis. Scaffold 4.6.2 was used to compare
identified proteins from mass spectrometry experiments and generate
Venn diagram. GeneGo 4.9.18 was used to generate network and
pathway maps of direct protein interactions from the 108 unique
proteins identified in MARCKS immunoprecipitation experiments and
not detected in IgG control. Link: https://portal.genego.com/
Orthotopic implantation, cranial
radiation and survival analysis
All animal studies were carried out in accordance
with the policies set by the University of Alabama (UAB)
Institutional Animal Care and Use Committee (IACUC) and performed
according to their guidelines. Moreover, the experimental protocols
were registered and approved by the UAB Occupational Health and
Safety (Project no. 14-124). Five- to six-week-old female athymic
nude mice (Charles River, Hartford, CT, USA) were started on
doxycycline chow 1 week prior to an intracranial injection of
500,000 cells per mouse with the aid of UAB’s Brain Tumor Animal
Model Core. A total of 40 mice (20 for data shown in Fig. 1A and 20 for data shown in Fig. S3) were used with an average weight
of 20-22 g per mouse. All mice were housed under the care and
maintenance of UAB’s fully accredited (AAALAC) animal resources
program (ARP) with routine monitoring by veterinarians. Mice were
housed no more than 6 to a cage and had 24-h access to food and
water maintained daily. The animal room was maintained at 21°C and
50% humidity with 12-h light-dark cycles. Intracranial injections
were carried out as previously described (36). In brief cells were suspended in a
(1:1) mixture of methylcellulose and loaded into a 1 ml Hamilton
syringe with a 12-gauge needle. Mice were anesthetized using an
intraperitoneal injection of ketamine (100 mg/kg) and xylazine (15
mg/kg), and a midline scalp incision was made and a burr hole was
drilled 1 mm posterior to the coronal suture and 2 mm right of the
midline. Subsequently, 5 µl of cell mixture was
stereotactically delivered to a depth of 2.5 mm into the right
caudate-putamen of each mouse. The data shown in Fig. 1A are composed of 4 groups with 5
mice in each (2 with CTL, 2 with WT+) with 1 group of each cell
type receiving either 12 Gy (6 by 2 Gy fractions) or sham
irradiation (0 Gy). Mice were euthanized at the first appearance of
neurological symptoms or at the request of veterinary staff using
CO2 exposure at 20% chamber volume displacement/minute
(1.5 l/min in an IACUC approved chamber with a flow meter) for ~5
min followed by secondary cervical dislocation as per the AVMA
guidelines. Brains were collected and halved through the injection
site for preservation by both formalin and liquid nitrogen.
Statistical analysis and data
reproducibility
All statistical analyses were calculated in Prism
8.0 (GraphPad) with P-values <0.05 considered to indicate
statistically significant differences. Significance in Fig. 1 was calculated with the Log-rank
(Mantel-Cox) test, and in Fig. 2,
with the similarity score log-transformed Pearson’s correlation. In
Fig. 3, significance (A and B) was
determined using one-way ANOVA with Dunnett’s multiple comparison
test, or (C) a two tailed t-test. In Fig. 4, significance was determined using
two-way ANOVA with Dunnett’s multiple comparison test. All
experiments were repeated at least 2 times.
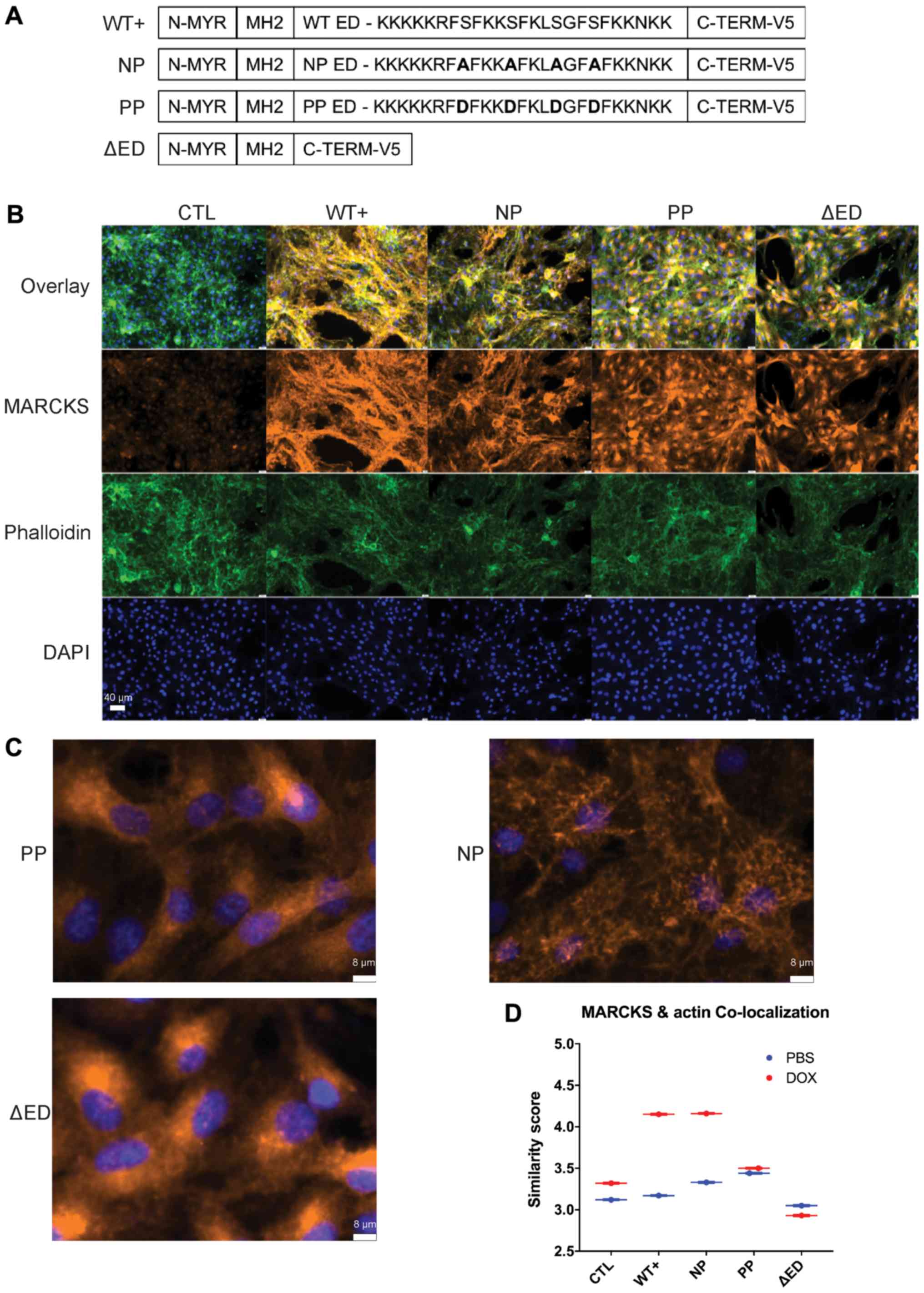 | Figure 2MARCKS ED phosphorylation regulates
actin binding and the cellular localization of MARCKS in GBM. (A)
Diagram of MARCKS effector domain mutants. WT+ has 4 PKC
phosphorylatable serine residues in the effector domain. NP mutant
replaces the 4 serine residues with alanine, PP replaces serines
with aspartic acid residue and ΔED has a deleted effector domain.
