Introduction
Ollier’s disease is characterized by the
hamartomatous proliferation of cartilage cells, producing masses
termed chondromas. A patient presented with Ollier’s disease which
was found to be associated with diffuse gliomas. Clinicians should
be alert to the possibility of brain tumors during the follow-up of
patients with Ollier’s disease.
Case report
A 4-year-old male was referred to our hospital with
gait impairment. A shortening of the left leg with a length
discrepancy of 3.5 cm was observed. The patient was using a 3.0 cm
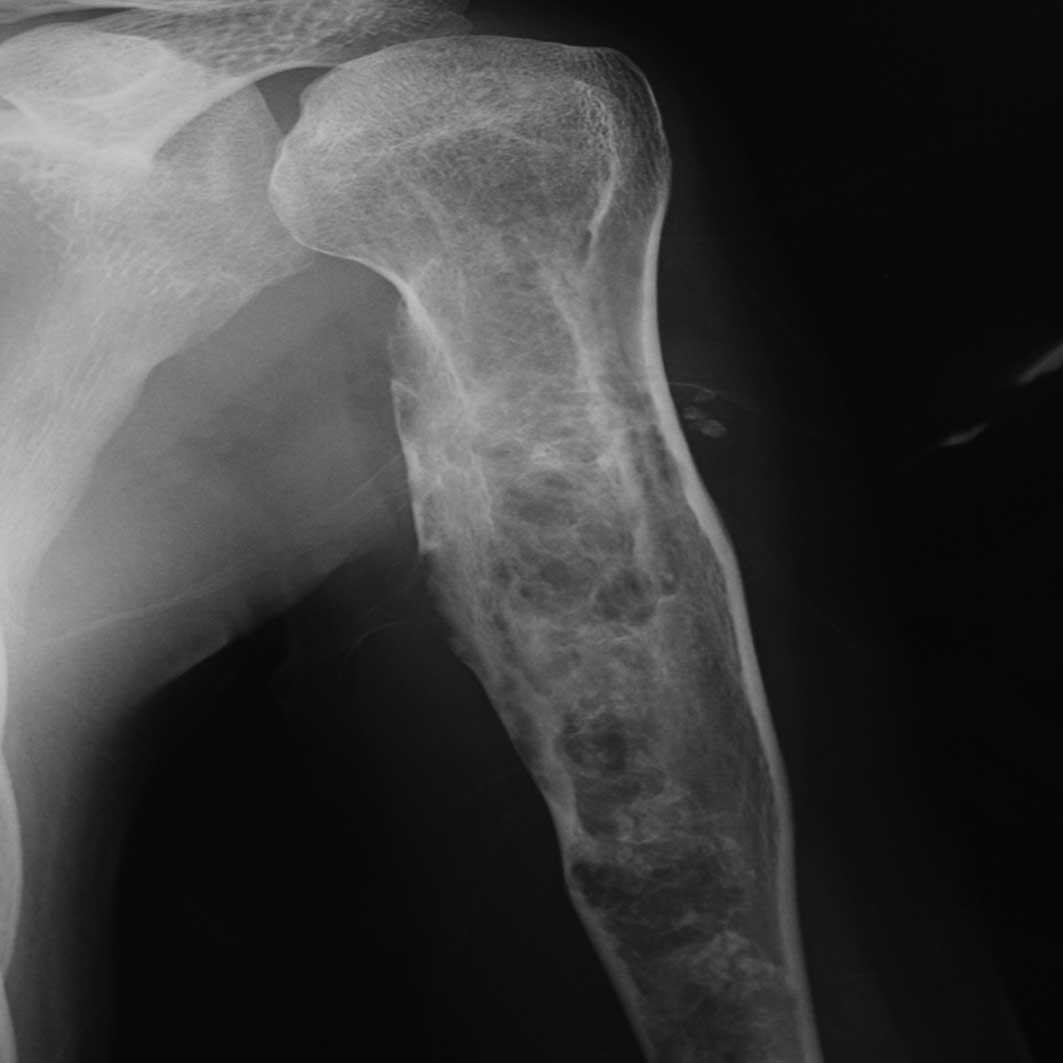
shoe insert. Radiographic findings showed multiple osteolytic
changes in his left humerus, femur, tibia and calcaneus (Fig. 1). A bone biopsy was performed from
the left distal tibia and the pathology report indicated
enchondroma. Subcutaneous hemangiomas were not found, and a
clinical diagnosis of Ollier’s disease was made. As valgus
deformity of the left ankle with a 9 cm shortening of the left limb
was evident, the patient underwent epiphysiodesis of the left ankle
and tibia lengthening with the Orthofix external fixator at 10
years of age. Nevertheless, an 11 cm limb length discrepancy
developed with the patient’s growth, and he underwent additional
limb lengthening with an intramedullary nail at the age of 13. He
received an annual follow-up at the outpatient clinic (Fig. 2).
