Introduction
Genetic mutations in cancer cells promote
preferential clonal proliferation and survival over normal cells.
In addition to genetic mutations which alter the inherent
characteristics of cancer cells, the physical and biological
properties of their microenvironment promote their growth (1,2). The
constitutive activation of glycolysis in cancer cells leads to
increased lactic acid production and tumor microenvironment
acidification by extracellular proton (H+) transport
through vacuolar V-ATPases. The functional expression of these
(V)-ATPases on the cancer cell plasma membrane facilitates the
maintenance of neutral intracellular and acidic extracellular pH
(2). One of the key mechanisms for
chemoresistance is the acidification of the tumor microenvironment
(3,4).
Previous studies have revealed the antitumor effects
and drug resistance reversal characteristics of proton pump
inhibitors (PPIs) in numerous cancer types (4-10).
It has been suggested that agents clinically used in peptic
diseases to suppress gastric acid production, such as lansoprazole
(LPZ), esomeprazole (EPZ), pantoprazole (PPZ), and omeprazole
(OPZ), act by inhibiting H+/K+-ATPases to
suppress V-ATPase-mediated H+ transport in tumor cells
(2,7). This inhibition reverses
extra-cellular acidification and suppresses formation of
intracellular acidic vesicles, including lysosomes, to enhance
tumor cell sensitivity to anticancer agents (4,5).
PPIs have been revealed to induce apoptosis and antimigratory
effects (4). PPI treatment
synergized with doxorubicin in breast cancer cell lines (5), and PPZ was revealed to induce
mitochondrial apoptosis and attenuate the NF-κB signaling pathway
in glioma cells (8). In addition,
PPIs reversed epithelial ovarian cancer paclitaxel resistance by
alkalinizing the acidic tumor microenvironment created by V-ATPase
D1 and inhibiting the multidrug resistance Yes-associated protein
(9). In another study, the
PPZ-mediated increased lysosomal pH caused inhibition of acid
phosphatase activity and sensitized the chemo-resistant oral
epidermoid carcinoma cells to vincristine (VCR) by reducing VCR
lysosomal sequestration (10).
These studies suggest combining PPIs with anticancer drugs as a
promising approach to further enhance chemotherapy efficacy
(3,4).
Autophagy recycles the cellular components and may
facilitate cell survival after chemotherapy (11). PPZ has been revealed to inhibit
autophagy in a time- and dose-dependent manner and sensitize cancer
cells to anticancer drugs (12).
PPZ was revealed to inhibit docetaxel-induced autophagy and reverse
docetaxel resistance to potentiate its in vitro toxicity.
This effect was confirmed in vivo in tumor sections with
increased γH2AX foci and cleaved caspase-3 expression and decreased
Ki67 expression (13). These
results confirmed the involvement of autophagy as the underlying
mechanism of docetaxel chemotherapy resistance. In contrast, EPZ
has been reported to induce autophagy as a survival response to
oxidative stress in human melanoma cells (14). Therefore, the role of PPIs in
autophagic flux is still controversial, and their precise
underlying molecular mechanisms are yet to be elucidated.
Our group as well as other research groups have
reported that macrolide antibiotics such as azithromycin (AZM) and
clarithromycin (CAM) potently inhibit autophagic flux as an
off-target effect (15-17). Combining AZM or CAM with the
epidermal growth factor receptor inhibitors (e.g., gefitinib and
erlotinib), which are potent inducers of autophagy, enhanced their
antitumor effect against pancreatic and non-small cell lung cancer
(NSCLC) cell lines (18,19). In addition, we revealed that
concurrent inhibition of the ubiquitin-proteasome and
autophagy-lysosome systems by bortezomib (proteasome inhibitor) and
macrolides synergistically induced endoplasmic reticulum
stress-mediated cytotoxicity in multiple myeloma and breast cancer
cell lines (15,20). Since the combination of PPIs and
macrolide antibiotics is a well-established clinical therapy for
Helicobacter pylori infection in chronic gastritis (21), in the present study, it was
investigated whether the LPZ + AZM drug combination could be
repurposed for cancer treatment.
Materials and methods
Reagents
LPZ and OPZ were purchased from Wako Pure Chemical
Industries and dissolved in dimethyl sulfoxide (DMSO) (Wako Pure
Chemical Industries) to prepare 50 mM stock solutions. AZM and CAM
were purchased from Tokyo Chemical Industry and dissolved in DMSO
to prepare 10 mM stock solutions. Z-VAD-fmk, a pan-caspase
inhibitor, was purchased from Peptide Institute, Inc. Necrostatin-1
(NEC-1), a specific inhibitor of receptor-interacting
serine/threonine-protein kinase 1 (RIPK1), was purchased from Enzo
Life Sciences. Thapsigargin was purchased from Nacalai Tesque, Inc.
Staurosporine, TNF-α, and gefitinib were purchased from Wako Pure
Chemical Industries. L-Leucyl-L-Leucine methyl ester
(hydrochloride) (LLOMe) was purchased from Cayman Chemical Company.
Cycloheximide was purchased from Calbiochem; Merck KGaA.
Cell lines and culture conditions
The human cancer cell lines, A549 (NSCLC), CAL 27
(oral squamous cell carcinoma), Detroit 562 (pharyngeal carcinoma),
PANC-1 (pancreatic cancer), and HT-29 (colon adenocarcinoma) were
obtained from the American Type Culture Collection. The A549 cell
line was cultured in Roswell Park Memorial Institute-1640 medium,
whereas all other cell lines were cultured in Dulbecco's modified
Eagle's medium (DMEM). Both media were supplemented with 10% fetal
bovine serum (FBS) (Biosera) and 1% penicillin/streptomycin (Wako
Pure Chemical Industries). Cell cultures were maintained at 37°C in
a humidified incubator under 5% CO2 and 95% air. All
cell line experiments were conducted within 10 passages after
thawing. Mycoplasma contamination was tested routinely using the
e-Myco™ Mycoplasma PCR Detection kit ver.2.0 (iNtRON Biotechnology,
Inc.).
Cell viability and proliferation
assays
The number of viable cells was assessed by the
CellTiter Blue cell viability assay kit (Promega Corporation)
according to the manufacturer's instructions. Briefly, cells were
plated in a 96-well flat-bottom culture plate at a density of
3×103 cells/well and cultured for up to 72 h at 37°C in
a CO2 incubator in the presence of LPZ or OPZ at various
concentrations with/without either AZM or CAM at 50 µM.