(B) Immunofluorescent imaging of U87 MARCKS effector domain mutants
72 h after doxycycline induction using the image cytometer Xcyto10
(×20 magnification). (C) Magnified view of MARCKS staining in NP,
and PP mutants. Note the ruffled appearance of NP in contrast to
the perinuclear staining of PP (×20 magnification, 400% zoom). (D)
Quantification of co-localization using similarity score to
phalloidin (F-actin) calculated in XcytoView. The similarity score
is calculated from the log-transformed Pearson’s correlation
between two separate fluorescent channels within the indicated
compartment, graphed as mean ± SEM. MARCKS, myristoylated
alanine-rich C-kinase substrate; ED, effector domain; WT+,
V5-tagged MARCKS vector; CTL, control; NP, non-phosphorylatable ED
mutant; PP, pseudo-phosphorylated ED mutant; ΔED, deleted effector
domain mutant. |
Results
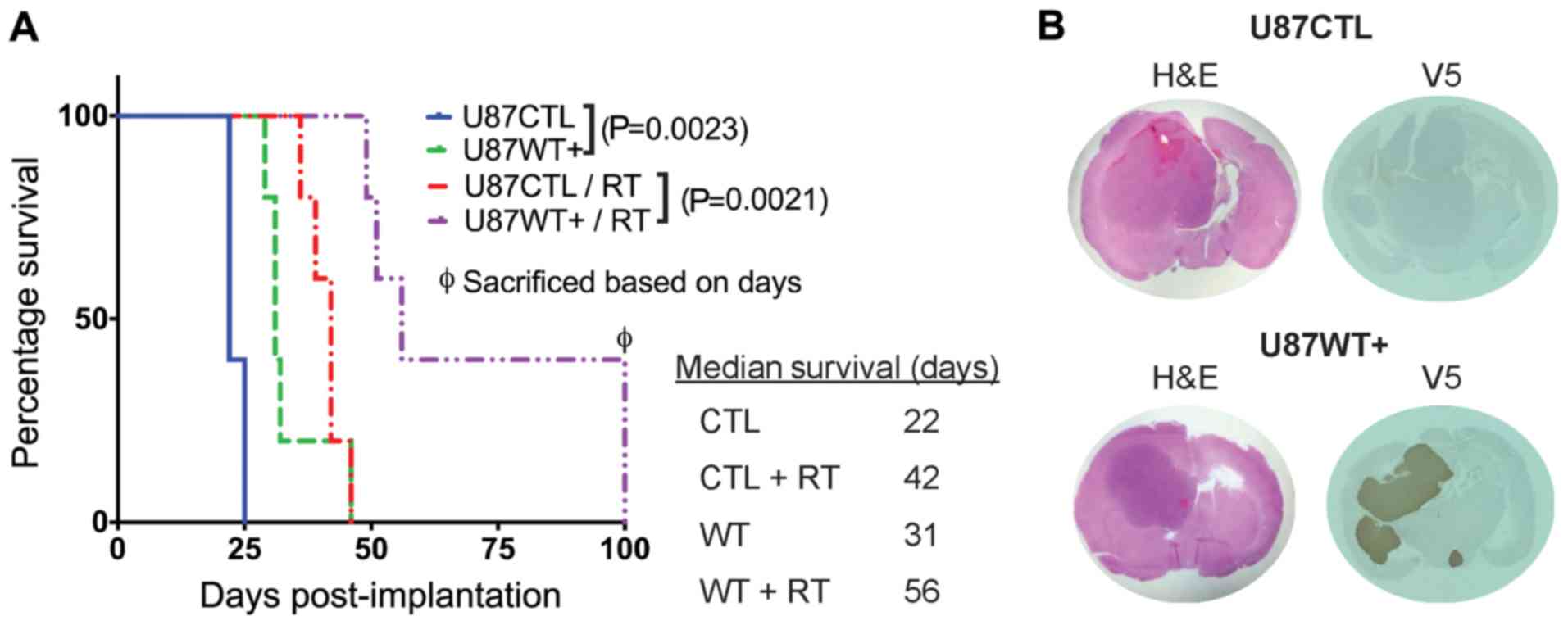
MARCKS overexpression prolongs survival
and enhances radiation sensitivity in GBM in vivo
We have previously shown that MARCKS protein
expression is inversely associated with GBM proliferation and
intracranial xenograft growth rates, with the knockdown of MARCKS
in the PTEN-null line, U251, resulting in an enhanced radiation
resistance (33). In this study,
to establish whether MARCKS overexpression can inhibit GBM growth
and enhance radiation sensitivity, we orthotopically implanted the
PTEN-null U87 cell line featuring a tetracycline-inducible,
V5-tagged MARCKS vector (WT+) (25) or an empty control vector (CTL) into
athymic nude mice and assessed the effects on survival. The WT+
mice were found to have a median survival of 31 days without
radiotherapy (RT) and a 56-day survival following RT at 12 Gy
(25-day enhancement), whereas the CTL mice had a median survival of
22 days without RT and a 42-day median survival with RT (20-day
enhancement) (Fig. 1A). MARCKS
overexpression increased the survival of the mice compared with the
CTL group by 40% (9 days), while the WT+ mice receiving RT had an
additional 25% (5 days) increase in survival compared with the CTL
mice (Fig. 1A). The successful
overexpression of MARCKS in vivo was verified in post-mortem
tumors by immunohistochemical staining (Fig. 1B). These data support the
hypothesis that the overexpression of MARCKS is capable of
suppressing growth and enhancing radiation sensitivity in PTEN-null
GBM.
MARCKS ED mutants mimic actin binding and
the cellular localization of MARCKS phosphorylation in GBM
We then investigated the mechanisms through which
the phosphorylation of the 4 serine residues present in MARCKS ED
affect the ability of MARCKS to suppress GBM growth and radiation
resistance by generating additional ED mutants: i) A
non-phosphorylatable ED mutant (NP) replaced the serine residues
with alanine, to prevent the loss of plasma membrane binding by
phosphorylation; ii) a pseudo-phosphorylated ED mutant (PP)
substituted the serine residues with aspartic acid, which prevented
membrane binding by mimicking negatively charged phosphorylation
groups; and iii) a deleted effector domain mutant (ΔED) that lacks
an ED (Fig. 2A). To evaluate the
cellular localization of the MARCKS mutants, immunofluorescent
imaging, and the analysis of the mutants 72 h following doxycycline
induction were performed using the image cytometer Xcyto10. An
unphosphorylated non-Ca2+/CaM bound ED is required for
MARCKS membrane binding and F-actin crosslinking (13,37)
allowing ΔED to serve as a cytoplasmic control. MARCKS that
co-localizes well with F-actin is consistent with an
unphosphorylated ED, whereas MARCKS that co-localizes poorly with
F-actin may indicate ED phosphorylation or binding to
Ca2+/CaM (14). Imaging
revealed WT+ and NP MARCKS to have substantial co-staining with
phalloidin (F-actin stain), while the PP and ΔED MARCKS lacked
co-staining with F-actin and appeared predominantly cytoplasmic
with perinuclear enrichment. Slight decreases in F-actin intensity
were observed in all MARCKS mutants compared with the control
(Fig. 2B). Fig. 2C highlights the differences in
MARCKS staining between PP and ΔED with minimal F-actin co-staining
and prominent perinuclear staining, while NP shows substantial
co-staining with F-actin (Fig.
2C). The quantification of MARCKS and F-actin co-staining
revealed that both WT+ and NP MARCKS co-stained strongly with
F-actin, while the CTL, PP and ΔED lines did not (Fig. 2D). The imaging of uninduced MARCKS
U87 mutants can be observed for comparison in Fig. S1. The overexpression of WT+ MARCKS
in an additional PTEN-null line (U373) revealed that MARCKS was
predominantly membrane-associated and perinuclear with a slight
increase in actin co-localization (Fig. S2). These data indicate that the
localization of WT+ and NP MARCKS mutants is consistent with an ED
that is unphosphorylated and membrane-bound, while the PP mutant
mimics the cytoplasmic localization of phosphorylated MARCKS.
