Introduction
Aggressive fibromatosis (AF), first identified by
Stout in 1954 (1), is a benign
non-encapsulated lesion of mesenchymal origin, with a tendency for
local spread along fascial planes. AF is locally invasive to bones,
organs and other tissues, which can be fatal. The tumors are rare,
observed in 2–4 cases per million individuals each year (2), and typically occur between puberty and
the age of 40, with a slight female preponderance. AF can arise in
the musculoaponeurotic tissue of any location, but is common in the
abdominal wall, extremities, head and neck, and shoulder girdle.
Individuals with familial adenomatous polyposis (FAP) or Gardner's
syndrome have a 1,000 times greater risk for developing the disease
due to inheritance of the adenomatous polyposis coli (APC)
gene (3). These patients may present
with intra-abdominal lesions following colonic resection (4). While AF does not metastasize, local
recurrence is common. Distant recurrence is extremely rare, but is
typically observed in those with a new primary tumor associated
with the APC mutation. The present study reports the case of
a 20-year-old female with sporadic contralateral recurrence of
clinically diagnosed AF and no familial predisposition.
Case report
A 20-year-old female first presented in April 1992
at Northeastern Pennsylvania Plastic Surgery Associates (Scranton,
PA, USA) with a pigmented lesion on the right shoulder overlying
the trapezius. The lesion had undergone a recent change in size and
color. Upon physical examination, an irregularly contoured brown
papule with portions of jet black speckling was noted. No cervical
or axillary adenopathy was present. The past medical history was
significant for a mitral valve prolapse and asthma. The patient's
family history was free of any familial disease. A biopsy of the
lesion was performed, which confirmed a 0.4-mm thick malignant
melanoma, Clark's level III. The patient underwent a negative
staging workup and the 1.7-cm tumor was excised with plastic
reconstruction. The margins were negative.
One year later, in July 1993, the patient returned
to Northeastern Pennsylvania Plastic Surgery Associates complaining
