Introduction
According to the current literature, breast cancer
is the most commonly diagnosed form of cancer and is the principal
cause of cancer-associated fatality among females. The disease
accounts for 14% of all cancer mortalities, particularly in
economically-developing countries, and 23% of all cancer cases
(1). The breast cancer 1
(BRCA1) gene is ~100 kb in length and is localized to
chromosome 17q21; it is composed of 24 exons, with 22 of these
translating into a 1,863 amino acid protein. It is currently
well-established that mutations in the BRCA1 gene result in
a significantly increased lifetime risk for the development of
breast cancer (2). Thus,
BRCA1, functioning as a tumor suppressor, may be regarded as
a strong candidate gene for breast cancer susceptibility.
The abnormal methylation of
cytosine-phosphate-guanine (CpG) islands in gene promoter regions
is the predominant epigenetic mechanism by which gene transcription
is effectively silenced; therefore, the transcriptional activity of
genes with CpG island promoters is suppressed upon methylation
(3). Notably, a previous study
concluded that microRNAs (miRNAs/miRs) may function as oncogenes
and tumor suppressors depending on the genetic variants in the
3′UTR binding sites, regulating gene expression
post-transcriptionally (4). For
example, miR-146a was observed to bind to the 3′UTR of BRCA1
and BRCA2 mRNA, potentially modulating their mRNA
expression, and a genetic polymorphism in the miR-146a gene
(rs2910164) was associated with a young diagnostic age of familial
ovarian and breast cancer (5).
However, the function of genetic variants in miRNA binding sites of
BRCA1 remains unclear. In the current study, the hypothesis
that the BRCA1 3′UTR variants are associated with its mRNA
expression was tested by performing bioinformatics analysis. In
addition, the CpG islands and transcription factor binding sites
(TFBSs) were predicted.
Materials and methods
Prediction of the CpG islands and
TFBSs in the promoter region
The human BRCA1 promoter region was obtained
by searching the UCSC Genome Bioinformatics online database
(www.genome.ucsc.edu/). The core
promoters were identified by Neural Network Promoter Prediction
(NNPP; www.fruitfly.org/seq_tools/promoter.html) and
Promoter2.0 (www.cbs.dtu.dk/services/promoter/), and the CpG
islands were predicted using the bioinformatics tool MethPrimer
(www.urogene.org/methprimer/). The
criteria used to define a CpG island was as follows: Island size,
>100 bp; GC percentage, >50.0; and observed/expected ratio,
>0.6. In addition, the TFBSs were predicted by JASPAR
(jaspar.genereg.net/).
Selection of polymorphisms in the
3′UTR and analysis of the genotype-phenotype association
The SNPs in the human BRCA1 3′UTR were
identified using the National Center for Biotechnology Information
SNP database (www.ncbi.nlm.nih.gov/SNP/). Subsequently, the
distribution of all BRCA1 genotypes among 11 distinctive
populations was calculated. The 11 populations studied were as
follows: Utah residents with Northern and Western European ancestry
from the Center for the Study of Human Polymorphisms collection
(CEU); Japanese individuals in Tokyo, Japan (JPT); members of the
Yoruba tribe in Ibadan, Nigeria (YRI); Han Chinese individuals in
Beijing, China (CHB); members of the Luhya tribe in Webuye, Kenya
(LWK); individuals of Mexican ancestry in Los Angeles, California
(MEX); Gujarati Indians in Houston, Texas (GIH); Chinese
individuals in Metropolitan Denver, Colorado (CHD); individuals of
African ancestry in Southwest USA (ASW); members of the Maasai
tribe in Kinyawa, Kenya (MKK); and Tuscan individuals in Italy
(TSI). SNP Function Prediction (snpinfo.niehs.nih.gov/snpinfo/snpfunc.htm) was
utilized to predict the possible miRNA binding sites in the
BRCA1 gene. Additionally, SNPs not in linkage disequilibrium
(LD; r2<0.8) were selected and LD maps of these SNPs in the
BRCA1 gene were plotted using the online program (snpinfo.niehs.nih.gov/snpinfo/snptag.htm). Additional
data was utilized regarding BRCA1 genotypes and mRNA levels,
which was available from the HapMap database (http://app3.titan.uio.no/biotools/tool.php?app=snpexp),
for the genotype-phenotype association analysis (6). The genotyping data from the HapMap phase
II release 23 dataset consists of ~4 million SNP genotypes from 270
individuals belonging to 4 different populations (7).