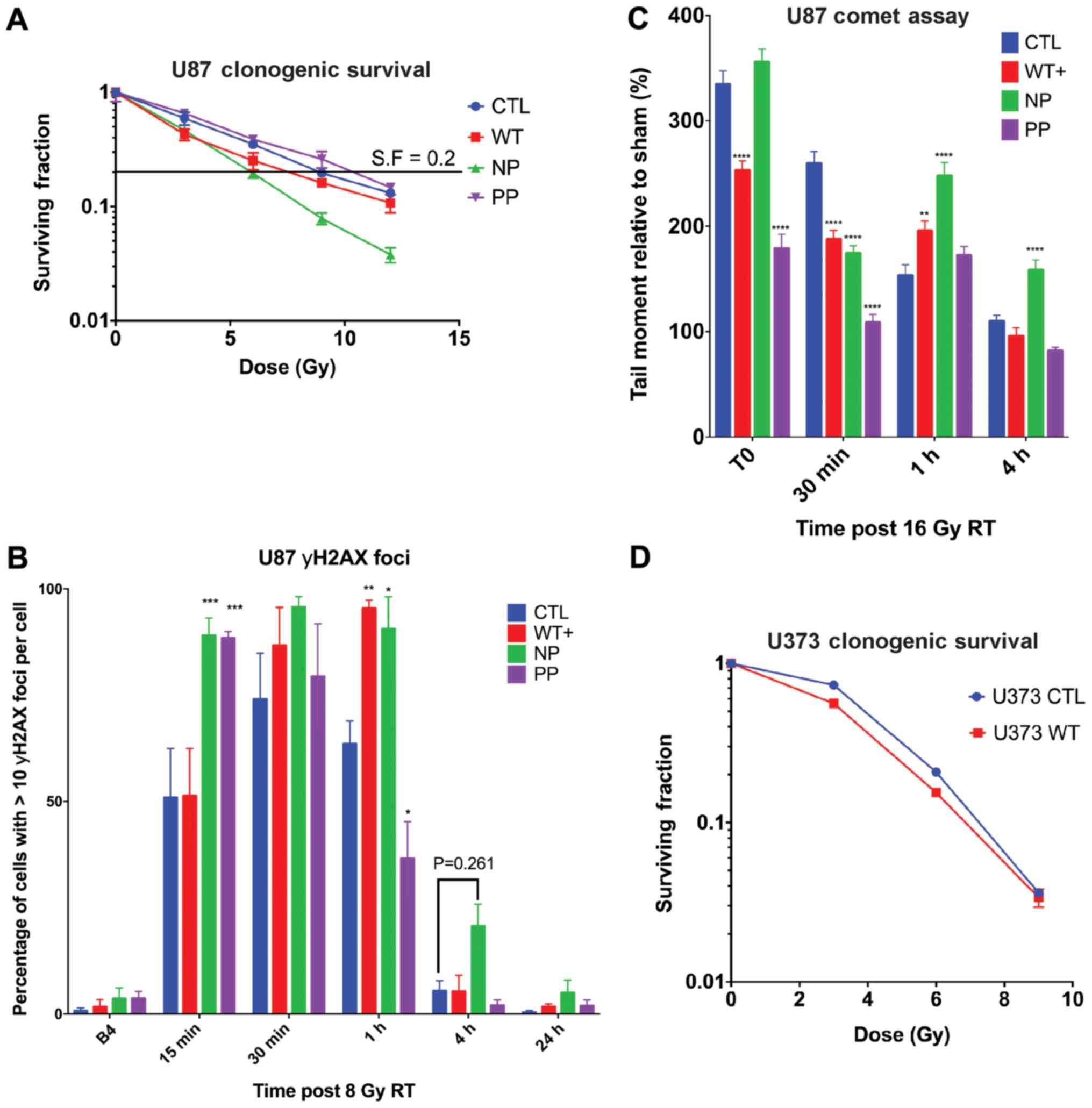
MARCKS ED phosphorylation overcomes
MARCKS growth suppression and promotes colony formation in
vitro
To identify differences in GBM growth with MARCKS
overexpression and the potential effects of ED phosphorylation, we
measured the growth of our MARCKS mutants 7 days following
doxycycline induction. Statistically significant (P<0.0001)
decreases in growth were observed in the WT+ and NP mutants, and no
decrease in growth in PP or ΔED compared to the CTL line (Fig. 3A). The comparison of mutants under
PBS and doxycycline conditions is available in Fig. S1 using ATPlite proliferation
assay. Colony formation assays revealed NP MARCKS to trend towards
(P=0.076) a decrease in colony number compared to CTL, while PP
exhibited significant (P=0.001) increases in the number of
colonies. WT+ (P=0.61) and ΔED (P=0.85) exhibited no significant
differences in colony number (Fig.
3B). Colonies formed by PP were also larger and contained more
cells per colony on average compared with CTL and other mutants
(Fig. 3C). A magnified view of NP
and PP colony differences (solid arrow) can be seen in Fig. 3D. The orthotopic implantation of
these ED mutants into mice, however, did not reveal significant
differences in survival between the ED mutants, suggesting that
MARCKS ED phosphorylation may not fully account for the MARCKS
survival benefit (Fig. S3). The
growth-suppressive effects of MARCKS overexpression (WT+) were
additionally observed in the PTEN-null U373 GBM cell line
(P=0.0010) (Fig. 3E), although we
lacked NP and PP ED mutants in U373 for additional validation.
Both AKT activation (6) and PKCα protein expression (38) are associated with enhanced cancer
growth, proliferation and survival signaling, and the knockdown of
MARCKS in GBM has been previously shown to enhance AKT
phosphorylation (33) and decrease
PKCα levels (32). In this study,
we examined the mechanisms through which the overexpression of
MARCKS ED mutants affect these features and found that WT+ and NP
MARCKS overexpression decreased the activation of AKT (T308 and
S473 phosphorylation) by 45 and 32%, respectively compared to the
PBS-treated cells, while CTL and PP exhibited negligible
suppression. No effects were observed on PKCα expression with the
overexpression of our MARCKS mutants (Fig. 3F). Overall, we found that MARCKS
overexpression (WT+) suppressed GBM growth and the data suggest
that it is the unphosphorylated ED (NP), which suppresses growth
and AKT activation, while the phosphorylated ED (PP) does not.
Since differences in AKT activation suggest differences in
radiation sensitivity (39), we
then investigated the effects of MARCKS ED phosphorylation on
radiation sensitivity.
MARCKS ED phosphorylation modifies GBM
sensitivity to radiation
Previous experiments in our laboratory have
demonstrated that the inhibition of MARCKS ED phosphorylation or
the overexpression of NP MARCKS in a lung cancer line sensitized
them to radiation (35,40). The GBM data in this study
demonstrated that the overexpression of WT+ MARCKS enhanced
radiation sensitization in vivo. To determine the mechanisms
through which the MARCKS ED phosphorylation state affects radiation
sensitivity, we first assessed our mutants using a clonogenic
assay. We found NP MARCKS mutants to have the lowest clonogenic
survival following escalating doses of radiation, showing radiation
sensitization compared with CTL. WT+ exhibited a mild enhancement
in radiation sensitivity compared with the control, while PP
exhibited slightly decreased sensitivity (dose enhancement ratios
at surviving fraction 0.2: CTL =1, WT+ =1.2, N =1.5 and PP =0.87)
(Fig. 4A). To investigate
potential alterations in DNA repair, we then examined the
phosphorylation of histone H2AX at S139 (γH2AX) as a surrogate for
DNA damage. At 1 h post-8 Gy single fraction radiotherapy, the WT+
(P=0.0021) and NP (P=0.0111) mutants exhibited prolonged increases
in γH2AX levels compared with the control, whereas PP (P=0.0116)
exhibited a slight decrease. At 4 h, no statistically significant
increases in γH2AX were observed compared with CTL; however, NP did
trend towards a significant increase (P=0.2609) (Fig. 4B). Due to the considerable
radiation resistance of U87 cells and variations in the cell cycle
that alter individual cell radiation sensitivity (41) we used an elevated 16 Gy dose of
radiotherapy to directly quantify the formation and resolution of
double-strand DNA (dsDNA) breaks using a neutral comet assay
(Fig. 4C). NP exhibited the
greatest and most significant increases in the tail moment compared
to CTL both at 1 h (P<0.0001) and 4 h (P=0.0012)
post-irradiation compared with CTL. WT+ had a lower basal amount of
DNA damage immediately following 16 Gy (T0) and at 30 min; however
at 1 h, WT+ displayed sustained DNA damage (P=0.0067) compared with
the control. PP also had a lower induction of double-strand DNA
damage immediately following RT and a similar return to baseline as
the control (Fig. 4C). The
overexpression of WT+ MARCKS in U373 GBM cells similarly revealed
increased yH2AX nuclear staining (Fig. S1C) and RT sensitivity measured by
clonogenic assay (Fig. 4D),
although we lacked ED mutants for additional validation. These data
support prior findings that MARCKS is involved in that DNA damage
response (33,40) and suggest that an unphosphorylated
ED promotes MARCKS radiation sensitization in vitro.
