Introduction
Besides surgery and radiotherapy, chemotherapy is
regarded as the most effective means of current clinical treatment
for cancer. A large group of chemotherapeutic drugs target
rapidly-dividing cancer cells by directly damaging genomic DNA,
thereby inhibiting tumor growth (1,2). Cancer
cells have high proliferation rates and replicate their DNA
rapidly, making them highly susceptible to DNA damage, as
replicating damaged DNA increases the likelihood of cell death
(1,3).
Commonly used drugs in this group include: Cisplatin, camptothecin,
etoposide, bleomycin, doxorubicin and gemcitabine (1).
Despite the success of chemotherapy, drug resistance
is a major obstacle to successful chemotherapy (4,5).
Resistance to chemotherapy results in increased tumor growth and
decreased patient survival. In addition, severe side effects
resulting from adverse toxicity to non-target tissues are often
observed in patients (4,6). These drawbacks pose a significant
impediment to the development of desired chemotherapy regimens. A
promising method of reducing severe side effects may be to develop
approaches that sensitize cancer cells to current
chemotherapies.
For chemotherapy that is based on inducing DNA
damage, modulating DNA damage response or DNA repair is highly
desirable for enhancement of the sensitivity of cancer cells to
these therapies (4,7). This may be achieved by specifically
targeting factors involved in damage response and DNA repair
pathways. Previous studies have demonstrated that the CTS telomere
maintenance complex component 1 (CTC1)-STN1-TEN1 (CST) complex may
have a role in maintaining genome stability (8–10). CTC1
and STN1 were originally identified as accessory factors of DNA
polymerase alpha (Polα) and were named as AAF132 and AAF44,
respectively (11). STN1 is also
known as oligonucleotide/oligosaccharide-binding fold containing 1
(OBFC1) (12). Deficiencies in
components of the CST complex induces DNA damage (8–10),
suggesting that CST may have an important role in safeguarding
genome stability. It appears that such a role is particularly
prominent in the presence of replication stress (9,10). After
cells are treated with hydroxyurea, which depletes the nucleotide
pool and induces replication stress, deficiency in CST leads to
delayed recovery of stalled replication (9,10). It has
been postulated that one significant function of the CST complex
may be to promote efficient replication of difficult-to-replicate
sequences throughout the genome, perhaps by facilitating efficient
restart of stalled replication (8–10).
Several previous studies have demonstrated that CST
also has a significant role in telomere protection (8–10,13–17).
Telomeres are highly complex nucleoprotein structures located at
the ends of linear eukaryotic chromosomes, which are conserved in
organisms ranging from unicellular eukaryotes to mammals (18,19). Human
telomeres consist of tandem repeats of the sequence TTA GGG, which
typically extend up to 10–15 kb. Potential functions of telomeres
include prevention of chromosome degradation, end-to-end fusions,
rearrangements and chromosome loss (18,20,21). In
normal somatic cells, progressive telomere shortening occurs during
each cell division (18,19). When telomere length becomes critically
short, cells senesce and growth arrests. Cancer cells require
mechanisms to maintain their telomeres in order to continue
dividing indefinitely (18,22). Disruption of telomere maintenance in
cancer cells prevents cells from uncontrolled proliferation
(23). Approximately 85–90% of cancer
cells activate the telomerase gene, which utilizes its reverse
transcriptase activity to add telomere repeats at chromosome ends
to counteract telomere shortening (18,19,24).
Telomerase inhibition or inactivation in telomerase-expressing
cancer cells drives telomere shortening, inducing growth arrest or
death of tumor cells (18).
The CST complex binds to telomeres and is important
for several aspects of telomere maintenance. It promotes efficient
replication of telomeric DNA. RNA interference (RNAi)-mediated
suppression of STN1 and CTC1 elevates defects in telomere
replication, leading to increased telomere loss (8–10). CST
interacts with Polα and is required for replenishing the telomere C
strand following replication. Loss of CST leads to elongated
G-overhangs (9,16,25). In
certain cancer cell lines, CST binds to single-stranded G-rich
overhangs at telomere ends and prevents telomerase from excessively
extending telomere DNA (15).
The multiple significant functions of CST in
maintaining genomic stability prompted the present study to
postulate that suppression of CST may act synergistically with
chemotherapeutic agents that induce genome instability, in
particular to those agents that cause DNA damage. Using various
cell proliferation assays, the present study observed that
depletion of STN1 enhanced the cytotoxicity of bleomycin,
camptothecin and etoposide in various cancer cells from diverse
origins including the lung, breast and cervix. Comet assay
additionally revealed that STN1 suppression significantly elevated
DNA damage levels in cells treated with damaging agents, suggesting
that CST may have an important role in repairing DNA damage.
Materials and methods
Materials
The following chemotherapeutic agents were used:
Camptothecin (Sigma-Aldrich, St. Louis, MO, USA), etoposide
(Sigma-Aldrich), bleomycin (Sigma-Aldrich) and meso-tetra
(N-methyl-4-pyridyl) porphine tetra tosylate (TMPyP4; EMD
Millipore, Darmstadt, Germany). All agents were dissolved in
dimethyl sulfoxide (DMSO) and stored under sterile conditions at
−20°C in the dark. The vehicle (DMSO) was utilized as a control
with a final concentration of <0.1%, which had no influence on
cell growth.