Statistical analysis
The genotype and phenotype correlation was analyzed
using the χ2 test. Statistical analysis was performed on
SPSS version 21 software (IMB SPSS, Armonk, NY, USA) All
statistical tests were two-sided, and P<0.05 was considered to
indicate a statistically significance difference.
Results
CpG islands and TFBSs in the BRCA1
promoter region
The online tools NNPP and Promoter2.0 indicated that
there were several core promoter regions located in the target
sequence. A total of 3 core promoter regions were determined by
NNPP, whilst Promoter2.0 identified 2 core promoter regions. The
bioinformatics software MethPrimer was used to predict the range of
the CpG islands in the BRCA1 gene. The results determined
that there were 3 CpG islands in the BRCA1 promoter region
(Fig. 1; Table I), which may serve a crucial role in
the expression of the BRCA1 gene. In total, 31 transcription
factors and 45 binding sites were predicted in the target sequence
regions when limiting the relative profile score threshold to
>95%.
 | Table I.CpG island prediction results. |
Table I.
CpG island prediction results.
| CpG island | Size, bp | Start | End |
|---|
| 1 | 129 | 412 | 540 |
| 2 | 244 | 608 | 851 |
| 3 | 221 | 950 | 1170 |
BRCA1 3′UTR selected variants and
their putative miRNA binding sites
A total of 28 SNPs were identified in the human
BRCA1 3′UTR; 13 of these SNPs had available minor allele
frequency (MAF) values, 2 of which (rs12516 and rs8176318) had MAF
values >0.05. SNP Function Prediction demonstrated that these 2
SNPs exhibited putative miRNA binding sites (Table II). As presented in Table II, the C-to-thymine (T) transition
mutation, rs12516, had 6 potential miRNA binding sites, namely,
hsa-miR-188-5p, hsa-miR-502-5p, hsa-miR-557, hsa-miR-623,
hsa-miR-637 and hsa-miR-639. The G-to-T transition mutation,
rs8176318, was observed to share hsa-miR-639 with rs12516, and
contain 4 other miRNA binding sites, namely, hsa-miR-1182,
hsa-miR-149, hsa-miR-345 and hsa-miR-544.
| Table II.Selected SNPs of 3′UTR and putative
miRNA binding sites. |
Table II.
Selected SNPs of 3′UTR and putative
miRNA binding sites.
| SNP | Alleles | MAF, % | Putative miRNA
binding sites |
|---|
| rs8176320 | G>A | 0.51 | hsa-miR-101,
hsa-miR-15a, hsa-miR-15b, has-miR-16, hsa-miR-194, hsa-miR-195,
hsa-miR-424, hsa-miR-450b-5p, hsa-miR-545 |
| rs184237074 | C>T | 0.05 | NA |
| rs189382442 | T>C | 0.05 | NA |
| rs182218567 | A>G | 0.05 | NA |
| rs12516 | C>T | 31.18 | hsa-miR-188–5p,
hsa-miR-502–5p, hsa-miR-557, hsa-miR-623, hsa-miR-637,
hsa-miR-639 |
| rs111791349 | C>T | 0.51 | NA |
| rs185966495 | G>C | 0.05 | NA |
| rs8176319 | C>T | 0.69 | NA |
| rs138782023 | T>C | 0.28 | NA |
| rs141850147 | G>A | 0.05 | NA |
| rs8176318 | G>T | 29.06 | hsa-miR-1182,
hsa-miR-149, hsa-miR-345, hsa-miR-544, hsa-miR-639 |
| rs56108540 | T>C | 0.23 | hsa-miR-125a-3p,
hsa-miR-224, hsa-miR-499–3p, hsa-miR-539, hsa-miR-548c-3p,
hsa-miR-767–5p |
| rs137892861 | G>A | 0.14 | NA |
Frequency distribution of selected
variants among distinct populations
As previously reported, the derived alleles at
rs12516 and rs8176318 in the BRCA1 3′UTR demonstrated a
positive association with familial ovarian and breast cancer in
Thai women, and the 2 SNPs were in strong linkage disequilibrium in
populations and varied by ethnicity (8). In the present study, in order to better
evaluate the global genotypes of rs12516 and rs8176318 in
BRCA1, the frequency distribution data of these variants
across 11 worldwide populations were summarized (Tables III and IV). The 2 SNPs presented differences in
genotype frequency distribution among the worldwide populations.