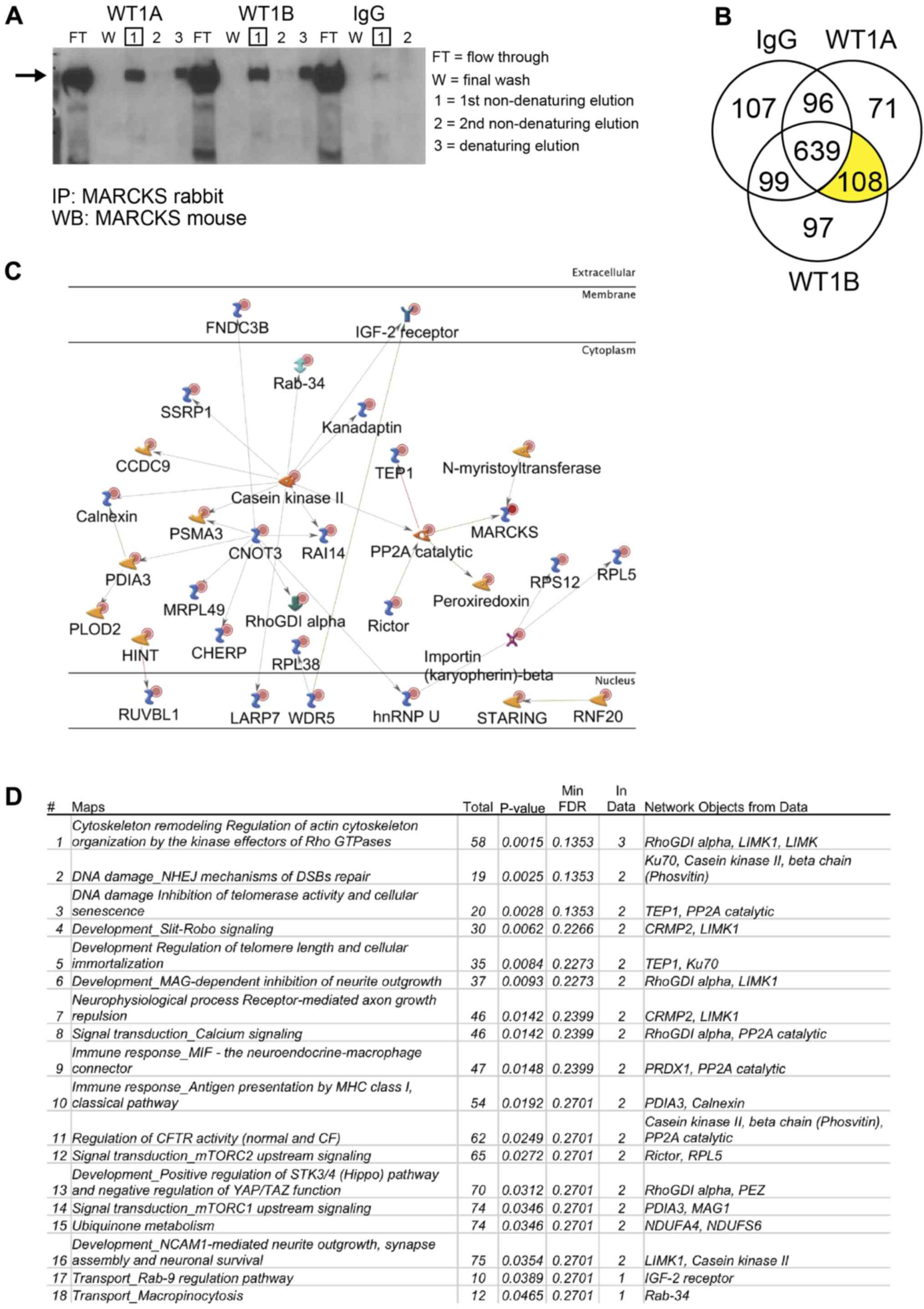
Identification of MARCKS protein-protein
interactions in GBM using immunoprecipitation coupled to mass
spectrometry
MARCKS is known to be phosphorylated by PKC and ROCK
and dephosphorylated by protein phosphatase 2 (PP2A), which alter
its ability to bind Ptdlns(4,5)P2, actin, and
Ca2+/CaM (10).
Recently, MARCKS ED has been shown to function as an NLS allowing
it to translocate into the nucleus of GBM and bind nuclear
Ptdlns(4,5)P2 (25). In this study, to identify novel
protein-protein interactions of MARCKS in GBM, we
immunoprecipitated MARCKS in the WT+ overexpressing U87 cell line
and detected protein interactions with high-resolution mass
spectrometry. The successful pulldown of MARCKS was verified by
western blot analysis before proceeding with in-gel digestion,
liquid chromatography and high-resolution mass spectrometry
(Fig. 5A). A total of 275 proteins
were detected in the two separate MARCKS IP that was not found in
the IgG control, 108 of which were identified in both fractions
(Fig. 5B). A GeneGo pathway
analysis map was constructed from these 108 proteins showing only
direct protein interactions (Fig.
5C). A common pathway map was similarly generated in GeneGo to
determine the potential signaling interactions of MARCKS (Fig. 5D). The top protein pathway
interactions found were involved in cytoskeletal remodeling and the
regulation of the actin cytoskeleton and non-homologous end joining
(NHEJ) DNA repair pathway. Notable direct protein interactions of
interest with MARCKS found using this technique include importin
β-2 (transportin-1), a nuclear import chaperone that binds nuclear
localization sequences, and Ku70, a protein involved in DNA repair.
However, additional validation of these targets at endogenously
expressed MARCKS levels is still required.
Discussion
GBM remains a devastating disease driven by high
rates of growth, therapeutic resistance and invasiveness, which
ultimately results in recurrence. Developing a better understanding
of the molecular mechanisms that contribute to this aggressiveness
is essential to developing future effective therapeutic strategies.
Over 80% of GBM cases contain activating mutations in RTKs/PI3K/AKT
or the loss of PTEN (6). These
mutations all contribute to dysregulations in Ptdlns (4,5)P2 signaling, which promotes
cell proliferation, differentiation (42,43),
invasion (32) and therapeutic
resistance (44-46) pathways, rendering this signaling
axis an ideal therapeutic target (6,47).
To date, however, small molecule inhibitors targeting this pathway
have had minimal success in GBM (48,49).
One potential reason for this failure lies in an incomplete
understanding of regulators of this pathway, such as MARCKS
(10). MARCKS is a well-known
regulator of Ptdlns(4,5)P2 levels (50-52),
which plays a vital role in AKT activation that drives cell
proliferation, chemo- and radiation resistance (45,46,53).
MARCKS expression has had a confusing association with both
positive and negative prognosis across different cancer types
(10,26-28,54,55),
and this ambiguity over the role of MARCKS in cancer has been
attributed to potential differences MARCKS ED phosphorylation
(10,30,56).
The phosphorylation of MARCKS ED by PKC or ROCK kinases results in
the translocation of MARCKS off the plasma membrane, prevents its
binding to PS (24),
Ca2+/calmodulin and crosslinking of actin filaments, and
releases sequestered Ptdlns(4,5)P2 (18,57,58).
MARCKS ED also serves as an NLS (25), and differences in its
phosphorylation are also likely to regulate nuclear import. Indeed,
we detected differences in the nuclear localization of our PP ED
mutant from WT+ and NP ED mutant that needs additional future
validation in a model system with endogenous MARCKS expression
(Fig. S4). Due to the important
role of Ptdlns(4,5)P2 in oncogenic signaling
(8), the frequent mutations of GBM
altering Ptdlns(4,5)P2 signaling (6), and the role of MARCKS in regulating
Ptdlns(4,5)P2 availability (59), in this study, we evaluated the
effects of MARCKS expression and ED phosphorylation on GBM growth
and therapeutic resistance.
The current study supports previous in vitro
findings that the loss of MARCKS enhances growth and radiation
resistance in a PTEN-null GBM line (33). Through the overexpression of WT+
MARCKS in an intracranial tumor model, we further established
MARCKS expression enhanced survival and radiation responses
compared to empty vector control (CTL) mice. To determine the
mechanisms through which the phosphorylation of MARCKS ED may alter
these tumor-suppressing effects, we mimicked ED phosphorylation or
prevented phosphorylation through substitution of the 4 ED serine
residues. MARCKS functions as an electrostatic switch based on it
ED phosphorylation status (13),
with F-actin and plasma membrane binding with an unphosphorylated
ED, and the loss of F-actin membrane binding after phosphorylation.