Fluorescence (560 nm excitation, 590 nm emission) was measured
using fluorometer SpectraMax iD3 (Molecular Devises, LLC). For the
positive control of RIPK1-dependent cell death, A549 cells were
treated with 25 µM gefitinib in amino acid-free DMEM (cat.
no. 048-33575; Wako Pure Chemical Industries) supplemented with 10%
FBS and 1% penicillin/streptomycin as previously described
(22). Cell confluency was used to
monitor cell proliferation and was evaluated using the IncuCyte
ZOOM 2016B software (Essen BioSciences).
Human ATG5 knockout by
CRISPR/Cas9-mediated genome editing
The target sequences for CRISPER interference were
as follows: Human ATG5 (exon 3), AAG AGT AAG TTA TTT GAC GT;
non-targeting control, GTA GCG AAC GTG TCC GGC GT (23). Two complementary oligonucleotides
with BpiI restriction sites for guide RNAs (gRNAs) were
synthesized at Fasmac, Inc., and were cloned into the pSpCas9
(BB)-2A-Puro (pX459) V2.0 plasmid vector (gift from Dr Feng Zhang;
plasmid cat. no. 48139; Addgene) (24) following an online protocol from the
Feng Zhang laboratory (https://www.addgene.org/62988/). A549 cells
(1×107 cells) were suspended in 100 µl of
Opti-MEM I (cat. no. 31985-070, Thermo Fisher Scientific, Inc.)
with 10 µg of pX459-gRNA and electroporated using the Super
Electroporator NEPA 21 (NEPA GENE Co. Ltd.) with a 2-mm gap cuvette
(cat.no. EC-002; NEPA GENE) under the following conditions: Poring
pulse: Voltage, 120 V; pulse interval, 50 ms; pulse width, 10 ms;
pulse number, 1 and attenuation rate 10%; transfer pulse: Voltage,
20 V; pulse interval, 50 ms; pulse width, 50 ms; pulse number, 5
and attenuation rate 40%. One day after electroporation, cells were
incubated for 2 days with 2 µg/ml of puromycin
dihydrochloride (Wako Pure Chemical Industries). The individual
puromycin-resistant clones were isolated using a cloning ring.
After successfully obtaining clones, A549/ATG5 knock out (KO) and
A549/control cells were used for the following experiments.
Morphological assessments
Cells were seeded onto a 60-mm dish at
1×106 cells /dish and treated with various reagents for
48 h. After treatment with LPZ and/or AZM, the adherent cells were
harvested by trypsinization. Cell spreads were prepared on glass
slides using a Cytospin 4 centrifuge (Thermo Fisher Scientific,
Inc.) [1,000 × g, for 5 min, at room temperature (RT)].
May-Grünwald-Giemsa staining was performed with May-Grünwald's
stain solution (without dilution; cat. no. 15053; Muto Pure
Chemicals) for 3 min at RT followed with Giemsa's stain solution (1
drop/1 ml H2O; cat. no. 15003; Muto Pure Chemicals) for
15 min at RT. Glass slides were examined under a digital light
microscope (BZ-8100; Keyence Corporation) (objective magnification,
×100). Representative images were selected.
Transmission electron microscopy
(TEM)
Cells were seeded onto a 60-mm dish at
1×106 cells/dish and treated with various reagents for
48 h. Then, the cells were fixed for 1 h at 4°C with 2.5%
glutaraldehyde (TAAB Laboratories Equipment, Ltd.) in 0.1 M
phosphate buffer (pH 7.3) (prepared with monobasic sodium phosphate
and dibasic sodium phosphate; Wako Pure Chemical Industries), and
fixed subsequently for 1 h at RT in 1% osmium tetroxide (Nisshin EM
Co., Ltd.), dehydrated in graded ethanol (30-100%), and embedded in
Quetol 812 epoxy resin (Nisshin EM Co., Ltd.) at 60°C for 2-4 days.
ultrathin sections (60 nm) were obtained using an Ultracut J
micro-tome (Reichert Jung), and the sections were stained with 4%
lead nitrate (RT, 5 min) and saturated uranium acetate (RT, 10 min)
and imaged using a transmission electron micro-scope JEM-1200EX II
(JEOL, Ltd.) (magnification ranging from ×1,000 to ×10,000). All
images were captured on films (Electron-microscopic film FG;
Fujifilm).
Flow cytometry
To assess apoptosis, cells were suspended at
1×106 cells/ml in Annexin V binding buffer and stained
using an Annexin V-FITC apoptosis detection kit (Nacalai Tesque,
Inc.) according to the manufacturer's instructions to detect
Annexin V and propidium iodide (PI) staining. Samples were analyzed
by flow cytometry using the Attune Acoustic Focusing Cytometer
(Life Technologies; Thermo Fisher Scientific, Inc.). Data analysis
were performed with Attune Cytometric software v2.1 (Life
Technologies; Thermo Fisher Scientific, Inc.).
Immunoblotting
Immunoblotting was performed as previously described
(17). Briefly, the cells were
seeded onto 60 mm dish at 1×106 cells/dish and treated
with various reagents for optimal duration. Thereafter, the cells
were lysed with RIPA lysis buffer (Nacalai Tesque, Inc.)
supplemented with a protease and phosphatase inhibitor cocktail
(Nacalai Tesque, Inc.). Protein concentrations were quantified by
the Bradford assay (Thermo Fisher Scientific, Inc.). Proteins (15
µg) were loaded and separated by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (7.5, 10, and
15% gels were used) and transferred to Immobilon-P membranes (EMD
Millipore). The membranes were probed at 4°C for overnight with the
following primary antibodies (Abs): Anti-caspase-3 Ab (1:1,000;
product no. 9662S), anti-poly (ADP-ribose) polymerase (PARP) Ab
(1:1,000; cat. no. 9542S), anti-RIPK1 Ab (1:1,000; product no.
4926S), and anti-phospho-RIPK1 (Ser166) mAb (1:1,000; cat. no.