For rs12516, the genotype frequencies of CC, CT and TT were highest
in YRI (69.0%), CHD (57.6%) and GIH (18.2%). The frequency
distributions of the GG, GT and TT genotypes of rs8176318 ranked
first among LWK (84.4%), CHD (56.6%) and GIH (18.2%). The T allele
frequencies of rs12516 in the various populations ranged between
44.9% in GIH and 17.3% in YRI. Notably, the frequency distributions
of the GG, GT and TT genotypes of rs8176318 were the same as the
data for CC, CT and TT genotypes of rs12516 among the CEU and CHB
populations.
| Table III.Genotype frequency of the breast
cancer 1 gene rs12516 polymorphism in different populations. |
Table III.
Genotype frequency of the breast
cancer 1 gene rs12516 polymorphism in different populations.
|
| Genotype frequency,
n (%) | Allele frequency,
% |
|
|---|
|
|
|
|
|
|---|
| Populations | n | CC | CT | TT | C | T | HWP |
|---|
| CEU | 226 | 100 (44.2) | 102 (45.1) | 24 (10.6) | 66.8 | 33.2 | 1.000 |
| JPT | 170 | 82 (48.2) | 76 (44.7) | 12 (7.1) | 70.6 | 29.4 | 0.527 |
| YRI | 226 | 156 (69.0) | 62 (27.4) | 8 (3.5) | 82.7 | 17.3 | 0.752 |
| CHB | 82 | 38 (46.3) | 32 (39.0) | 12 (14.6) | 65.9 | 34.1 | 0.403 |
| LWK | 180 | 112 (62.2) | 62 (34.4) | 6 (3.3) | 79.4 | 20.6 | 0.655 |
| MEX | 98 | 62 (63.3) | 26 (26.5) | 10 (10.2) | 76.5 | 23.5 | 0.100 |
| GIH | 176 | 50 (28.4) | 94 (53.4) | 32 (18.2) | 55.1 | 44.9 | 0.479 |
| CHD | 170 | 46 (27.1) | 98 (57.6) | 26 (15.3) | 55.9 | 44.1 | 0.150 |
| ASW | 98 | 44 (44.9) | 50 (51.0) | 4 (4.1) | 70.4 | 29.6 | 0.150 |
| MKK | 286 | 172 (60.1) | 104 (36.4) | 10 (3.5) | 78.3 | 21.7 | 0.403 |
| TSI | 174 | 64 (36.8) | 88 (50.6) | 22 (12.6) | 62.1 | 37.9 | 0.527 |
| Table IV.Genotype frequency of the breast
cancer 1 gene rs8176318 polymorphism in different populations. |
Table IV.
Genotype frequency of the breast
cancer 1 gene rs8176318 polymorphism in different populations.
|
| Genotype frequency,
n (%) | Allele frequency,
% |
|
|---|
|
|
|
|
|
|---|
| Populations | n | GG | GT | TT | G | T | HWP |
|---|
| CEU | 226 | 100 (44.2) | 102 (45.1) | 24 (10.6) | 66.8 | 33.2 | 1.000 |
| JPT | 172 | 88 (51.2) | 72 (41.9) | 12 (7.0) | 72.1 | 27.9 | 0.752 |
| YRI | 226 | 180 (79.6) | 42 (18.6) | 4 (1.8) | 88.9 | 11.1 | 0.584 |
| CHB | 82 | 38 (46.3) | 32 (39.0) | 12 (14.6) | 65.9 | 34.1 | 0.403 |
| LWK | 180 | 152 (84.4) | 26 (14.4) | 2 (1.1) | 91.7 | 08.3 | 0.655 |
| MEX | 98 | 62 (63.3) | 28 (28.6) | 8 (8.2) | 77.6 | 22.4 | 0.251 |
| GIH | 176 | 52 (29.5) | 92 (52.3) | 32 (18.2) | 55.7 | 44.3 | 0.584 |
| CHD | 170 | 48 (28.2) | 96 (56.5) | 26 (15.3) | 56.5 | 43.5 | 0.200 |
| ASW | 98 | 56 (57.1) | 42 (42.9) | 0 (0) | 78.6 | 21.4 | 0.251 |
| MKK | 286 | 206 (72.0) | 72 (25.2) | 8 (2.8) | 84.6 | 15.4 | 0.752 |
| TSI | 176 | 68 (38.6) | 88 (50.0) | 20 (11.4) | 63.6 | 36.4 | 0.479 |
LD of all SNPs in the BRCA1 gene
calculation
SNPs not in LD (r2<0.8) were selected and LD maps
of those SNPs in the BRCA1 gene were plotted using the SNP
Function Prediction program to identify the potential functional
relevance of all selected SNPs (Fig.