Additionally, ΔED functions as a cytoplasmic control as the ED
contains both the actin-binding domain and the poly-lysine (+13)
electrostatic attraction to the plasma membrane (13). The ED mutants were found to
appropriately mimic MARCKS ED phosphorylation state with NP
co-localizing with F-actin at the plasma membrane, while PP and ΔED
mutants localized to the cytoplasmic and perinuclear region. WT+
closely mimicked NP with high levels of F-actin co-localization and
similar morphologic appearance suggesting the majority of
overexpressed MARCKS was membrane-bound. Although slight decreases
in filamentous actin staining were observed following the
overexpression of MARCKS in all our mutant lines compared with
control or uninduced group, no substantial differences existed
between the ED mutants. This suggested cytoskeletal impairment was
not a major factor in the phenotypic differences of our
mutants.
We then examined the effects of MARCKS ED
phosphorylation on GBM growth by measuring its effects on cell
viability using ATP luminescence (60). To control for potential metabolic
disturbances occurring from lentiviral transduction or doxycycline
exposure we used the empty vector CTL line for comparison of the
effects of MARCKS ED mutant expression. We found the WT+ and NP
MARCKS overexpression led to significant decreases in cell
viability (P<0.0001), while the PP or ΔED mutants did not
suppress viability. The significant suppression of cell viability
in WT+ and NP (P=0.004 and P=0.0093, respectively) was dependent on
exposure of the mutants to doxyxcline, and its resulting expression
of mutant MARCKS protein, with no significant decreases in
viability seen in CTL or PP as observed in Fig. S1B. The cytotoxic effects of MARCKS
expression were not observed; thus, this decreased cell viability
was attributed to decreased proliferation. Although ATP levels are
a reliable measure of metabolic viability and typically, cell
number, ATP can also be altered by circadian rhythms, proliferation
and differentiation, which we did not differentiate from (61). We utilized the colony formation
assay as a second assay for investigating MARCKS growth
effects.
Colony formation assay, which estimates the
proportion of cells capable of ‘unlimited’ replication, revealed
that NP trended towards a decrease in colony forming ability, while
PP significantly enhanced colony formation. This increased number
and size of the colonies would indicate that PP MARCKS may enhance
the proliferative capacity of GBM. Differential growth effects of
MARCKS have previously described in epithelial and vascular smooth
muscle cells, although differences in ED phosphorylation status
were not investigated (62).
However, testing in vivo failed to reveal statistically
significant differences in the median survival (one-way ANOVA
P=0.0879) or mitotic counts (one-way ANOVA P=0.1587) between MARCKS
mutants and control. This suggests ED phosphorylation may have a
less definitive roll in affecting overall survival. The weak
correlation of mitotic counts to survival time is confounded by the
fact mitotic counts were acquired at the time of sacrificing
opposed to a similar time-point. Potential reasons for these in
vivo, in vitro differences, include MARCKS ED
phosphorylation may function more in releasing MARCKS inhibition on
the proliferative capacity than directly driving proliferation,
especially under the different microenvironmental conditions, such
as limited nutrient and oxygen availability of the intracranial
growth environment. Additionally, MARCKS may enhance survival in
GBM ways not directly altered by ED phosphorylation. The
investigation of MARCKS ED phosphorylation on p-AKT (T308/S473)
revealed WT+ and NP MARCKS overexpression suppressed p-AKT levels,
while PP MARCKS did not in GBM. These data support other findings
that ED phosphorylation is an important regulator of this pathway
(10). No effect was observed on
PKCα expression with or MARCKS overexpression model as previously
reported in a GBM EGFR-VIII line, suggesting that EGFR-VIII
expression may be essential for MARCKS-driven PKCα expression
differences (32).
MARCKS knockdown was previously shown to enhance
the NHEJ DNA repair mechanism and radiation resistance in GBM
(33). Consistent with this, in
this study, we found that MARCKS overexpression (WT+) in U87 and
U373 cells increased radiation sensitivity. Investigating the
effects of ED phosphorylation on MARCKS radiation sensitization, we
found that the NP mutant was radiation-sensitive by clonogenic
assay, γH2AX, and comet assay, while the PP mutant was not. The
clonogenic assay measures the cumulative effects of radiation on
survival, including cell death, senescence, metabolic disturbances,
that may take generations to develop and is considered the most
sensitive in vitro radiation sensitivity assay (63). NP was found to have a prolonged
presence of γH2AX quantified as a percentage of cells with >10
foci per cell. However, the PP mutant revealed prolonged γH2AX
staining in some instances when quantified by mean nuclear
intensity (Fig. S1), although
this result was inconsistent with the comet assay, clonogenic assay
findings, and traditional foci count methodology. The high level of
radiation resistance by U87 cells (41) and the differences in sensitivity
between γH2AX quantification methods may account for these
differences, which can be minimized with higher doses or radiation
(64). Higher 16 Gy doses of
radiotherapy were needed for significant neutral comet assay
results. NP also exhibited the most prolonged increase in dsDNA
breaks following RT, most closely reflecting the clonogenic assay
findings in suggesting it is radiation sensitive. Immediately
following irradiation (T0), WT and PP showed lower levels of dsDNA,
however, at 1 h WT+ also exhibited a significant (P=0.0067)
elevation of DNA damage relative to the control, while PP did not.
As such, we propose that the membrane-bound, unphosphorylated form
of MARCKS promotes radiation sensitization.
Utilizing IP/MS of MARCKS in U87 cells, we
identified known MARCKS interactors including PP2A, known to
dephosphorylate MARCKS ED, and N-myristoyltransferase (NMT) which
preferentially myristoylated MARCKS over many other myristoylated
proteins (65). These exploratory
studies suggest a number of new interacting partners that can be
validated in future studies. Rho GTPases including LIMK1 and Rho
GDP-dissociation inhibitor 1 were also detected and are important
regulators of the actin cytoskeleton promoting cell migration
(66). We detected a notable
interaction with transportin-1a nuclear import protein that
regulates nuclear-cytoplasmic transport through adapter proteins
(67). Transportin-1 and MARCKS
interactions are previously unreported, but are consistent with
emerging data that MARCKS is selectively imported into the nucleus
in specific cell types (68), and
our findings that only the PP mutant was not enriched in the
nucleus suggests ED phosphorylation may inhibit its translocation
through the nuclear membrane. Interaction with casein kinase 2
(CK2) and its central location in MARCKS protein network suggest it
may also directly regulate MARCKS at the ED or other
phosphorylation domains. CK2 has previously been shown to
phosphorylate proteins regulating their nuclear localization
(69), regulate the cell cycle,
NHEJ DNA repair and WNT signaling (70), and mediate non-canonical WNT
signaling through PKCΔ (71).
Several interactions with nuclear proteins were detected that were
involved in DNA repair including XRCC6 (Ku70), TEP1 and SSRP1,
suggesting that MARCKS may play a direct role in DNA repair
mechanism beyond Ptdlns(4,5)P2 sequestration. Perinuclear
proteins involved in endoplasmic reticulum calcium homeostasis
including CHERP and TRAPG were identified, along with Myoferlin, a
calcium/phospholipid binding-protein with a role in plasmalemma
repair. Lastly, MARCKS has previously been shown to become
phosphorylated by elevated levels of H2O2 in
a PKCΔ dependent manner (72). The
potential interaction with peroxiredoxin 1a, a regulator of
intracellular H2O2 signaling previously shown
to be involved in promoting invasion, radiation and chemotherapy
resistance (73), further
strengthens the relationship of H2O2
signaling and MARCKS. Future studies investigating
H2O2 effects on GBM growth and invasion
should be considered due to H2O2 use during
GBM resection for tumoricidal effects (74). H2O2 use at
subtherapeutic levels may in theory trigger invasive and
proliferative effects seen with MARCKS phosphorylation (32).