At the age of 19, he complained of appetite loss and
dysarthria. A neurological evaluation showed GCS E3V3M4, pupil
inequality, motor aphasia and right hemiparesis. A head computed
tomography scan showed a diffuse low-density area in his cerebrum
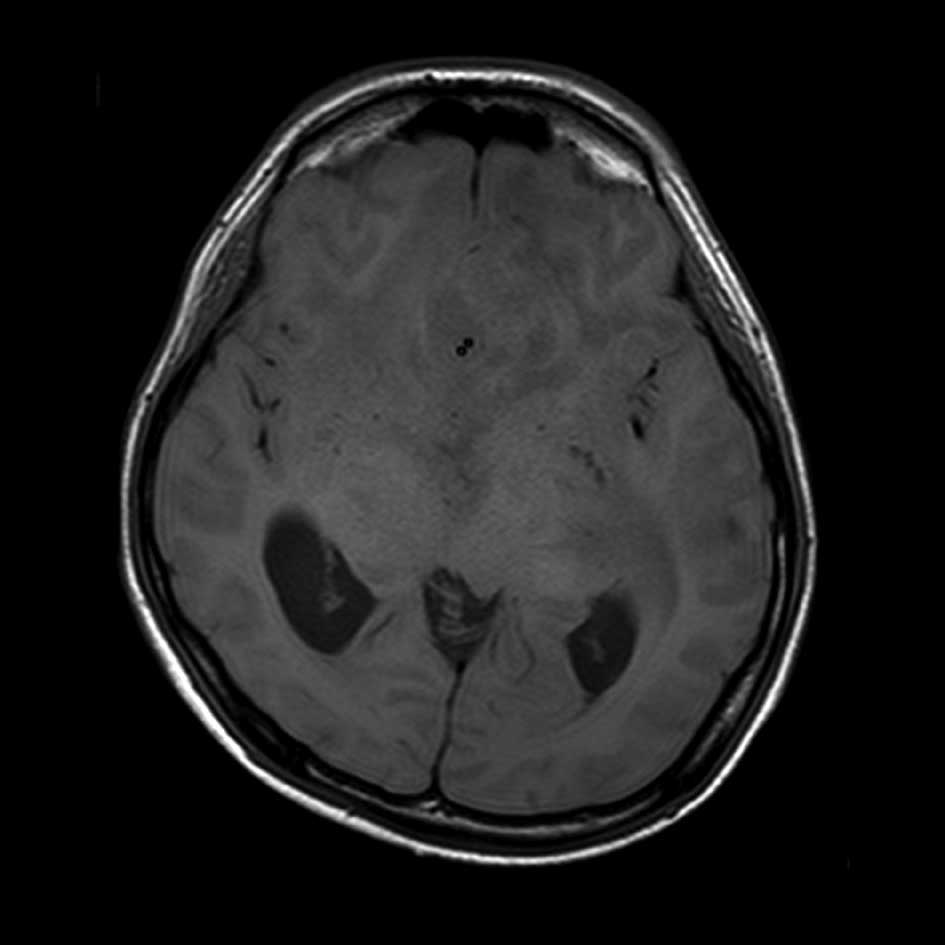
with an enlarged ventricle. On magnetic resonance imaging,
T1-weighted images revealed a diffuse area with low
signal-intensity in the brain stem and T2-weighted images showed
widespread diffuse infiltrated areas in the brain stem, medulla and
cortex, suggesting glioma involvement (Fig. 3A and B). Neurosurgical excision of
the brain tumor and ventricular drainage were performed due to
evidence of advanced brain herniation and a decerebrate state.