2). The degree of pairwise LD between all SNPs was estimated as
quantified by the disequilibrium coefficient (D') and r2, which
represented the proportion of disequilibrium and the maximum
possible disequilibrium given for observed allele frequencies,
respectively. The higher the D' value, the greater the association
between the two loci being studied. The color of each SNP spot
reflects its D' value, which changes from red to white as it
decreases. The MAF of each aforementioned allele was >0.05. As
for the SNPs in the BRCA1 3′UTR, only rs12516 and rs8176318
were included in the LD plot, but neither were the predicted tag
SNP.
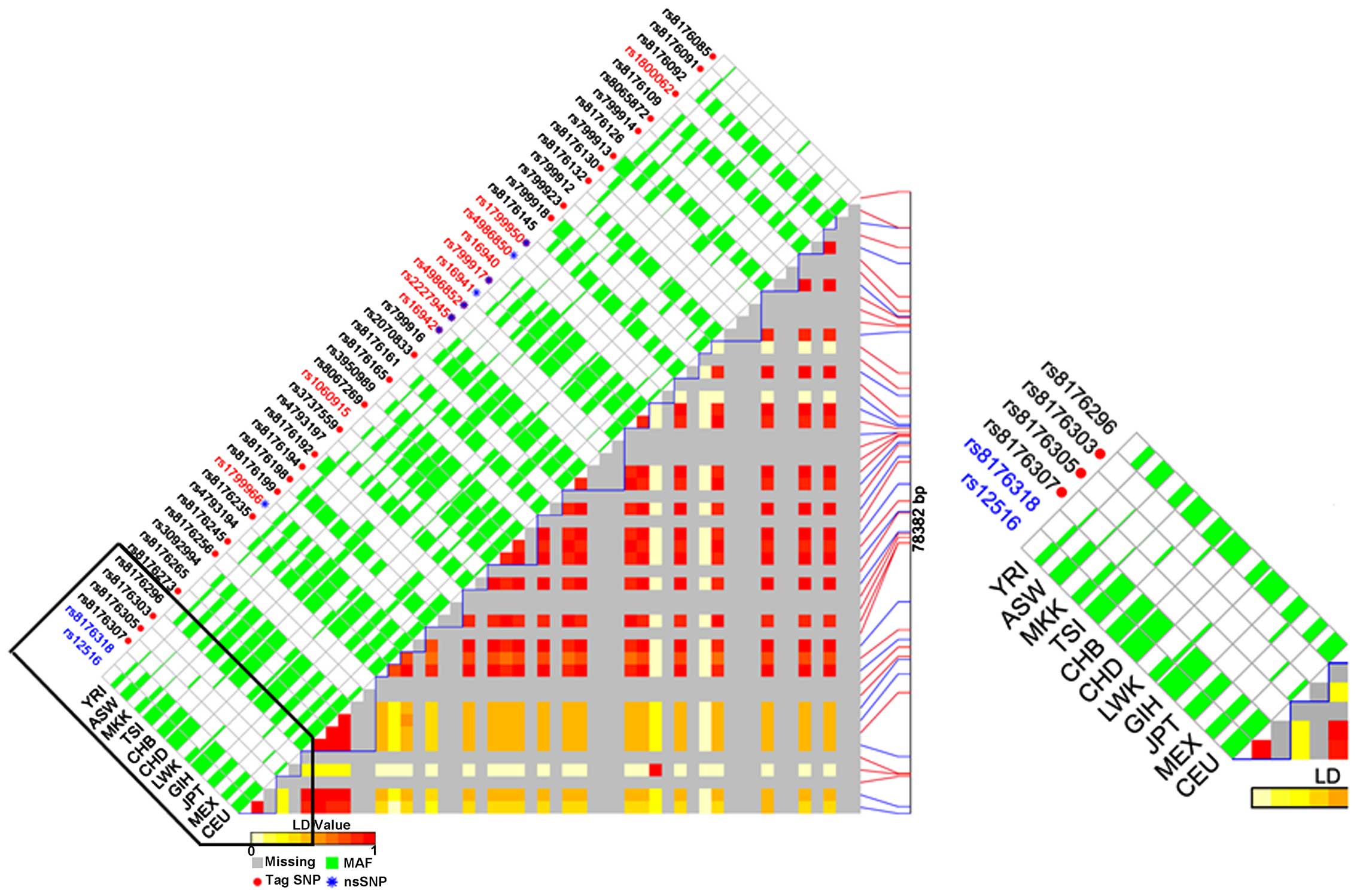 | Figure 2.Linkage disequilibrium plot of the
breast cancer 1 gene region using SNP function prediction. The
color of each SNP spot reflects its D' value, which changes from
red to white as the D' value decreases. SNP, single nucleotide
polymorphism; LWK, members of the Luhya tribe in Webuye, Kenya;
CHD, Chinese individuals in Metropolitan Denver, Colorado; YRI,
members of the Yoruba tribe in Ibadan, Nigeria; MEX, individuals of
Mexican ancestry in Los Angeles, California; JPT, Japanese
individuals in Tokyo, Japan; GIH, Gujarati Indians in Houston,