65746S). All the aforementioned antibodies were purchased from Cell
Signaling Technology, Inc. Anti-p62/SQSTM1 mAb (1:1,000; cat. no.
sc-28359), anti-H+/K+ ATPase-β (C-4) mAb
(1:1,000; cat. no. sc-374094), anti-GAPDH mAb (1:1,000; cat. no.
sc-32233), and anti-β-actin mAb (1:1,000; cat. no. sc-47778) were
obtained from Santa Cruz Biotechnology, Inc. and anti-LC3B Ab
(1:4,000; cat. no. NB600-1384) was purchased from Novus
Biologicals, LLC. Anti-mixed lineage kinase domain-like protein
(MLKL) Ab (1:1,000; product code ab183770) and anti-phospho-MLKL
(Ser358) Ab (1:1,000; product code ab187091) were purchased from
Abcam. Immunoreactive proteins were detected at RT for 1 h with
horseradish peroxidase-conjugated secondary Abs (anti-mouse: cat.
no. 115-035-003, at 1:5,000 dilution; anti-rabbit: cat. no.
711-035-152, at 1:5,000 dilution; Jackson ImmunoResearch
Laboratories, Inc.) and visualized with an enhanced
chemiluminescence reagent (EMD Millipore). Protein bands were
imaged and analyzed using the ChemiDoc XRS system (Bio-Rad
Laboratories, Inc.). For positive control of necroptosis, HT-29
cells were pre-treated with Z-VAD-fmk (20 µM) for 30 min,
and subsequently treated with cycloheximide (10 µg/ml) and
TNF-α (20 ng/ml) for 8 h as previously described (22). For positive control of cleaved
caspase-3 and cleaved PARP, CAL 27 cells were treated with 1
µM staurosporine for 4 h.
Fluorescent dextran uptake
Cells were cultured in CELLview 35-mm glass-bottom
cell culture dishes with four compartments (cat. no. 627870;
Greiner Bio-One). Two days after seeding (8×104
cells/dish), cells were incubated for 8 h with 50 µg/ml
Alexa Fluor 488 dextran (molecular weight: 10,000; cat. no. D22910;
Thermo Fisher Scientific, Inc.) in a CO2 incubator at
37°C. The medium containing fluorescent dextran was subsequently
replaced with fresh medium containing LysoTracker Red (50 nM) (Life
Technologies; Thermo Fisher Scientific, Inc.) and cultured further
for up to 6 h with LPZ or AZM alone or in combination at 37°C, 5%
CO2 under humidified conditions on a stage top incubator
(Carl Zeiss AG). Fluorescence images were captured by confocal
micros-copy (LSM 700; Carl Zeiss) (objective magnification, ×63)
and analyzed using the ZEN 2.3 SP1 Black Edition software (Carl
Zeiss AG). Alexa488 fluorescence intensity or number of particles
of each cell at 0 and 6 h were analyzed by ImageJ (1.50i; National
Institutes of Health), and the fold change of fluorescence
intensity or number of particles were calculated.
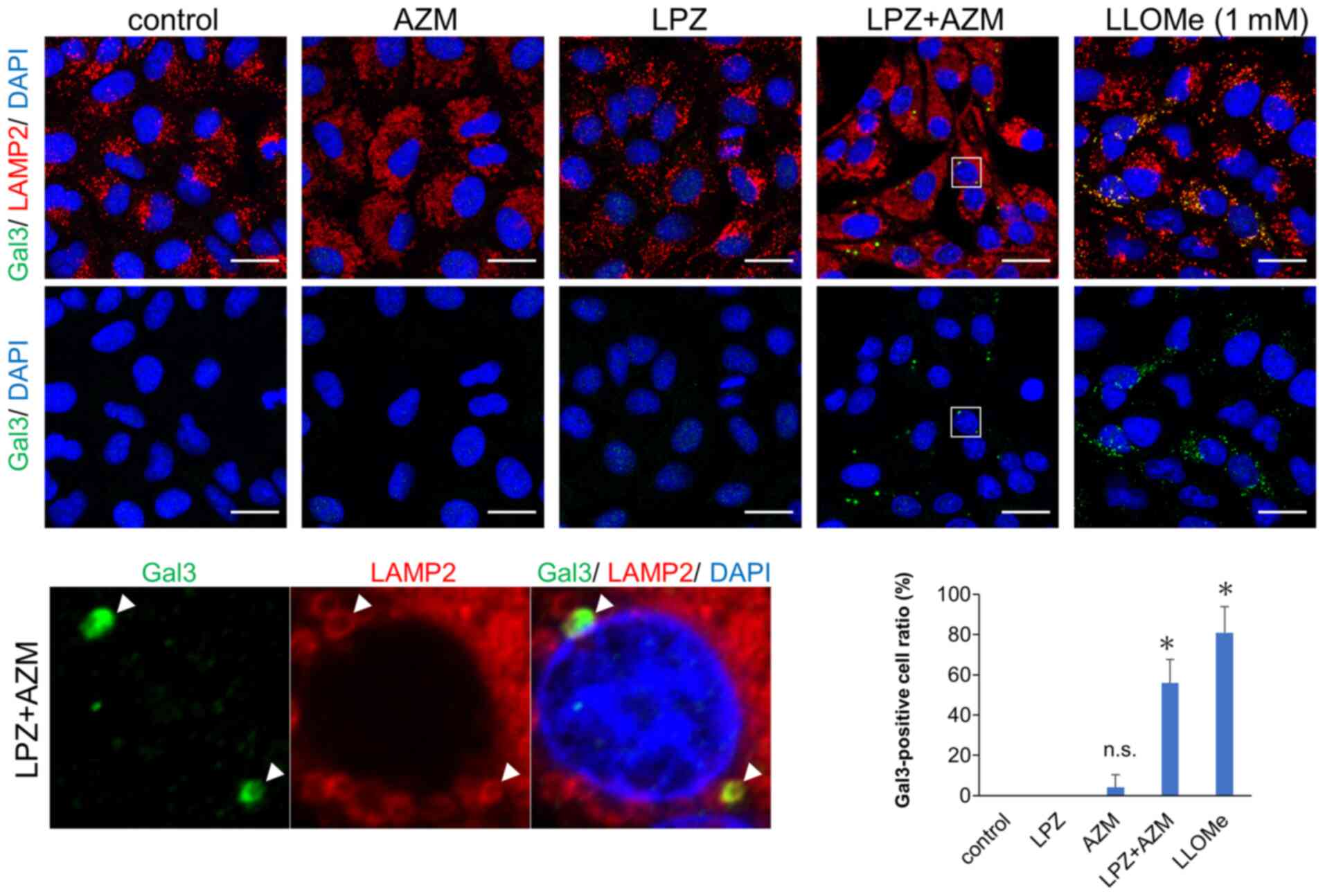
Galectin puncta assay for lysosomal
membrane permeabilization detection
The lysosomal membrane permeabilization was assessed
by fluorescence immunocytochemistry. Cells were fixed with methanol
at -20°C for 10 min, blocked with 10% normal goat serum in TBST,
and incubated with anti-galectin-3 (1:100 at 4°C, overnight; cat.