Histological specimens from the brain showed increased numbers of
glial cells with nuclear atypia and without necrosis or vascular
proliferation, establishing the diagnosis of World Health
Organization (WHO) grade 3 anaplastic astrocytoma (Fig. 4). Postoperative radiotherapy with
chemotherapy (temozolamide, 75mg/m2) were administered.
The disease currently remains stable according to the findings of
clinical and radiological examinations. Family and patient consent
was obtained. Additionally, the patient sample was obtained with
the approval of the ethics committee of the Mie University Graduate
School of Medicine.
Discussion
In 1899, Ollier described a developmental anomaly
termed ‘De la dyschondroplasi’ which presented with predominantly
metaphyseal non-ossifying cartilaginous masses (1). He noted severe asymmetric, bilateral
deformities of the extremities. Ollier’s disease, also known as
multiple enchondromatosis, is regarded as a congenital disease with
unknown etiology. This rare disease is characterized by the
hamartomatous growth of cartilage cells, producing masses termed
chondromas. Chondromas primarily affect long tubular bones, but
involvement of the skull and pelvis is uncommon. Radiographs show
large radiolucent lesions within the metaphyses (and to a lesser
degree in the epiphyses and diaphyses), leading to the bowing and
shortening of the affected bones. The disease is considered to be
non-hereditary.
Recent studies with long-term follow-ups of patients
with Ollier’s disease show a 25–30% risk of sarcomatous
transformation of the cartilaginous lesions (2). Patients with Ollier’s disease are
generally considered to be less prone to extra-osseous disease.
However, Schwartz et al reported that out of 37 patients
with Ollier’s disease, a low-grade chondrosarcoma developed in
four, an astrocyoma developed in one and a granulose-cell ovarian
tumor developed in another patient (3). Kimmel reported that among 76 cases
with Ollier’s disease, 29 (35%) developed non-skeletal tumors.
Intracranial neoplasms were noted in 15 patients, including 4 (5%)
with gliomas, 3 with pituitary adenomas, 3 with neuromas and 2
patients with chordomas (4).
Therefore, evidence in the literature indicates that gliomas may be
relatively frequent in patients with Ollier’s disease.
To our knowledge, 14 cases of Ollier’s disease
associated with gliomas have been reported, including the present
case (5–15). The patients were predominantly male
and the median age at diagnosis of the glioma was 26.4 years
(14–46). No significant differences were found with regard to
ethnicity or tumor location. The gliomas were classified as
astrocytomas is surgery in combination with radiotherapy and
chemotherapy. Outcomes were: 1 continually disease-free patient, 4
no evidence of disease patients, 1 alive with disease and 1
deceased of disease. The results indicated a poor prognosis for
patients with glioma and Ollier’s disease.
It has been suggested that enchondromas result from
abnormalities in signaling pathways controlling proliferation and
differentiation of chondrocytes. Consistent with this hypothesis, a
functionally deleterious mutation in parathyroid hormone
(PTH)/parathyroid hormone-related peptide (PTHrP) type 1 receptor
(PTHR1)(R150C) was identified in enchondromas in 2 out of 6
unrelated patients with enchondromatosis (16). PTHR1 interacts with the parathyroid
hormone and PTHrP and appears to play a local role in the
regulation of chondrocyte differentiation and enchondral
ossification (17). On the other
hand, PTHrP was found to be expressed in astrocytic tumors
(18) and to regulate
glioma-associated oncogene transcriptional activation (18). Therefore, there may be a common
pathogenesis concerning the PTH/PTHrP receptor and ligand with
regard to enchondromatosis and glioma. Further research is required
to understand the pathophysiology of this rare disease.
The importance of this disease lies in the high risk
of sarcomatous transformation of the skeletal lesions as well as in
the increased risk of developing extra-osseous malignancies
(19,20). Only a limited number of studies
address systematic screening for early diagnosis. However, early
diagnosis of a brain tumor would improve the prognosis of patients.
Since the signs and symptoms of a brain tumor may initially be
vague, a neurological examination and magnetic resonance imaging,
if necessary, should be conducted during the follow-up period.