Texas; CHB, Han Chinese individuals in Beijing, China; CEU, Utah
residents with Northern and Western European ancestry from the
Center for the Study of Human Polymorphismscollection; LD, linkage
disequilibrium; MAF, minor allele frequency; nsSNP, non-synonymous
SNP. |
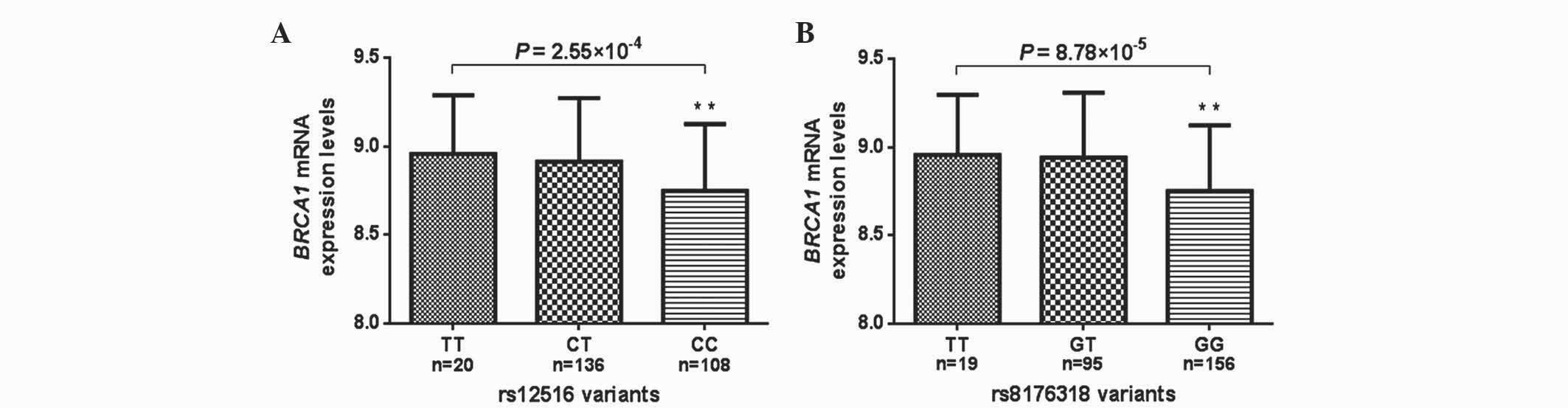
BRCA1 mRNA expression by genotypes in
lymphoblastoid cell lines
The present study took advantage of the available
HapMap-complementary DNA expression database for the correlation
analysis between BRCA1 genotype and mRNA expression in 270
lymphoblastoid cell lines. Excluding the 6 cell lines with
unavailable values for rs12516, 20 (7.6%) cell lines exhibited the
TT genotype, 108 (40.9%) cell lines exhibited the CT genotype and
136 (51.5%) cell lines exhibited the CC genotype. For rs8176318, 19
(7.0%) cell lines exhibited the TT genotype, 95 (35.2%) exhibited
the GT genotype and 156 (57.8%) exhibited the GG genotype. The
effect of the rs12516 and rs8176318 genotypes on BRCA1 mRNA
expression levels is presented in Fig.
3. For rs12516, the BRCA1 mRNA expression level was
significantly decreased in the CC genotype compared with the CT and
TT genotypes (P=2.55×10-4; Fig. 3A).