of pain at the surgical site. The area was profoundly painful upon
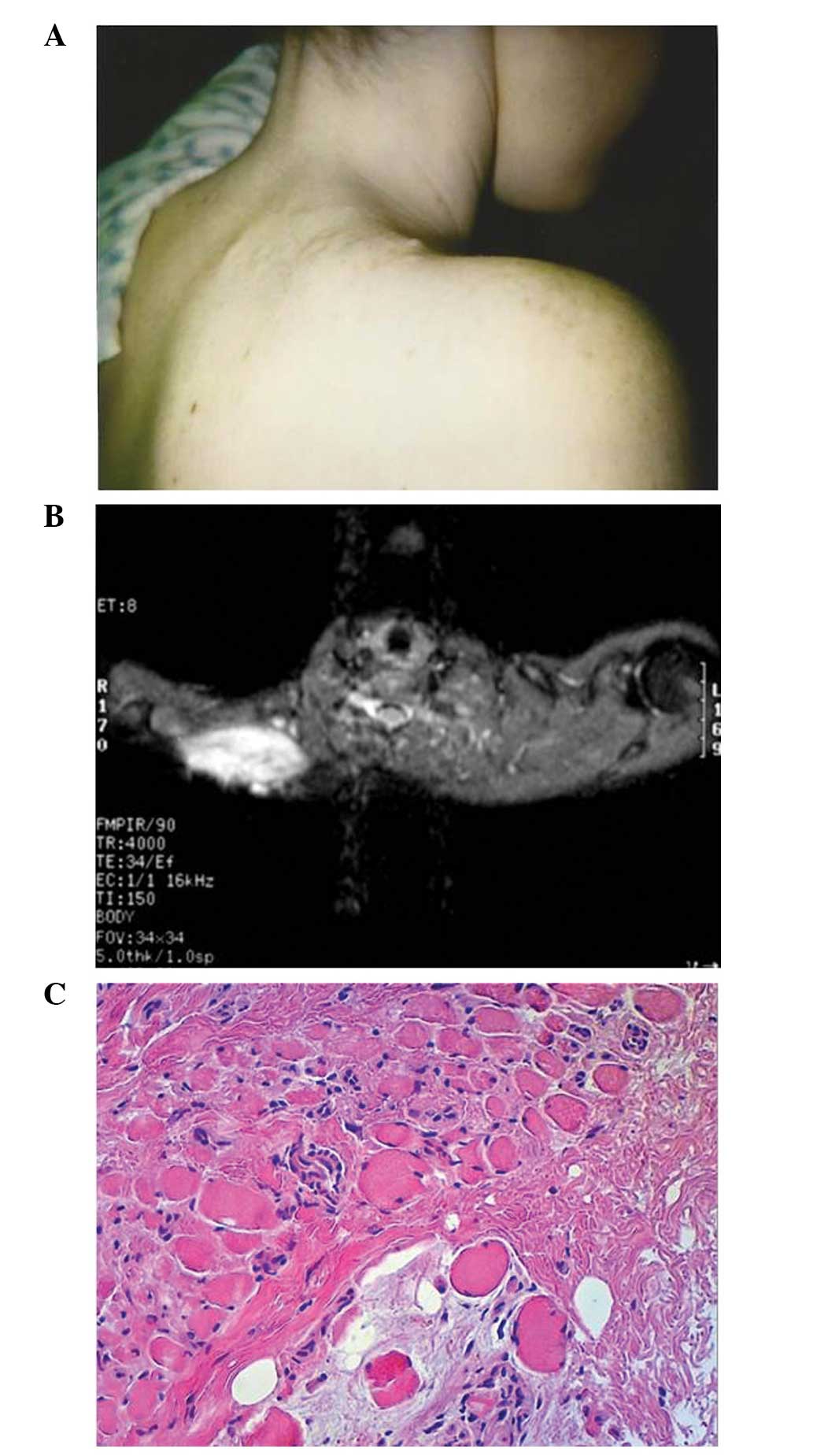
palpation and a discrete mass was identified deep to the incision
(Fig. 1A). Two enlarged axillary
lymph nodes were noted. Fearing a recurrent melanoma, magnetic
resonance imaging (MRI) and biopsies of the lymph nodes and
shoulder mass were obtained. MRI showed a 7×3×3-cm area of altered
signal extending from the skin into the trapezius muscle. The
lesion was isodense to muscle on T1-weighted imaging, with
increased intensity on T2-weighted imaging (Fig. 1B). On biopsy, the nodes were benign
and reactive in nature. The lesion itself showed a proliferation of
spindle cells, with no evidence of increased mitotic rate or
necrosis, in a collagenous matrix infiltrating into the adjacent
muscle fibers (Fig. 1C). S-100 and
HMB-45 stains were negative. A diagnosis of AF was made and
confirmed by independent pathologists at the University of
Pennsylvania (Philadelphia, PA, USA) and the Roswell Park Cancer
Institute (Buffalo, NY, USA) (Fig.
1A). The patient initially refused surgery or radiation therapy
and was treated with tamoxifen. Tamoxifen was started at 20 mg once
daily beginning in June 1995 following recovery from surgery and
radiation treatments No improvement was noted and the patient
eventually authorized surgical excision due to the rapid growth and
infiltrative properties of the lesion. The mass was resected along
with the right trapezius muscle due to tumor invasion and the
shoulder girdle was resuspended with the levator scapulae muscle.
Due to a positive margin, the patient was referred for radiation
therapy and tamoxifen treatment was continued: Tamoxifen treatment
continued at 20 mg once daily until January 1996, when the
treatment was discontinued at the request of the patient.
Radiotherapy consisted of 4,500 cGy in 180-cGy fractions, with 6
MeV photons to the right shoulder and scapula. A boost of 1,620 cGy
in 180-cGy fractions, with 9 MeV electrons, was administered to the
surrounding scar. The treatment was successful and follow up MRI
scans showed no evidence of recurrent disease.