The limitations of this study include the use of a
doxycycline-inducible model with overexpression of MARCKS ED
mutants, as opposed to testing at endogenous MARCKS levels with
true serine phosphorylation, and the lack of additional NP and PP
ED mutants that could be tested in PTEN null cell lines. Due to the
dynamic and differential effects MARCKS ED can have in regulating
cellular functions (62), the
impact of MARCKS expression and ED phosphorylation should be
carefully considered in other model systems and signaling
environments, such as serum-free growth conditions to establish the
effects of MARCKS.
In conclusion, the intrinsically unstructured
nature and electrostatic properties of MARCKS allow it to have a
broad range of cellular interactions regulated by its centrally
located 25 amino acid ED. At a minimum, MARCKS ED binds or responds
to: Ptdlns(4,5)P2 (19), phosphatidylserine (24), F-actin crosslinking (58), phosphorylation by PKC and ROCK
kinases and binding to Ca2+/calmodulin (10), intracellular calcium levels
(13), and
H2O2 signaling (72) allowing it to coordinate cellular
functions, such as vesicle release (75), cell migration (76), proliferation and differentiation
(42,43,62).
MARCKS plays a potential role in cancer stemness that needs future
evaluation (77). Prior to this
study, MARCKS knockdown was associated with enhanced GBM growth and
radiation resistance (33), and
the phosphorylation of MARCKS ED was associated with enhanced
invasion (32) with an
undetermined effect on growth and radiation sensitivity in GBM.
This study investigated the mechanisms through which ED
phosphorylation regulates MARCKS cellular localization, and in
turn, examined the effects of MARCKS on growth suppression and
radiation sensitivity in PTEN-null GBM. We find that NP MARCKS to
exhibited a similar suppression of growth and AKT activation and
enhancement radiation sensitivity as WT+ overexpression, while the
PP mutant did not exhibit these features. This study suggests that
MARCKS ED phosphorylation may be a method with which to overcome
MARCKS growth-suppressing and radiation-sensitizing effects and
that the determination of the ED phosphorylation status is vital to
understanding the potential effects of MARCKS expression.
Supplementary Materials
Funding
This study was supported by funding from the
National Institutes of Health (the UAB MSTP training grant:
T32GM008361 and the UAB Training Program in Brain Tumor Biology:
T32NS048039), the American Cancer Society through a Research
Scholar Grant (Grant no. RSG-14-071-01-TBG), as well as by an
intramural research grant from the UAB Department of Radiation
Oncology.
Availability of data and materials
All data generated or analyzed during this study
are included in this published article.
Authors’ contributions
CDW, NJE and GYG conceived and designed the study.
CDW, ABH, GYG, NJE, CPL, JAM, JRH, RTP, JCA, JSJ and JAB developed
the methodology. GYG, NJE, CPL, HQT, KC, JAM, JRH, JCA and PHH
acquired the data. CDW, ABH, GYG, NJE, CPL, HQT, JAM, JRH, RTP,
JCA, PHH and JAB analyzed and interpreted the data. All authors
reviewed the manuscript. All authors have read and approved the
final manuscript.
Ethics approval and consent to
participate
All animal studies were carried out in accordance
with the policies set by the University of Alabama (UAB)
Institutional Animal Care and Use Committee (IACUC) and performed
according to their guidelines. The experimental protocols were
registered and approved by the UAB Occupational Health and Safety
(project no. 14-124).
Patient consent for publication
Not applicable.
Competing interests
RTP is an employee at ChemoMetec and assisted in
the use of Xcyto10, aided in Xcyto10 data analysis, and reviewed
the manuscript. ChemoMetec provided the Xcyto10 microscope on loan
and also provided microscope slides, Blue MASK, and DAPI reagents
but no financial compensation. The other authors declare no
competing interests.
Acknowledgments
The authors would like to thank Brandon Young at
the UAB comprehensive cancer center mass spectrometry core for his
assistance with the mass spectrometry analysis.
References
|
1
|
Li YM, Suki D, Hess K and Sawaya R: The
influence of maximum safe resection of glioblastoma on survival in
1229 patients: Can we do better than gross-total resection? J
Neurosurg. 124:977–988. 2016. View Article : Google Scholar
|
|
2
|
Johnson DR and O’Neill BP: Glioblastoma
survival in the United States before and during the temozolomide
era. J Neurooncol. 107:359–364. 2012. View Article : Google Scholar
|
|
3
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn
U, et al European Organisation for Research and Treatment of Cancer
Brain Tumor and Radiotherapy Groups; National Cancer Institute of
Canada Clinical Trials Group: Radiotherapy plus concomitant and
adjuvant temozolomide for glioblastoma. N Engl J Med. 352:987–996.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mao H, Lebrun DG, Yang J, Zhu VF and Li M:
Deregulated signaling pathways in glioblastoma multiforme:
Molecular mechanisms and therapeutic targets. Cancer Invest.
30:48–56. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ceccarelli M, Barthel FP, Malta TM,
Sabedot TS, Salama SR, Murray BA, Morozova O, Newton Y, Radenbaugh
A, Pagnotta SM, et al: TCGA Research Network: Molecular Profiling
Reveals Biologically Discrete Subsets and Pathways of Progression
in Diffuse Glioma. Cell. 164:550–563. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li X, Wu C, Chen N, Gu H, Yen A, Cao L,
Wang E and Wang L: PI3K/Akt/mTOR signaling pathway and targeted
therapy for glioblastoma. Oncotarget. 7:33440–33450.
2016.PubMed/NCBI
|
|
7
|
Jhanwar-Uniyal M, Amin AG, Cooper JB, Das
K, Schmidt MH and Murali R: Discrete signaling mechanisms of mTORC1
and mTORC2: Connected yet apart in cellular and molecular aspects.
Adv Biol Regul. 64:39–48. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Denley A, Gymnopoulos M, Kang S, Mitchell
C and Vogt PK: Requirement of
phosphatidylinositol(3,4,5)trisphosphate in phosphatidylinositol
3-kinase-induced oncogenic transformation. Mol Cancer Res.
7:1132–1138. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ramos AR, Elong Edimo W and Erneux C:
Phosphoinositide 5-phosphatase activities control cell motility in
glioblastoma: Two phosphoinositides PI(4,5)P2 and PI(3,4)P2 are
involved. Adv Biol Regul. 67:40–48. 2018. View Article : Google Scholar
|
|
10
|
Fong LWR, Yang DC and Chen CH:
Myristoylated alanine-rich C kinase substrate (MARCKS): A multirole
signaling protein in cancers. Cancer Metastasis Rev. 36:737–747.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Guan Y, Zhu Q, Huang D, Zhao S, Jan Lo L
and Peng J: An equation to estimate the difference between
theoretically predicted and SDS PAGE-displayed molecular weights
for an acidic peptide. Sci Rep. 5:133702015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ramsden JJ: MARCKS: A case of molecular
exaptation. Int J Biochem Cell Biol. 32:475–479. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Arbuzova A, Schmitz AA and Vergères G:
Cross-talk unfolded: MARCKS proteins. Biochem J. 362:1–12. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Brudvig JJ and Weimer JM: X MARCKS the
spot: Myristoylated alanine-rich C kinase substrate in neuronal
function and disease. Front Cell Neurosci. 9:4072015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tapp H, Al-Naggar IM, Yarmola EG, Harrison
A, Shaw G, Edison AS and Bubb MR: MARCKS is a natively unfolded
protein with an inaccessible actin-binding site: Evidence for
long-range intramolecular interactions. J Biol Chem. 280:9946–9956.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Arbuzova A, Wang J, Murray D, Jacob J,
Cafiso DS and McLaughlin S: Kinetics of interaction of the
myristoylated alanine-rich C kinase substrate, membranes, and
calmodulin. J Biol Chem. 272:27167–27177. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hartwig JH, Thelen M, Rosen A, Janmey PA,
Nairn AC and Aderem A: MARCKS is an actin filament crosslinking
protein regulated by protein kinase C and calcium-calmodulin.