no. 87985; Cell Signaling Technology, Inc.) and
anti-lysosome-associated membrane protein-2 (LAMP-2; 1:100 at 4°C,
overnight; cat. no. sc-18822, Santa Cruz Biotechnology, Inc.); the
latter is a lysosomal and late endosomal marker (25). Then, cells were incubated with
anti-mouse IgG Alexa 488 and anti-Rabbit IgG Alexa 555 antibodies
(cat. nos. A11029 and A21428; Molecular Probes; Thermo Fisher
Scientific, Inc.) (1:250 at RT for 1 h). Confocal microscopic
observation was performed using an LSM 700 confocal laser scanning
microscope (Carl Zeiss AG) (objective magnification, ×63). As a
positive control for lysosomal membrane permeabilization (LMP),
cells were treated with 1 mM LLOMe for 3 h.
Statistical analysis
All the quantitative data were expressed as the mean
± standard deviation (SD). Statistical analysis except for the
galectin-3 puncta assay and band intensity analysis of immunoblots
were performed with two-way ANOVA, followed by Bonferroni's
multiple comparison test. For the galectin-3 puncta assay and
immunoblotting analysis, one-way ANOVA followed by Bonferroni's
multiple comparison test was used. A P-value <0.05 was
considered to indicate a statistically significant difference. All
analyses were performed with GraphPad Prism 5 software (GraphPad
Software, Inc.).
Results
Combination treatment with LPZ and AZM
reveals enhanced cytotoxicity in cancer cells
Western blot results from all the tested cell lines
revealed the presence of H+/K+-ATPase-β
subunit protein, a PPI target (Fig.
1A). Cells were treated with varying concentrations (0, 1, 10,
50, 100 and 250 µM) of LPZ and OPZ for 24 and 48 h, and the
cell viability was assessed. LPZ and OPZ inhibited the growth of
all cell lines in both time- and dose-dependent manners (Fig. 1B), with a greater growth inhibition
observed with LPZ-treated cells. It has been suggested that
PPI-treatment increases the pH of the tumor microenvironment
(3,4). However, no pH differences between
LPZ- or OPZ-treated and untreated culture media were observed after
monitoring for over 72 h (data not shown). In addition, changing
the culture media pH from 6.0 to 8.0 did not affect the PPI
cytotoxicity profile of tumor cell lines (data not shown). Hence,
subsequent experiments with LPZ were conducted with cells cultured
in pH 7.3 media.
 | Figure 1PPIs inhibit the growth of
H+/K+-ATPase-β-expressing cancer cell lines.
(A) Immunoblots revealing H+/K+-ATPase-β
expression. Cancer cell line proteins were separated on an 11.25%
SDS-PAGE gel and probed with
anti-H+/K+-ATPase-β Ab. (B) A549, CAL 27,
Detroit, and PANC-1 cells were incubated with LPZ or OPZ at various
concentrations (0, 1, 10, 50, 100, 250 µM) for 24 and 48 h.
Cell viability was assessed using a CellTiter Blue viability assay,
as described in the Materials and methods section, and the
viability of cells (control) without drug treatment was set as
100%. (n=5; mean ± SD). *P<0.05 vs. the control.
PPIs, proton pump inhibitors; LPZ, lansoprazole; OPZ,
omeprazole. |
Next, we examined the drug combination treatment
effects by adding either LPZ + CAM, LPZ + AZM, or LPZ alone.
Combination treatment potentiated the cytotoxicity effects as
compared to that by LPZ alone (Fig.
2A). The increased cytotoxicity after 48-h incubation with LPZ
+ AZM or LPZ + CAM was observed in CAL 27 and A549 cells, and this
effect was confirmed by cell confluency assays with the IncuCyte
ZOOM (Fig. 2B). In addition, the
enhancing effect of AZM was greater than that of CAM (Fig. 2A and B). We examined the morphology
of May-Grünwald-Giemsa-stained A549 and CAL 27 cells after LPZ and
AZM treatments to evaluate the cytotoxic vs. cytostatic effect.
A549 and CAL 27 cells did not exhibit the typical apoptotic
morphological features of cells, such as condensed chromatin,
nuclear fragments, and apoptotic bodies; however, they exhibited
cytoplasmic and nuclear swelling with reduced plasma membrane
integrity (Fig. 2C), thus
indicating a non-apoptotic mode of cell death. Cell cycle analysis
after treatment with LPZ and AZM revealed no cytostatic effects
(data not shown).
Combination treatment with LPZ and AZM
causes accumulation of autolysosomes containing undigested
cytoplasmic debris
The cell-death phenotype after LPZ and AZM treatment
was verified by conducting TEM of A549 cells incubated with either
AZM (50 µM), LPZ (100 µM), or both for 48 h.
AZM-treated cells exhibited a significantly increased number of
swollen autolysosomes in the cytoplasm (Fig. 3A and C). Notably, in LPZ-treated
cells, the lysosomes and autolysosomes were localized to the
perinuclear regions, whereas a majority of the swollen
autolysosomes were distributed evenly in the cytoplasm of
AZM-treated cells (Fig. 3B and C).
In addition, the autolysosomes in LPZ-treated cells were smaller
than those in AZM-treated cells. The combination treatment with LPZ
+ AZM caused an increased number of swollen autolysosomes
containing undigested cellular components, including
autophagosomes/lysosomes, and were distributed in the cytoplasm of
adherent cells (Fig. 3D).