Nearly 5 years after the diagnosis of right shoulder
AF, in March 1999, the patient presented at Northeastern
Pennsylvania Plastic Surgery Associate with left forearm pain and
failure to pronate or supinate the extremity >10°. Flexion and
extension were preserved. There was exquisite tenderness upon
palpation between the radius and ulna proximally. Upper extremity
sensation was intact and reflexes were 2+ bilaterally using the
National Institute of Neurological Disordersand Stroke (NINDS)
scale developed by Hallett in 1993 (5). An MRI scan was obtained showing a thin
5-cm area of enhancement located between the proximal radius and
ulna, possibly involving the periosteum of each. The area was
isodense to muscle on T1-weighted imaging, with increased intensity
on T2-weighted imaging. A monophasic bone scan showed areas of
increased intensity in the proximal radius and ulna. A computed
tomography-guided needle biopsy demonstrated a collagenous fibrous
stroma with plump spindle cells devoid of increased mitotic
activity, consistent with AF. The patient underwent surgical
resection of the forearm lesion, but declined further radiation or
medical therapy. Although grossly and clinically the lesion behaved
as AF, the final pathological report was equivocal, noting
fibroadipose tissue with myxoid change (Fig. 2). However, a clinical diagnosis of AF
was made and the patient was treated accordingly.
The patient still attends follow-up examinations
nearly 20 years after the initial diagnosis of melanoma. The
patient is in a good condition and has experienced no further
recurrence of either lesion. However, shoulder girdle suspension
using the levator scapulae is a continual source of pain and
weakness, and may require further stabilization in the future
(Fig. 3).
Discussion
Sporadic AF arises from somatic mutations in the
APC gene or in CTNNB1, which codes for β-catenin
(6,7).
Germline APC mutations found in individuals with FAP
predispose the patient to AF. APC acts to downregulate the
expression of β-catenin as part of the canonical Wnt pathway. In
the absence of activation, a complex of APC, axin and glycogen
synthase kinase 3β phosphorylates β-catenin resulting in
ubiquitin-mediated degradation (8).
Mutation prevents this complex from forming. β-catenin accumulates
in the cytoplasm and nucleus where it subsequently binds
transcription factors of the T cell/lymphoid enhancer-binding
factor family, inducing gene expression (9). Hedgehog signaling, which modulates the
Wnt pathway and β-catenin expression, is also involved in the
pathogenesis (10). The buildup of
β-catenin within mesenchymal progenitor cells likely maintains an
undifferentiated fibroblast-like state, producing uncontrolled
proliferation and stromal expansion (11).
The tumor exhibits phases of progression, latency
and occasional spontaneous regression. Extra-abdominal tumors
produce symptoms, including pain, weakness and parasthesia.
Intra-abdominal lesions can cause intestinal obstruction or
ischemia (12). MRI is the most
useful imaging technique for diagnosis, showing intermediate
intensity on T1-weighted imaging and high signal intensity on
T2-weighted imaging. Intravenous contrast agents produce moderate-
to high-grade enhancement in highly cellular regions. Non-enhancing
bands of low intensity dominate, representing densely packed
fibrous tissue (13,14). Once AF is identified, a tissue biopsy
should be performed. Fine-needle aspiration is acceptable if an
open biopsy is contraindicated (15).
Histological analysis shows a scattered proliferation of spindle
cells with bland, occasionally bipolar, nuclei. Neoplastic cells
are dispersed among a dense collagenous, keloid-like or myxoid
matrix. Staining for nuclear β-catenin is positive, while staining
for S-100, c-kit, cluster of differentiation 34, estrogen receptor
α and desmin is negative (15). A
cytogenic association with trisomy 8 and 20 is occasionally noted
(16).
For stable or slow growing masses, close monitoring
is now advocated as the primary treatment strategy, as AF has a
tendency to regress. Studies have shown no difference in survival
between patients treated with surgery or radiation therapy compared
with no treatment in this population (17,18).
Surgical resection is indicated in those individuals with
aggressive lesions. The 5-year risk of recurrence following
resection is 17.6%, regardless of whether or not microscopic
margins remain (19). Radiation
therapy at a dose of 50–60 Gy is also an effective primary therapy
(20). Neoadjuvant or adjuvant
radiation is occasionally performed, but its use is controversial.