Nature. 356:618–622. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yarmola EG, Edison AS, Lenox RH and Bubb
MR: Actin filament cross-linking by MARCKS: Characterization of two
actin-binding sites within the phosphorylation site domain. J Biol
Chem. 276:22351–22358. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang J, Arbuzova A, Hangyás-Mihályné G and
McLaughlin S: The effector domain of myristoylated alanine-rich C
kinase substrate binds strongly to phosphatidylinositol
4,5-bispho-sphate. J Biol Chem. 276:5012–5019. 2001. View Article : Google Scholar
|
|
20
|
Zhang W, Crocker E, McLaughlin S and Smith
SO: Binding of peptides with basic and aromatic residues to bilayer
membranes: Phenylalanine in the myristoylated alanine-rich C kinase
substrate effector domain penetrates into the hydrophobic core of
the bilayer. J Biol Chem. 278:21459–21466. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Thelen M, Rosen A, Nairn AC and Aderem A:
Regulation by phosphorylation of reversible association of a
myristoylated protein kinase C substrate with the plasma membrane.
Nature. 351:320–322. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tanabe A, Kamisuki Y, Hidaka H, Suzuki M,
Negishi M and Takuwa Y: PKC phosphorylates MARCKS Ser159 not only
directly but also through RhoA/ROCK. Biochem Biophys Res Commun.
345:156–161. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Denisov G, Wanaski S, Luan P, Glaser M and
McLaughlin S: Binding of basic peptides to membranes produces
lateral domains enriched in the acidic lipids phosphatidylserine
and phosphatidylinositol 4,5-bisphosphate: An electrostatic model
and experimental results. Biophys J. 74:731–744. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nakaoka T, Kojima N, Ogita T and Tsuji S:
Characterization of the phosphatidylserine-binding region of rat
MARCKS (myristoylated, alanine-rich protein kinase C substrate).
Its regulation through phosphorylation of serine 152. J Biol Chem.
270:12147–12151. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rohrbach TD, Shah N, Jackson WP, Feeney
EV, Scanlon S, Gish R, Khodadadi R, Hyde SO, Hicks PH, Anderson JC,
et al: The effector domain of MARCKS is a nuclear localization
signal that regulates cellular PIP2 levels and nuclear PIP2
localization. PLoS One. 10:e01408702015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bickeböller M, Tagscherer KE, Kloor M,
Jansen L, Chang-Claude J, Brenner H, Hoffmeister M, Toth C,
Schirmacher P, Roth W, et al: Functional characterization of the
tumor-suppressor MARCKS in colorectal cancer and its association
with survival. Oncogene. 34:1150–1159. 2015. View Article : Google Scholar
|
|
27
|
Brooks G, Brooks SF and Goss MW: MARCKS
functions as a novel growth suppressor in cells of melanocyte
origin. Carcinogenesis. 17:683–689. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hanada S, Kakehashi A, Nishiyama N, Wei M,
Yamano S, Chung K, Komatsu H, Inoue H, Suehiro S and Wanibuchi H:
Myristoylated alanine-rich C-kinase substrate as a prognostic
biomarker in human primary lung squamous cell carcinoma. Cancer
Biomark. 13:289–298. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Manenti S, Malecaze F, Chap H and Darbon
JM: Overexpression of the myristoylated alanine-rich C kinase
substrate in human choroidal melanoma cells affects cell
proliferation. Cancer Res. 58:1429–1434. 1998.PubMed/NCBI
|
|
30
|
Chen CH, Cheng CT, Yuan Y, Zhai J, Arif M,
Fong LW, Wu R and Ann DK: Elevated MARCKS phosphorylation
contributes to unresponsiveness of breast cancer to paclitaxel
treatment. Oncotarget. 6:15194–15208. 2015.PubMed/NCBI
|
|
31
|
Chen CH, Fong LWR, Yu E, Wu R, Trott JF
and Weiss RH: Upregulation of MARCKS in kidney cancer and its
potential as a therapeutic target. Oncogene. 36:3588–3598. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Micallef J, Taccone M, Mukherjee J, Croul
S, Busby J, Moran MF and Guha A: Epidermal growth factor receptor
variant III-induced glioma invasion is mediated through
myristoylated alanine-rich protein kinase C substrate
overexpression. Cancer Res. 69:7548–7556. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jarboe JS, Anderson JC, Duarte CW, Mehta
T, Nowsheen S, Hicks PH, Whitley AC, Rohrbach TD, McCubrey RO, Chiu
S, et al: MARCKS regulates growth and radiation sensitivity and is
a novel prognostic factor for glioma. Clin Cancer Res.
18:3030–3041. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cerami E, Gao J, Dogrusoz U, Gross BE,
Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et
al: The cBio cancer genomics portal: An open platform for exploring
multidimensional cancer genomics data. Cancer Discov. 2:401–404.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rohrbach TD, Jones RB, Hicks PH, Weaver
AN, Cooper TS, Eustace NJ, Yang ES, Jarboe JS, Anderson JC and
Willey CD: MARCKS phosphorylation is modulated by a peptide mimetic
of MARCKS effector domain leading to increased radiation
sensitivity in lung cancer cell lines. Oncol Lett. 13:1216–1222.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bradley JD, Kataoka Y, Advani S, Chung SM,
Arani RB, Gillespie GY, Whitley RJ, Markert JM, Roizman B and
Weichselbaum RR: Ionizing radiation improves survival in mice
bearing intracranial high-grade gliomas injected with genetically
modified herpes simplex virus. Clin Cancer Res. 5:1517–1522.
1999.PubMed/NCBI
|
|
37
|
Arbuzova A, Murray D and McLaughlin S:
MARCKS, membranes, and calmodulin: Kinetics of their interaction.
Biochim Biophys Acta. 1376:369–379. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cameron AJ, Procyk KJ, Leitges M and
Parker PJ: PKC alpha protein but not kinase activity is critical
for glioma cell proliferation and survival. Int J Cancer.
123:769–779. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li HF, Kim JS and Waldman T:
Radiation-induced Akt activation modulates radioresistance in human
glioblastoma cells. Radiat Oncol. 4:432009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Rohrbach TD, Jarboe JS, Anderson JC,
Trummell HQ, Hicks PH, Weaver AN, Yang ES, Oster RA, Deshane JS,
Steele C, et al: Targeting the effector domain of the myristoylated
alanine rich C-kinase substrate enhances lung cancer radiation
sensitivity. Int J Oncol. 46:1079–1088. 2015. View Article : Google Scholar
|
|
41
|
Naidu MD, Mason JM, Pica RV, Fung H and
Peña LA: Radiation resistance in glioma cells determined by DNA
damage repair activity of Ape1/Ref-1. J Radiat Res (Tokyo).
51:393–404. 2010. View Article : Google Scholar
|
|
42
|
Moon SH, Kim DK, Cha Y, Jeon I, Song J and
Park KS: PI3K/Akt and Stat3 signaling regulated by PTEN control of
the cancer stem cell population, proliferation and senescence in a
glioblastoma cell line. Int J Oncol. 42:921–928. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Peltier J, O’Neill A and Schaffer DV:
PI3K/Akt and CREB regulate adult neural hippocampal progenitor
proliferation and differentiation. Dev Neurobiol. 67:1348–1361.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Golding SE, Morgan RN, Adams BR, Hawkins
AJ, Povirk LF and Valerie K: Pro-survival AKT and ERK signaling
from EGFR and mutant EGFRvIII enhances DNA double-strand break
repair in human glioma cells. Cancer Biol Ther. 8:730–738. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Toulany M, Lee KJ, Fattah KR, Lin YF,
Fehrenbacher B, Schaller M, Chen BP, Chen DJ and Rodemann HP: Akt
promotes post-irradiation survival of human tumor cells through
initiation, progression, and termination of DNA-PKcs-dependent DNA
double-strand break repair. Mol Cancer Res. 10:945–957. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Toulany M and Rodemann HP:
Phosphatidylinositol 3-kinase/Akt signaling as a key mediator of
tumor cell responsiveness to radiation. Semin Cancer Biol.
35:180–190. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Fan QW and Weiss WA: Inhibition of
PI3K-Akt-mTOR signaling in glioblastoma by mTORC1/2 inhibitors.