Similarly, the rs8176318 GG genotype had a significantly lower
expression level than the GT and TT genotypes
(P=8.78×10−5; Fig.
3B).
Discussion
Since the identification of the BRCA1 gene 20
years ago (9), the biological basis
underlying the high breast cancer risk in women with BRCA1
mutations has gained increasing attention. BRCA1 operates in
a series of cellular processes, including DNA repair, chromatin
remodeling, protein ubiquitination, regulation of transcription,
apoptosis and cell cycle checkpoint control. As observed in
carriers of germline BRCA1 mutations, the disruption of any
of the aforementioned processes may result in an increased risk for
carcinogenesis (10). Dacheux et
al (11) demonstrated that
BRCA1 modified the translational regulation of ~7% of genes
expressed in MCF7 cells, including structural maintenance of
chromosomes 6, thyroid hormone receptor α and topoisomerase I. It
was proposed that BRCA1 may serve a direct transcriptional
role in the regulation of p27Kip1 resulting in S-phase
arrest; this suggested that the BRCA1-mediated
transcriptional regulation of p27Kip1 may function as a
mechanism for BRCA1-induced growth inhibition (12).
BRCA1 promoter methylation is considered to
serve a key role in the etiology of human breast cancer. Iwamoto
et al (13) reported that
BRCA1 promoter methylation in peripheral blood cells may
establish a novel risk factor for the development of breast cancer.
Xu et al (14) demonstrated
that methylation of the BRCA1 promoter was associated with
increased mortality rates among women with breast cancer. The
promoter region of the BRCA1 gene was methylated in a large
proportion of Taiwanese patients with early-stage breast cancer,
and patients with BRCA1-methylated tumors exhibited poorer
survival outcomes (15). A
meta-analysis provided evidence that methylation of the
BRCA1 promoter was associated with the poor survival of
patients with breast cancer (16). To
directly investigate BRCA1 gene regulation on a
transcriptional level, the present study predicted 3 CpG islands
and several putative TFBSs in the BRCA1 promoter region
using bioinformatics analysis.
Previous studies have demonstrated that BRCA1
3′UTR miRNA binding site variants are associated with breast cancer
risk post-translationally. For example, miR-24 was observed to
directly target the 3′UTR of BRCA1 and resulted in
significant repression of the BRCA1 gene (17). In addition, it was demonstrated that
the BRCA1 polymorphism, rs799917, was associated with breast
cancer risk (18). By contrast, Hasan
et al (19) reported that
rs799917 demonstrated no significant association with breast cancer
in 100 patients with breast cancer in Saudi Arabia (19). Therefore, the role of genetic variants
in the BRCA1 3′UTR and its post-transcriptional regulation
remains unclear. Such differences may be due to reproductive
patterns, in addition to exposure to particular environmental
carcinogens, different lifestyles and different genetic backgrounds
(19). The present study focused on
the polymorphisms in the BRCA1 3′UTR, and identified that
rs12516 and rs8176318 had potential miRNA binding sites.
Furthermore, rs12516 and rs8176318 exhibited differences in
genotype frequency distribution among populations worldwide. Each
SNP had a significant association with BRCA1 mRNA
expression, therefore implying that these SNPs may partially
contribute to BRCA1 post-transcriptional regulation.
In conclusion, the current study predicted 3 GpG
islands, 45 TFBSs located in the promoter region and 13 SNPs
located in the 3′UTR of the BRCA1 gene. A total of 4 SNPs
(rs8176320, rs12516, rs8176318 and rs56108540) were confirmed to
have putative miRNA binding sites, among which only 2 (rs12516 and
rs8176318) had MAF values >0.05. These 2 SNPs, rs12516 and
rs8176318, each demonstrated a significant association with
BRCA1 mRNA expression. The results from the present study
have raised the possibility that rs12516 and rs8176318 may be
associated with an increased risk of breast cancer by altering the
BRCA1 mRNA level. However, such results require
substantiation by experimental data, therefore further
investigations are warranted to confirm the function of these
polymorphisms. In addition, further functional analysis is
necessary to validate the promoter CpG islands and SNPs in the
3′UTR to allow for investigation of BRCA1 gene regulation as
a potential therapy for breast cancer.