Conflicting studies have shown either no difference in recurrence
rate following adjuvant therapy (21)
or a delayed time to recurrence with no impact on overall survival
(22). Patients with
contraindications to surgery or radiotherapy can undergo medical
treatment. Non-steroidal anti-inflammatory drug therapy with
sulindac or indomethacin has proven effective due to tumor
overexpression of cyclooxygenase-2 (23). A number of small trials and case
reports indicate that anti-estrogen therapy, including tamoxifen or
toremifine, appears to improve or stabilize AF lesions (24). A large randomized trial is required to
corroborate these reports. Systemic chemotherapy also may be used,
although specific guidelines are lacking. Tyrosine kinase
inhibitors or anthracyclines are popular, but the use of a variety
of other agents is possible (25).
The present study reports a case with a presumptive
recurrence of AF at a site distant and contralateral to the initial
lesion. Local recurrence of AF is common and is usually caused by
residual tumor cells following treatment failure. There have also
been reports of patients with local multicentric tumors present at
diagnosis. These include a patient with masses on the right thigh
and buttock (26), and another with
masses in the left hip and popliteal fossa (27). In these cases, microscopic contiguous
invasion of the initial lesion probably led to the formation of the
second focus. Even rarer are reports of distant recurrence
occurring on the same side of the body. A 2002 report by Watanabe
et al reported the case of a patient with AF of the right
dorsal foot. The tumor recurred on the right knee, followed later
by the right thigh and right shoulder (28). To the best of our knowledge, there has
been only one published case of AF present on contralateral sides
of the body. Contralateral lesions were found in multiple members
of a family with FAP secondary to an inherited APC gene
mutation. The index case presented with multifocal AF of the
paraspinal muscles, which later recurred intra-abdominally.
Affected relatives include a woman with recurrent AF of the right
occiput and bilateral breasts, as well as a male with cutaneous AF
of each arm, the right occiput and multiple paraspinal masses
(29). In this previous study, the
germline APC gene mutation predisposed the family to form
multiple AF lesions. The patient described in the present study,
however, had no family history of AF or FAP. It is likely that the
clinically diagnosed contralateral distant recurrence was the
result of the development of a second primary tumor. Contiguous
spread from the initial site on the right shoulder to the left
forearm five years later is improbable.
Contralateral recurrence of AF is extremely rare.
However, a low index of suspicion for recurrent AF should be
maintained in patients presenting with symptoms of muscle weakness,
pain, parasthesia and a palpable mass.
References
|
1
|
Stout AP: Juvenile fibromatoses. Cancer.
7:953–978. 1954. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hosalkar HS, Torbert JT, Fox EJ, Delaney
TF, Aboulafia AJ and Lackman RD: Musculoskeletal desmoid tumors. J
Am Acad Orthop Surg. 16:188–198. 2008.PubMed/NCBI
|
|
3
|
Gurbuz AK, Giardiello FM, Petersen GM, et
al: Desmoid tumours in familial adenomatous polyposis. Gut.
35:377–381. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hartley JE, Church JM, Gupta S, McGannon E
and Fazio VW: Significance of incidental desmoids identified during
surgery for familial adenomatous polyposis. Dis Colon Rectum.
47:334–338. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hallet M: NINDS myotatic reflex scale.
Neurology. 43:27231993. View Article : Google Scholar
|
|
6
|
Tejpar S, Nollet F, Li C, et al:
Predominance of beta-catenin mutations and beta-catenin
dysregulation in sporadic aggressive fibromatosis (desmoid tumor).
Oncogene. 18:6615–6620. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Alman BA, Li C, Pajerski ME, Diaz-Cano S
and Wolfe HJ: Increased beta-catenin protein and somatic APC
mutations in sporadic aggressive fibromatoses (desmoid tumors). Am
J Pathol. 151:329–334. 1997.PubMed/NCBI
|
|
8
|
Aberle H, Bauer A, Stappert J, Kispert A
and Kemler R: β-Catenin is a target for the ubiquitin-proteasome
pathway. EMBO J. 16:3797–3804. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rubinfeld B, Albert I, Porfiri E, Fiol C,
Munemitsu S and Polakis P: Binding of GSK3β to the APC-β-catenin
complex and regulation of complex assembly. Science. 272:1023–1026.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ghanbari-Azarnier R, Sato S, Wei Q,
Al-Jazrawe M and Alman BA: Targeting stem cell behavior in desmoid
tumors (aggressive fibromatosis) by inhibiting hedgehog signaling.