Methods Mol Biol. 821:349–359. 2012. View Article : Google Scholar :
|
|
48
|
Westhoff MA, Karpel-Massler G, Brühl O,
Enzenmüller S, La Ferla-Brühl K, Siegelin MD, Nonnenmacher L and
Debatin KM: A critical evaluation of PI3K inhibition in
Glioblastoma and Neuroblastoma therapy. Mol Cell Ther. 2:322014.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Opel D, Westhoff MA, Bender A, Braun V,
Debatin KM and Fulda S: Phosphatidylinositol 3-kinase inhibition
broadly sensitizes glioblastoma cells to death receptor- and
drug-induced apoptosis. Cancer Res. 68:6271–6280. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Trovò L, Ahmed T, Callaerts-Vegh Z, Buzzi
A, Bagni C, Chuah M, Vandendriessche T, D’Hooge R, Balschun D and
Dotti CG: Low hippocampal PI(4,5)P(2) contributes to reduced
cognition in old mice as a result of loss of MARCKS. Nat Neurosci.
16:449–455. 2013. View Article : Google Scholar
|
|
51
|
Wang J, Gambhir A, Hangyás-Mihályné G,
Murray D, Golebiewska U and McLaughlin S: Lateral sequestration of
phos-phatidylinositol 4,5-bisphosphate by the basic effector domain
of myristoylated alanine-rich C kinase substrate is due to
nonspecific electrostatic interactions. J Biol Chem.
277:34401–34412. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Glaser M, Wanaski S, Buser CA, Boguslavsky
V, Rashidzada W, Morris A, Rebecchi M, Scarlata SF, Runnels LW,
Prestwich GD, et al: Myristoylated alanine-rich C kinase substrate
(MARCKS) produces reversible inhibition of phospho-lipase C by
sequestering phosphatidylinositol 4,5-bisphosphate in lateral
domains. J Biol Chem. 271:26187–26193. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Vadlakonda L, Pasupuleti M and Pallu R:
Role of PI3K-AKT-mTOR and Wnt Signaling Pathways in Transition of
G1-S Phase of Cell Cycle in Cancer Cells. Front Oncol. 3:852013.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Jarboe JS, Anderson JC, Duarte CW, Mehta
T, Nowsheen S, Hicks PH, Whitley AC, Rohrbach TD, McCubrey RO, Chiu
S, et al: MARCKS regulates growth and radiation sensitivity and is
a novel prognostic factor for glioma. Clin Cancer Res.
18:3030–3041. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Masaki T, Tokuda M, Yoshida S, Nakai S,
Morishita A, Uchida N, Funaki T, Kita Y, Funakoshi F, Nonomura T,
et al: Comparison study of the expressions of myristoylated
alanine-rich C kinase substrate in hepatocellular carcinoma, liver
cirrhosis, chronic hepatitis, and normal liver. Int J Oncol.
26:661–671. 2005.PubMed/NCBI
|
|
56
|
Chen CH, Statt S, Chiu CL, Thai P, Arif M,
Adler KB and Wu R: Targeting myristoylated alanine-rich C kinase
substrate phosphor-ylation site domain in lung cancer. Mechanisms
and therapeutic implications. Am J Respir Crit Care Med.
190:1127–1138. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Li H, Chen G, Zhou B and Duan S: Actin
filament assembly by myristoylated alanine-rich C kinase
substrate-phosphati-dylinositol-4,5-diphosphate signaling is
critical for dendrite branching. Mol Biol Cell. 19:4804–4813. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Nairn AC and Aderem A: Calmodulin and
protein kinase C cross-talk: The MARCKS protein is an actin
filament and plasma membrane cross-linking protein regulated by
protein kinase C phosphorylation and by calmodulin. Ciba Found
Symp. 164:145–161. 1992.PubMed/NCBI
|
|
59
|
Sundaram M, Cook HW and Byers DM: The
MARCKS family of phospholipid binding proteins: Regulation of
phospholipase D and other cellular components. Biochem Cell Biol.
82:191–200. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Adan A, Kiraz Y and Baran Y: Cell
proliferation and cytotoxicity sssays. Curr Pharm Biotechnol.
17:1213–1221. 2016. View Article : Google Scholar
|
|
61
|
Posimo JM, Unnithan AS, Gleixner AM, Choi
HJ, Jiang Y, Pulugulla SH and Leak RK: Viability assays for cells
in culture. J Vis Exp. 83:506452014.
|
|
62
|
Yu D, Makkar G, Dong T, Strickland DK,
Sarkar R and Monahan TS: MARCKS signaling differentially regulates
vascular smooth muscle and endothelial cell proliferation through a
KIS-, p27kip1-dependent mechanism. PLoS One. 10:e01413972015.
View Article : Google Scholar
|
|
63
|
Franken NA, Rodermond HM, Stap J, Haveman
J and van Bree C: Clonogenic assay of cells in vitro. Nat Protoc.
1:2315–2319. 2006. View Article : Google Scholar
|
|
64
|
Borràs M, Armengol G, De Cabo M,
Barquinero JF and Barrios L: Comparison of methods to quantify
histone H2AX phosphorylation and its usefulness for prediction of
radiosensi-tivity. Int J Radiat Biol. 91:915–924. 2015. View Article : Google Scholar
|
|
65
|
Thinon E, Serwa RA, Broncel M, Brannigan
JA, Brassat U, Wright MH, Heal WP, Wilkinson AJ, Mann DJ and Tate
EW: Global profiling of co- and post-translationally
N-myristoylated proteomes in human cells. Nat Commun. 5:49192014.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Ridley AJ: Rho GTPases and cell migration.
J Cell Sci. 114:2713–2722. 2001.PubMed/NCBI
|
|
67
|
Lott K and Cingolani G: The importin β
binding domain as a master regulator of nucleocytoplasmic
transport. Biochim Biophys Acta. 1813:1578–1592. 2011. View Article : Google Scholar
|
|
68
|
Vargova J, Vargova K, Dusilkova N, Kulvait
V, Pospisil V, Zavadil J, Trneny M, Klener P and Stopka T:
Differential expression, localization and activity of MARCKS
between mantle cell lymphoma and chronic lymphocytic leukemia.
Blood Cancer J. 6:e4752016. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Herhaus L, Perez-Oliva AB, Cozza G,
Gourlay R, Weidlich S, Campbell DG, Pinna LA and Sapkota GP: Casein
kinase 2 (CK2) phosphorylates the deubiquitylase OTUB1 at Ser16 to
trigger its nuclear localization. Sci Signal. 8:ra352015.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Gao Y and Wang HY: Casein kinase 2 Is
activated and essential for Wnt/beta-catenin signaling. J Biol
Chem. 281:18394–18400. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Tu X, Joeng KS, Nakayama KI, Nakayama K,
Rajagopal J, Carroll TJ, McMahon AP and Long F: Noncanonical Wnt
signaling through G protein-linked PKCdelta activation promotes
bone formation. Dev Cell. 12:113–127. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Jin BY, Lin AJ, Golan DE and Michel T:
MARCKS protein mediates hydrogen peroxide regulation of endothelial
permeability. Proc Natl Acad Sci USA. 109:14864–14869. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Chen WC, McBride WH, Iwamoto KS, Barber
CL, Wang CC, Oh YT, Liao YP, Hong JH, de Vellis J and Shau H:
Induction of radioprotective peroxiredoxin-I by ionizing
irradiation. J Neurosci Res. 70:794–798. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Mesiwala AH, Farrell L, Santiago P, Ghatan
S and Silbergeld DL: The effects of hydrogen peroxide on brain and
brain tumors. Surg Neurol. 59:398–407. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Xu XH, Deng CY, Liu Y, He M, Peng J, Wang
T, Yuan L, Zheng ZS, Blackshear PJ and Luo ZG: MARCKS regulates
membrane targeting of Rab10 vesicles to promote axon development.
Cell Res. 24:576–594. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Yu D, Makkar G, Strickland DK, Blanpied
TA, Stumpo DJ, Blackshear PJ, Sarkar R and Monahan TS:
Myristoylated alanine-rich protein kinase substrate (MARCKS)
regulates small GTPase Rac1 and Cdc42 activity and is a critical
mediator of vascular smooth muscle cell migration inintimal
hyperplasia formation. J Am Heart Assoc. 4:e0022552015. View Article : Google Scholar
|
|
77
|
Patel AP, Tirosh I, Trombetta JJ, Shalek
AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT,
Martuza RL, et al: Single-cell RNA-seq highlights intratumoral
heterogeneity in primary glioblastoma. Science. 344:1396–1401.
2014. View Article : Google Scholar : PubMed/NCBI
|