Acknowledgements
This study was supported by the Foundation for
Clinical Medicine Science and Technology Special Project of the
Jiangsu Province, China (grant no. BL2014071) to X.G.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ayoub N, Lucas C and Kaddoumi A: Genomics
and pharmacogenomics of breast cancer: Current knowledge and
trends. Asian Pac J Cancer Prev. 12:1127–1140. 2011.PubMed/NCBI
|
|
3
|
Zeisberg EM and Zeisberg M: The role of
promoter hypermethylation in fibroblast activation and
fibrogenesis. J Pathol. 229:264–273. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hook L, Hancock M, Landais I, Grabski R,
Britt W and Nelson JA: Cytomegalovirus microRNAs. Curr Opin Virol.
7:40–46. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shen J, Ambrosone CB, DiCioccio RA, Odunsi
K, Lele SB and Zhao H: A functional polymorphism in the miR-146a
gene and age of familial breast/ovarian cancer diagnosis.
Carcinogenesis. 29:1963–1966. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Holm K, Melum E, Franke A and Karlsen TH:
SNPexp - A web tool for calculating and visualizing correlation
between HapMap genotypes and gene expression levels. BMC
Bioinformatics. 11:6002010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
International HapMap Consortium: The
International HapMap Project. Nature. 426:789–796. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pelletier C, Speed WC, Paranjape T, Keane
K, Blitzblau R, Hollestelle A, Safavi K, van den Ouweland A,
Zelterman D, Slack FJ, et al: Rare BRCA1 haplotypes including 3′UTR
SNPs associated with breast cancer risk. Cell Cycle. 10:90–99.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Narod SA, Feunteun J, Lynch HT, Watson P,
Conway T, Lynch J and Lenoir GM: Familial breast-ovarian cancer
locus on chromosome 17q12-q23. Lancet. 338:82–83. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang ES and Xia F: BRCA1 16 years later:
DNA damage-induced BRCA1 shuttling. FEBS J. 277:3079–3085. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dacheux E, Vincent A, Nazaret N, Combet C,
Wierinckx A, Mazoyer S, Diaz JJ, Lachuer J and Venezia ND:
BRCA1-Dependent Translational Regulation in Breast Cancer Cells.
PLoS One. 8:e673132013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Williamson EA, Dadmanesh F and Koeffler
HP: BRCA1 transactivates the cyclin-dependent kinase inhibitor
p27(Kip1). Oncogene. 21:3199–3206. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Iwamoto T, Yamamoto N, Taguchi T, Tamaki Y
and Noguchi S: BRCA1 promoter methylation in peripheral blood cells
is associated with increased risk of breast cancer with BRCA1
promoter methylation. Breast Cancer Res Treat. 129:69–77. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xu X, Gammon MD, Zhang Y, Bestor TH,
Zeisel SH, Wetmur JG, Wallenstein S, Bradshaw PT, Garbowski G,
Teitelbaum SL, et al: BRCA1 promoter methylation is associated with
increased mortality among women with breast cancer. Breast Cancer
Res Treat. 115:397–404. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hsu NC, Huang YF, Yokoyama KK, Chu PY,
Chen FM and Hou MF: Methylation of BRCA1 promoter region is
associated with unfavorable prognosis in women with early-stage
breast cancer. PLoS One. 8:e562562013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu L, Wang F, Xu R, Zhang S, Peng X, Feng
Y, Wang J and Lu C: Promoter methylation of BRCA1 in the prognosis
of breast cancer: A meta-analysis. Breast Cancer Res Treat.
142:619–627. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lal A, Navarro F, Maher CA, Maliszewski
LE, Yan N, O'Day E, Chowdhury D, Dykxhoorn DM, Tsai P, Hofmann O,
et al: miR-24 Inhibits cell proliferation by targeting E2F2, MYC
and other cell-cycle genes via binding to ‘seedless’ 3′UTR microRNA
recognition elements. Mol Cell. 35:610–625. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huo X, Lu C, Huang X, Hu Z, Jin G, Ma H,
Wang X, Qin J, Wang X, Shen H and Tang J: Polymorphisms in BRCA1,
BRCA1-interacting genes and susceptibility of breast cancer in
Chinese women. J Cancer Res Clin Oncol. 135:1569–1575. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hasan TN, Shafi G, Syed NA, Alsaif MA,
Alsaif AA and Alshatwi AA: Lack of association of BRCA1 and BRCA2
variants with breast cancer in an ethnic population of Saudi
Arabia, an emerging high-risk area. Asian Pac J Cancer Prev.
14:5671–5674. 2013. View Article : Google Scholar : PubMed/NCBI
|