Non-adherent A549 cells, mostly dead cells that lost their ability
to adhere, contained the largest autolysosomes containing
undigested materials (Fig. 3E),
and some cells also had condensed chromatin. However, the dying
cells were swollen and had enlarged organelles and reduced plasma
membrane integrity. These observations are consistent with the
necrotic cell death processes along with increased autolysosome
numbers, although no morphological changes associated with
apoptosis, such as fragmented nuclei and apoptotic bodies, were
observed (26).
 | Figure 3Transmission electron microscopy of
A549 cells treated with LPZ or AZM or both. A549 cells were
cultured for 48 h in complete culture medium containing (A) 0.1%
DMSO as a control, treated with (B) 50 µM AZM, or with (C)
100 µM LPZ for 48 h. (D) Adherent A549 cells after 48
h-treatment with AZM (50 µM) and LPZ (100 µM). (E)
Non-adherent A549 cells after 48 h-treatment with the LPZ + AZM
combination. Scale bar represents the magnification. The right
panels reveal enlarged images of the section indicated by the
square box in the left panels. N, nucleus; Mt, mitochondria; open
(white) arrowhead, lysosome; closed (black) arrow, autolysosome;
open (white) arrow, autophagosome. LPZ, lansoprazole; AZM,
azithromycin; DMSO, dimethyl sulfoxide. |
Combination treatment with LPZ and AZM
exhibits atypical cell death phenotypes
To confirm that cells treated with the combination
of LPZ and AZM undergo non-apoptotic cell death, flow cytometric
analysis of PI and Annexin V-stained cells was performed. As
revealed in Fig. 4A, after LPZ +
AZM treatment, there were more PI+/Annexin V−
cells, as compared to PI−/Annexin V+ cells,
thus indicating cells did not undergo early apoptosis (26). Immunoblots of protein expression
did not detect caspase-3 or PARP cleavage (Fig. 4B). Furthermore, treatment with
Z-VAD-fmk, a pan-caspase inhibitor, did not rescue cells from LPZ +
AZM treatment-induced cytotoxicity (Fig. 4C). Collectively, these results
suggest that LPZ + AZM treatment induced a non-apoptotic form of
cell death. Hence, it was investigated whether LPZ + AZM treatment
induces necroptosis instead. However, following LPZ + AZM
treatment, phosphorylation of RIPK1 and MLKL was undetectable
(Fig. 4D). Additionally, cell
death could not be prevented by co-culturing cells with the RIPK1
inhibitor NEC-1, although NEC-1 exhibited the significant
cancellation of GEF-induced cell death under amino-acid-depleted
culture conditions, which was used as a positive control for
RIPK1-dependent cell death induction (22) (Fig.
4E). Thus, the involvement of necroptosis in LPZ + AZM-induced
cell death can also be excluded.
 | Figure 4Mechanism of cell death after
treatment with LPZ or AZM or both in A549 cells. (A) Flow cytometry
of Annexin V/PI double-stained A549 cells 24 and 48 h after
treatment with 0.1% DMSO (control), LPZ (100 µM), AZM (50
µM), or LPZ + AZM combination. The vertical axis indicates
the log fluorescence intensity of PI, and the horizontal axis
indicates the log fluorescence intensity of Annexin V. The numbers
indicate the percentage of cells in each area. The percentage of
cells in each area were summarized and presented in the right
panel. (n=3; mean ± SD). *P<0.05 vs. the control,
#P<0.05 vs. LPZ. (B) Immunoblots of caspase-3 and
PARP expression in A549 cells cultured in control medium, LPZ (100
µM), AZM (50 µM), or LPZ + AZM for 24, 48, and 72 h.
CAL 27 cells cultured in complete culture medium with 1 µM
staurosporine for 4 h were used as the positive control. Band
intensities were standardized by β-actin. (C) The effect of
Z-VAD-fmk (25 and 50 µM) on the viability of A549 cells
treated with or without LPZ (100 µM) or AZM (50 µM)
or both for 48 h, as measured by a CellTiter Blue viability assay.
The viability of cells (control) without drug treatment was set as
100%. (n=3, mean ± SD). (D) Immunoblots of phospho-RIPK1, RIPK1,
phospho-MLKL, and MLKL expression in A549 cells cultured in control
medium, LPZ (100 µM), AZM (50 µM), or LPZ + AZM for
24 and 48 h. HT-29 cells treated with Z-VAD-fmk, cycloheximide, and
TNF-α indicated as ZCT were used as a positive necroptosis control
as previously described (22).
Relative band intensities of pRIPK1/RIPK1 and pMLKL/MLKL were
summarized in the right panel. (n=3; mean ± SD).
*P<0.05 vs. the control. (E) The effect of NEC-1 (25
and 50 µM) on the viability of A549 cells treated with or
without LPZ (100 µM) or AZM (50 µM) or both for 48 h.
GEF-treated A549 cells in amino acid-depleted culture conditions
were used as positive controls for RIPK1-dependent cell death as
previously described (22). The
viability of cells (control) without rug treatment was set as 100%.
(n=3, mean ± SD). *P<0.05 vs. without NEC-1. LPZ,
lansoprazole; AZM, azithromycin; DMSO, dimethyl sulfoxide; PI,
propidium iodide; RIPK1, receptor-interacting
serine/threonine-protein kinase 1; MLKL, mixed lineage kinase
domain-like protein; NEC-1, necrostatin-1; GEF, gefitinib, |
We previously reported that macrolide antibiotics,
such as AZM and CAM, suppress autophagic flux (15,17),
whereas EPZ was revealed to induce autophagy in melanoma cells as a
survival response to oxidative stress (14). However, recent studies revealed
that PPIs suppress autophagy in various cancer types (12,13).
As revealed in Fig. 5A, AZM
induced the autophagic substrate p62 accumulation and increased the
autophagosome marker LC3B-II expression in A549 cells (27). These changes indicated that AZM
treatment blocked the autophagic flux, leading to
autophagosome/autolysosome accumulation, as previously demonstrated
(15,17), and is consistent with the TEM
observations revealing accumulation of swollen autolysosomes
(Fig. 3C). After LPZ treatment,
the expression levels of p62 and LC3B-II were almost equivalent to
those in control cells. Co-administration of AZM and LPZ for 48 h
further increased LC3B-II expression compared with AZM alone. Thus,
the present results indicated that LPZ exhibited a minimal effect
on autophagic induction.