References
|
1
|
Shapiro F: Ollier’s Disease. An assessment
of angular deformity, shortening, and pathological fracture in
twenty-one patients. J Bone Joint Surg Am. 64:95–103. 1982.
|
|
2
|
Herbert S and Schwartz MD: AAOS Orthopedic
Knowledge Update. Musculoskeletal Tumors. 2:108–113. 2007.
|
|
3
|
Schwartz HS, Zimmerman NB, Simon MA,
Wroble RR, Millar EA and Bonfiglio M: The malignant potential of
enchondromatosis. J Bone Joint Surg Am. 69:269–274. 1987.PubMed/NCBI
|
|
4
|
Kimmel DW, Cheng TM and Mokri B: Primary
intracranial neoplasms in Ollier’s disease. J Neurooncol.
39:1621998.
|
|
5
|
Walid MS and Troup EC: Cerebellar
anaplastic astrocytoma in a teenager with Ollier Disease. J
Neurooncol. 89:59–62. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mahafza WS: Multiple enchondromatosis
Ollier’s disease with two primary brain tumors. Saudi Med J.
25:1261–1263. 2004.
|
|
7
|
Van Nielen KM and de Jong BM: A case of
Ollier’s disease associated with two intracerebral low-grade
gliomas. Clin Neurol Neurosurg. 11:106–110. 1999.
|
|
8
|
Frappaz D, Ricci AC, Kohler R, Bret P and
Mottolese C: Diffuse brain stem tumor in an adolescent with
multiple enchondromatosis (Ollier’s disease). Childs Nerv Syst.
15:222–225. 1999.PubMed/NCBI
|
|
9
|
Balcer LJ, Galetta SL, Cornblath WT and
Liu GT: Neuro-ophthalmologic manifestations of Maffucci’s syndrome
and Ollier’s disease. J Neuroophthalmol. 19:62–66. 1999.
|
|
10
|
Hofman S, Heeg M, Klein JP and Krikke AP:
Simultaneous occurrence of a supra- and an infratentorial glioma in
a patient with Ollier’s disease: more evidence for non-mesodermal
tumor predisposition in multiple enchondromatosis. Skeletal Radiol.
27:688–691. 1998.PubMed/NCBI
|
|
11
|
Chang S and Prados MD: Identical twins
with Ollier’s disease and intracranial gliomas: case report.
Neurosurgery. 34:903–906. 1994.
|
|
12
|
Bendel CJ and Gelmers HJ: Multiple
enchondromatosis (Ollier’s disease) complicated by malignant
astrocytoma. Eur J Radiol. 12:135–137. 1991.
|
|
13
|
Patt S, Weigel K and Mayer HM: A case of
dyschondroplasia associated with brain stem glioma: diagnosis by
stereotactic biopsy. Neurosurgery. 27:487–491. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mellon CD, Carter JE and Owen DB: Ollier’s
disease and Maffucci’s syndrome: distinct entities or a continuum.
Case report: enchondromatosis complicated by an intracranial
glioma. J Neurol. 235:376–378. 1988.
|
|
15
|
Rawlings CE III, Bullard DE, Burger PC and
Friedman AH: A case of Ollier’s disease associated with two
intracranial gliomas. Neurosurgery. 21:400–403. 1987.
|
|
16
|
Hopyan S, Gokgoz N, Poon R, et al: A
mutant PTH/PTHrP type 1 receptor in enchondromatosis. Nat Genet.
30:306–310. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Couvineau A, Wouters V, Bertrant G, et al:
PTHR1 mutations associated wirh Ollier disease result in receptor
loss of function. Hum Mol Genet. 17:2766–2775. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Alman BA and Wunder JS: Parathyroid
hormone-related protein regulates glioma-associated oncogene
transcriptional activation: lessons learned from bone development
and cartilage neoplasia. Ann NY Acad Sci. 1144:35–41. 2008.
View Article : Google Scholar
|
|
19
|
Sato K, Hayashi M, Katsumura H, Ishii H
and Kubota T: A case of Maffucci’s syndrome with brain-stem tumor.
No To Shinkei. 41:631–634. 1989.
|
|
20
|
Goto H, Ito Y, Hirayama A, Sakamoto T and
Kowada M: Maffucci’s syndrome associated with primary brain tumor:
report of a case. No Shinkei Geka. 15:971–975. 1987.
|