Neoplasia. 15:712–719. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wu C, Nik-Amini S, Nadesan P, Stanford WL
and Alman BA: Aggressive fibromatosis (desmoid tumor) is derived
from mesenchymal progenitor cells. Cancer Res. 70:7690–7698. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Escobar C, Munker R, Thomas JO, Li BD and
Burton GV: Update on desmoid tumors. Ann Oncol. 23:562–569. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rhim JH, Kim JH, Moon KC, et al: Desmoid
type fibromatosis in the head and neck: CT and MR imaging
characteristics. Neuroradiology. 55:351–359. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nishio J, Aoki M, Nabeshima K, Iwasaki H
and Naito M: Imaging features of desmoid-type fibromatosis in the
teres major muscle. In Vivo. 27:555–559. 2013.PubMed/NCBI
|
|
15
|
Owens CL, Sharma R and Ali SZ: Deep
fibromatosis (desmoid tumor): Cytopathologic characteristics,
clinicoradiologic features and immunohistochemical findings on
fine-needle aspiration. Cancer. 111:166–172. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Qi H, Cal Cin P, Hernandez JM, et al:
Trisomies 8 and 20 in desmoid tumors. Cancer Genet Cytogenet.
92:147–149. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fiore M, Rimareix F, Mariani L, et al:
Desmoid-type fibromatosis: A front-line conservative approach to
select patients for surgical treatment. Ann Surg Oncol.
16:1587–1593. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bonvalot S, Eldweny H, Haddad V, et al:
Extra-abdominal primary fibromatosis: Aggressive management could
be avoided in a subgroup of patients. Eur J Surg Oncol. 34:462–468.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
van Broekhoven DL, Verhoef C, Elias SG, et
al: Local recurrence after surgery for primary extra-abdominal
desmoid type fibromatosis. Br J Surg. 100:1214–1219. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ballo MT, Zagars GK, Pollack A, Pisters PW
and Pollack RA: Desmoid Tumor: Prognosis factors and outcome after
surgery, radiation therapy, or combined surgery and radiation
therapy. J Clin Oncol. 17:158–167. 1999.PubMed/NCBI
|
|
21
|
Gluck I, Griffith KA, Biermann JS, Feng
FY, Lucas DR and Ben-Josef E: Role of radiotherapy in the
management of desmoid tumors. Int J Radiat Oncol Biol Phys.
80:787–792. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shin SH, Ko KR, Cho SK, Choi YL and Seo
SW: Surgical outcome of desmoid tumors: adjuvant radiotherapy
delayed the recurrence, but did not affect long-term outcomes. J
Surg Oncol. 108:28–33. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Klein WA, Miller HH, Anderson M and
DeCosse JJ: The use of indomethacin, sulindac and tamoxifen for the
treatment of desmoid tumors associated with familial polyposis.
Cancer. 60:2863–2868. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Janinis J, Patriki M, Vini L, Aravantinos
G and Whelan JS: The pharmacological treatment of aggressive
fibromatosis: A systematic review. Ann Oncol. 14:181–190. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Devata S and Chugh R: Desmoid tumors: A
comprehensive review of the evolving biology, unpredictable
behavior and myriad of management options. Hematol Oncol Clin North
Am. 27:989–1005. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sundaram M, Duffrin H, McGuire MH and Vas
W: Synchronous multicentric desmoid tumors (aggressive
fibromatosis) of the extremities. Skeletal Radiol. 17:16–19. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Antal I, Szendroi M, Kovacs G, Nagykalnai
T and Entz L: Multicentric extraabdominal desmoid tumor: a case
report. J Cancer Res Clin Oncol. 120:190–193. 1994. View Article : Google Scholar
|
|
28
|
Watanabe K, Ogura G, Tajino T and Suzuki
T: Extra-abdominal desmoid fibromatosis: Two familial cases with
synchronous and metachronous multicentric hyalinizing nodules.
Histopathology. 41:118–121. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Eccles DM, van der Lujit R, Breukel C, et
al: Hereditary desmoid disease due to a frameshift mutation at
codon 1924 of the APC gene. Am J Hum Genet. 59:1193–201.
1996.PubMed/NCBI
|