Since the number of autolysosomes increased in A549
cells after treatment with LPZ + AZM (Fig. 3D and E), the ATG5-knockout A549
cell line was next established to exclude autophagy involvement
(Fig. 5B). The cytotoxic response
to LPZ + AZM combination treatment did not differ between
ATG5-knockout and the parental cell lines (Fig. 5C), indicating a lack of involvement
of autophagic cell death in drug cytotoxicity. Collectively, LPZ
and AZM combination treatment induced a potent antitumor cytotoxic
response independent of apoptosis, necroptosis, or
autophagy-dependent cell death.
Combination treatment with LPZ and AZM
induces lysosomal membrane permeabilization in A549 cells
To identify the molecular mechanism responsible for
combination treatment-induced cytotoxicity, TEM images were
obtained revealing the necrosis-like phenotype with increased
autolysosomes containing undigested debris (Fig. 3E). Alexa 488-labeled dextran was
used to monitor the lysosomal degradation processes. In control
cells, numerous Alexa 488-dextran particles co-localized with
LysoTracker Red, and the Alexa 488 signal diminished after 6 h
(Fig. 6). This represents the
process by which Alexa 488-dextran is endocytosed, fused with
lysosomes, and subsequently digested by acidic hydrolases as part
of the lysosomal content. The cytoplasmic LysoTracker intensity was
reduced in the presence of AZM, and Alexa 488-dextran particles
were visible 6 h after AZM addition. The intensity and/or the
number of undigested Alexa 488-dextran particles was increased when
cells were treated with a combination of AZM + LPZ. These results
are consistent with the TEM images that revealed that the number of
autolysosomes increased in A549 cells after treatment with LPZ +
AZM and strongly suggest the impairment of lysosomal function along
with increased lysosomal pH (Fig.
3D).
Recent studies have revealed leakage of
intra-lysosomal hydrolases such as cathepsins due to several types
of stress-induced lysosomal membrane permeabilization (LMP),
resulting in lysosome-dependent cell death (LDCD) (28,29).
Thus, it is likely that accumulation of autolysosomes with
undigested contents induced LMP-associated cell death. A galectin
puncta assay involving double-immunostaining with anti-galactin-3
and anti-LAMP-2 Abs revealed that LMP, which is shown as
colocalized puncta of galectin-3 and LAMP-2, was induced by LPZ +
AZM combination treatment (Fig. 7)
(25). Notably, treatment with
either LPZ or AZM alone did not affect lysosomal galectin-3 puncta
expression. Therefore, the induction of non-apoptotic cell death by
LPZ + AZM drug combination appears to be mediated by LMP-associated
necrosis.
Discussion
In the present study, it is reported that LPZ + AZM
combination treatment induced a potent cytotoxic effect in several
cancer cell lines. Since AZM alone was not cytotoxic, AZM enhanced
LPZ-induced cell death acting as an agonist. However, the cell
death induced by this drug combination was not due to apoptosis,
necroptosis, or autophagy-dependent cell death and appears to be
unique. Necrosis along with a substantial number of autolysosomes
and lysosomes containing undigested materials were identified. The
impaired lysosomal function was also supported by the long-term
retention of Alexa 488-dextran in the endosomes. Additionally, LMP
was also induced.
Lysosomes recycle cellular components and contain
over 50 different acid hydrolases. These acid hydrolases are active
at relatively low pH (approximately 4-5) and can degrade most
cellular macromolecules (30).
Cytosolic galectins act as sensors for lysosomal damage by binding
to lysosomal β-galactosidases and are localized to the luminal side
of the lysosomal membrane and become accessible to galectins during
LMP (25,28,29).
LMP and the consequent cytosolic release of lysosomal acid
hydrolases result in an uncontrolled breakdown of cell components,
which leads to cell death by necrosis and LDCD (28). Thus, the cell death phenotype
observed in the present study could be categorized as 'LDCD', LDCD
can be invoked when the cell death execution is dependent on
cathepsin activity. Hence, cell death induction should be
suppressed by pharmacologic or genetic blockade of cathepsin
activities (28,29). However, no difference in LPZ +
AZM-mediated cytotoxicity was observed when CA-074, a cathepsin B
inhibitor, was added to the cultures (data not shown). Since
lysosomes contain numerous different acid hydrolases other than
cathepsin B, their role in the cell death processes by inhibiting
other cathepsins require further evaluation. Hence, the involvement
of necrosis along with LMP in the treatment-associated cytotoxicity
was surmised.
Next, it was determined whether LPZ + AZM
combination treatment enhances LMP-associated necrosis. As
demonstrated in the results, a galectin-3 puncta assay revealed
detectable LMP after concurrent treatment with AZM and LPZ, but not
with AZM or LPZ alone. In addition, significant lysosomal
dysfunction was observed after treatment with both drugs. AZM
blocks the later stage of autophagic flux and leads to the
accumulation of cytoplasmic autolysosomes (17,18).
However, ATG5 knockout in the A549 cell line resulted in complete
inhibition of autophagosome formation and did not affect LPZ + AZM
treatment-induced cytotoxicity. In addition, unlike previous
studies (12-14), we could not detect any autophagic
changes in LPZ-treated cells, regardless of whether it promotes or
suppresses autophagy. Therefore, the present results suggest that
autophagy did not play a significant role in the LPZ+AZM treatment
cytotoxicity.
Notably, AZM treatment increased the number of
LAMP-2-positive vesicles, which suggests that lysosomal biogenesis
was upregulated. AZM impaired lysosomal function and increased
lysosomal pH. PPIs, including LPZ, have been reported to inhibit
lysosomal enzyme activities, including acid phosphatase and
β-N-acetylglucosaminidase, both in vitro and in vivo
(31,32). The combination of enzyme activity
inhibition with lysosomal alkalization leads to lysosomal
impairment (33). Using the
LysoTracker Red reagent, decreased lysosomal acidification in cells
after AZM or LPZ + AZM treatments, were detected but not after
treatment with LPZ alone. Thus, the LPZ + AZM combination induced
significant accumulation of damaged lysosomes, leading to LMP.
Additionally, the cytosolic lysosomal membrane surface acts as a
signaling platform for the interaction of the mammalian target of
rapamycin complex 1 (mTORC1) with its cofactors in response to
stress and other cellular factors (34,35).
Impaired lysosomal accumulation signals mTORC1 release from the
lysosomal membranes. Subsequently, dephosphorylation of the master
regulator of lysosomal biogenesis transcription factor EB (TFEB)
occurs, causing its translocation to the nucleus, transcriptional
activation, and de novo lysosomal biogenesis (30). Concurrently, the damaged lysosomes
are removed by lysophagy (36).
Thus, the present results indicated that after AZM blocked the
autophagic flux, the feedback loop described below may explain the
resulting increase in impaired lysosome numbers, leading to LMP and
necrosis: LPZ and AZM induced lysosomal damage → lysosomal
biogenesis by TFEB → lysosomal accumulation due to AZM blocking
lysophagy → a considerable number of lysosomes with LMP →
pronounced LMP-mediated necrosis induction. Although further
studies are required to elucidate the underlying molecular
mechanisms, this hypothesis adequately explains our TEM
findings.
In the present study, it was reported that AZM
potently enhanced the antitumor effects of LPZ in various cancer
cell lines via necrosis induction and LMP. The present results
indicate the potential of AZM and LPZ for use in cancer
therapeutics by the induction of LMP-mediated tumor cell death.
Although these drugs are in clinical use, caution is advised to
minimize adverse events such as necrosis-induced inflammatory
response. Further studies on cancer cell specificity and to reduce
the likelihood of the non-specific targeting of normal cells are
warranted.
Acknowledgments
We thank Ms Yumiko Yamada, Ms Ayako Hirota, and Ms
Hiromi Kazama (Department of Biochemistry, Tokyo Medical
University, Tokyo, Japan) for their technical assistance, helpful
advice, and fruitful discussions.
Abbreviations:
|
PPIs
|
proton pump inhibitors
|
|
LPZ
|
lansoprazole
|
|
EPZ
|
esomeprazole
|
|
PPZ
|
pantoprazole
|
|
OPZ
|
omeprazole
|
|
VCR
|
vincristine
|
|
AZM
|
azithromycin
|
|
CAM
|
clarithromycin
|
|
NSCLC
|
non-small cell lung cancer
|
|
DMSO
|
dimethyl sulfoxide
|
|
RIPK1
|
receptor-interacting
serine/threonine-protein kinase 1
|
|
PARP
|
poly(ADP-ribose) polymerase
|
|
LMP
|
lysosomal membrane
permeabilization
|
|
LDCD
|
lysosome-dependent cell death
|
|
mTORC1
|
mammalian target of rapamycin complex
1
|
|
TFEB
|
transcription factor EB
|
Funding
The present study was supported by funds provided by
the Strategic Research Foundation at Private Universities (grant
no. S1411011, 2014-2018) from the Ministry of Education, Culture,
Sports, Science, and Technology (MEXT) of Japan awarded to KM, and
by JSPS KAKENHI grants (no. 17K08771) to KM, (no. 17K15031) to NT,
and (no. 18K15031) to HH.
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
NT and KM designed the experiments. AT performed
most of the experiments and analyzed the data. HK performed
trans-mission electron microscopy. NT, HH, SM, and AA assisted AT
for acquisition and analysis of data. NT, MH, and KT were involved
in the conception and mentoring, as well as in critical
interpretation and evaluation of the data. AT, NT, MH, and KM were
involved in writing, reviewing, and editing the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient's consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kato Y, Ozawa S, Miyamoto C, Maehata Y,
Suzuki A, Maeda T and Baba Y: Acidic extracellular microenvironment
and cancer. Cancer Cell Int. 13:892013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Luciani F, Spada M, De Milito A, Molinari
A, Rivoltini L, Montinaro A, Marra M, Lugini L, Logozzi M, Lozupone
F, et al: Effect of proton pump inhibitor pretreatment on
resistance of solid tumors to cytotoxic drugs. J Natl Cancer Inst.
96:1702–1713. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lu ZN, Tian B and Guo X: Repositioning of
proton pump inhibitors in cancer therapy. Cancer Chemother
Pharmacol. 80:925–937. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Spugnini EP and Fais S: Drug repurposing
for anticancer therapies. A lesson from proton pump inhibitors.
Expert Opin Ther Pat. 30:15–25. 2020. View Article : Google Scholar
|
|
5
|
Ihraiz WG, Ahram M and Bardaweel SK:
Proton pump inhibitors enhance chemosensitivity, promote apoptosis,
and suppress migration of breast cancer cells. Acta Pharm.
70:179–190. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang B, Ling T, Zhaxi P, Cao Y, Qian L,
Zhao D, Kang W, Zhang W, Wang L, Xu G and Zou X: Proton pump
inhibitor pantoprazole inhibits gastric cancer metastasis via
suppression of telomerase reverse transcriptase gene expression.
Cancer Lett. 452:23–30. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Spugnini EP, Citro G and Fais S: Proton
pump inhibitors as Anti-vacuolar-ATPases drugs: A novel anticancer
strategy. J Exp Clin Cancer Res. 29:442010. View Article : Google Scholar
|
|
8
|
Geeviman K, Babu D and Prakash Babu P:
Pantoprazole induces mitochondrial apoptosis and attenuates NF-κB
signaling in glioma cells. Cell Mol Neurobiol. 38:1491–1504. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
He J, Shi XY, Li ZM, Pan XH, Li ZL, Chen
Y, Yan SJ and Xiao L: Proton pump inhibitors can reverse the YAP
mediated paclitaxel resistance in epithelial ovarian cancer. BMC
Mol Cell Biol. 20:492019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lu ZN, Shi ZY, Dang YF, Cheng YN, Guan YH,
Hao ZJ, Tian B, He HW and Guo XL: Pantoprazole pretreatment
elevates sensitivity to vincristine in drug-resistant oral
epidermoid carcinoma in vitro and in vivo. Biomed Pharmacother.
120:1094782019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tan Q, Wang M, Yu M, Zhang J, Bristow RG,
Hill RP and Tannock IF: Role of autophagy as a survival mechanism
for hypoxic cells in tumors. Neoplasia. 18:347–355. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tan Q, Joshua AM, Wang M, Bristow RG,
Wouters BG, Allen CJ and Tannock IF: Up-regulation of autophagy is
a mechanism of resistance to chemotherapy and can be inhibited by
pantoprazole to increase drug sensitivity. Cancer Chemother
Pharmacol. 79:959–969. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tan Q, Joshua AM, Saggar JK, Yu M, Wang M,
Kanga N, Zhang JY, Chen X, Wouters BG and Tannock IF: Effect of
pantoprazole to enhance activity of docetaxel against human tumour
xenografts by inhibiting autophagy. Br J Cancer. 112:832–840. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Marino ML, Fais S, Djavaheri-Mergny M,
Villa A, Meschini S, Lozupone F, Venturi G, Della Mina P, Pattingre
S, Rivoltini L, et al: Proton pump inhibition induces autophagy as
a survival mechanism following oxidative stress in human melanoma
cells. Cell Death Dis. 1:e872010. View Article : Google Scholar
|
|
15
|
Moriya S, Che XF, Komatsu S, Abe A,
Kawaguchi T, Gotoh A, Inazu M, Tomoda A and Miyazawa K: Macrolide
antibiotics block autophagy flux and sensitize to bortezomib via
endoplasmic reticulum stress-mediated CHOP induction in myeloma
cells. Int J Oncol. 42:1541–1550. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Renna M, Schaffner C, Brown K, Shang S,
Tamayo MH, Hegyi K, Grimsey NJ, Cusens D, Coulter S, Cooper J, et
al: Azithromycin blocks autophagy and may predispose cystic
fibrosis patients to mycobacterial infection. J Clin Invest.
121:3554–3563. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hirasawa K, Moriya S, Miyahara K, Kazama
H, Hirota A, Takemura J, Abe A, Inazu M, Hiramoto M, Tsukahara K
and Miyazawa K: Macrolide antibiotics exhibit cytotoxic effect
under amino Acid-depleted culture condition by blocking autophagy
flux in head and neck squamous cell carcinoma cell lines. PLoS One.
11:e01645292016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mukai S, Moriya S, Hiramoto M, Kazama H,
Kokuba H, Che XF, Yokoyama T, Sakamoto S, Sugawara A, Sunazuka T,
et al: Macrolides sensitize EGFR-TKI-induced non-apoptotic cell
death via blocking autophagy flux in pancreatic cancer cell lines.
Int J Oncol. 48:45–54. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sugita S, Ito K, Yamashiro Y, Moriya S,
Che XF, Yokoyama T, Hiramoto M and Miyazawa K: EGFR-independent
autophagy induction with gefitinib and enhancement of its cytotoxic
effect by targeting autophagy with clarithromycin in non-small cell
lung cancer cells. Biochem Biophys Res Commun. 461:28–34. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Komatsu S, Miyazawa K, Moriya S, Takase A,
Naito M, Inazu M, Kohno N, Itoh M and Tomoda A: Clarithromycin
enhances Bortezomib-induced cytotoxicity via endoplasmic reticulum
Stress-mediated CHOP (GADD153) induction and autophagy in breast
cancer cells. Int J Oncol. 40:1029–1039. 2012. View Article : Google Scholar
|
|
21
|
Yang JC, Lu CW and Lin CJ: Treatment of
Helicobacter pylori infection: Current status and future concepts.
World J Gastroenterol. 20:5283–5293. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Saito Y, Moriya S, Kazama H, Hirasawa K,
Miyahara K, Kokuba H, Hino H, Kikuchi H, Takano N, Hiramoto M, et
al: Amino acid starvation culture condition sensitizes
EGFR-expressing cancer cell lines to gefitinib-mediated
cytotoxicity by inducing atypical necroptosis. Int J Oncol.
52:1165–1177. 2018.PubMed/NCBI
|
|
23
|
O'Prey J, Sakamaki J, Baudot AD, New M,
Van Acker T, Tooze SA, Long JS and Ryan KM: Application of
CRISPR/Cas9 to autophagy research. Methods Enzymol. 588:79–108.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ran FA, Hsu PD, Wright J, Agarwala V,
Scott DA and Zhang F: Genome engineering using the CRISPR-Cas9
system. Nat Protoc. 11:2281–2308. 2013. View Article : Google Scholar
|
|
25
|
Aits S, Kricker J, Liu B, Ellegaard AM,
Hämälistö S, Tvingsholm S, Corcelle-Termeau E, Høgh S, Farkas T,
Holm Jonassen A, et al: Sensitive detection of lysosomal membrane
permeabilization by lysosomal galectin puncta assay. Autophagy.
11:1408–1424. 2015. View Article : Google Scholar :
|
|
26
|
Galluzzi L, Vitale I, Aaronson SA, Abrams
JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews
DW, et al: Molecular mechanisms of cell death: Recommendations of
the nomenclature committee on cell death 2018. Cell Death Differ.
25:486–541. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mizushima N and Yoshimori T: How to
interpret LC3 immunoblotting. Autophagy. 3:542–545. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang F, Gómez-Sintes R and Boya P:
Lysosomal membrane permeabilization and cell death. Traffic.
19:918–931. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Aits S and Jäättelä M: Lysosomal cell
death at a glance. J Cell Sci. 126:1905–1912. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Andrea B and Juan SB: Lysosomes as dynamic
regulators of cell and organismal homeostasis. Nat Rev Mol Cell
Biol. 21:101–118. 2020. View Article : Google Scholar
|
|
31
|
Zhang S, Wang Y and Li SJ: Lansoprazole
induces apoptosis of breast cancer cells through inhibition of
intracellular proton extrusion. Biochem Biophys Res Commun.
448:424–429. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu W, Baker SS, Trinidad J, Burlingame
AL, Baker RD, Forte JG, Virtuoso LP, Egilmez NK and Zhu L:
Inhibition of lysosomal enzyme activities by proton pump
inhibitors. J Gastroenterol. 48:1343–1352. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Česen MH, Pegan K, Spes A and Turk B:
Lysosomal pathways to cell death and their therapeutic
applications. Exp Cell Res. 318:1245–1251. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhitomirsky B, Yunaev A, Kreiserman R,
Kaplan A, Stark M and Assaraf YG: Lysosomotropic drugs activate
TFEB via lysosomal membrane fluidization and consequent inhibition
of mTORC1 activity. Cell Death Dis. 9:11912018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ballabio A and Bonifacino JS: Lysosomes as
dynamic regulators of cell and organismal homeostasis. Nat Rev Mol
Cell Biol. 21:101–118. 2020. View Article : Google Scholar
|
|
36
|
Papadopoulos C, Kravic B and Meyer H:
Repair or lysophagy: Dealing with damaged lysosomes. J Mol Biol.
432:231–239. 2020. View Article : Google Scholar
|