Cell culture
Cells obtained from American Type Culture Collection
(Manassas, VA, USA) were passaged in Dulbecco's modified Eagle's
medium (DMEM; GE Healthcare Life Sciences, Logan, UT, USA)
supplemented with 10% cosmic calf serum (GE Healthcare Life
Sciences) at 37°C in a humidified atmosphere containing 5%
CO2 for <6 months. No antibiotics were added to the
medium to avoid stress.
Antibodies
The following primary antibodies were utilized:
Polyclonal rabbit anti-OBFC1 (1:500 dilution; cat no. sc-135364;
Santa Cruz Biotechnology, Inc., Dallas, TX, USA) and monoclonal
mouse anti-β-actin (1:60,000 dilution; cat no. A2228;
Sigma-Aldrich). The secondary antibody was horseradish
peroxidase-conjugated polyclonal goat anti-mouse immunoglobulin
(Ig)G (cat no. 554002; BD Biosciences, San Jose, CA, USA) or
anti-rabbit IgG (cat no. PI-1000; Vector Laboratories, Inc.,
Burlingame, CA, USA).
RNAi
STN1 small hairpin (sh)RNA sequences targeting
GCUUAACCUCACAACUUAA (shStn1-2) (9)
and GGACUGCCAGAAACCAAAT (shStn1-4) were cloned into
pSIREN-retro-puro (Clontech Laboratories, Inc., Mountainview, CA,
USA). Control shRNA targeted luciferase and the sequence was CGU
ACG CGG AAU ACU UCG A (shLuc) (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). Infection and selection were performed as
previously described (9).
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay
MTT assay was employed to evaluate cell viability.
Briefly, cells were seeded into 96-well multiplates at a density of
1×104/ml. Following overnight incubation, cells in
triplicate wells were treated with camptothecin, etoposide or
bleomycin at the indicated concentrations (5, 50 and 500 nM) for 5
days, and subsequently incubated with 100 µl of 0.5 µg/ml MTT for
an additional 4 h at 37°C. MTT was subsequently removed, and DMSO
was added to dissolve the resulting formazan crystals. The light
absorption was measured at 570 nm with a microplate
spectrophotometer (BioTek Instruments, Inc., Winooski, VT, USA).
Effects of chemicals on cell survival were assessed by half maximal
inhibitory concentration (IC50) values (the
concentration resulting in 50% inhibition of cell growth).
Colony formation assay
H1299 shLuc, shSTN1-2 and shSTN1-4 cells, and HeLa
shLuc, shSTN1-2 and shSTN1-4 cells (2 days after puromycin
selection) were seeded into 6-well plates at a density of 100
cells/well and incubated overnight. On the following day, cells
were treated with various concentrations (500, 50 and 5 nM) of each
testing drug (camptothecin, etoposide and bleomycin) at 37°C.
Identical treatments were repeated every 4 days. After 10 days of
incubation, the medium was removed and cell colonies were fixed and
stained with crystal violet solution (0.1% crystal violet, 1%
methanol and 1% formaldehyde). Colonies with >50 cells were
counted and the percentage of drug-treated colonies relative to
DMSO-treated control colonies was calculated.
Soft agar colony formation assay
Soft agar assays were performed in 6-well plates.
Equal volumes of 1.2% agar were mixed with 2X DMEM and 1 ml of this
mixture was added into each well as the base layer. Plates were
chilled at 4°C until the agar solidified. Subsequently,
1×104 cells that were suspended in 1X DMEM containing a
gradient concentration (5, 50, 500 nM) of drugs, and then mixed
with equal volume of 0.7% agar. A total of 1 ml of the mixture was
poured as the top layer. Once the growth layer congealed, cells
were permitted to grow at 37°C for 10 days and total colonies were
counted and normalized against untreated cells.
Comet assay
DNA damage levels in wild-type and STN1 knockdown
cells following exposure to drugs were assessed using the comet
assay. H1299 cells were treated with 1 µM of each testing agent for
6 h, while HeLa cells were treated with 3 µM of each agent for 8 h.
Immediately subsequent to treatment, 10 µl cell suspension was
mixed with 50 µl 0.5% low-melting agarose, spread onto a Cometslide
(Trevigen, Gaithersburg, MD, USA), and pre-coated with 100 µl of 1%
normal-melting agarose. Cells were lysed by immersing the slides in
a freshly prepared lysis solution [2.5 M NaCl, 100 mM
ethylenediaminetetraacetic acid (EDTA), 10 mM Tris, 1% Triton
X-100, 10% DMSO; pH 10] at 4°C for 2 h. Following lysis, slides
were washed 3 times in 1X phosphate-buffered saline and placed in a
gel electrophoresis apparatus with freshly prepared electrophoresis
buffer (1 mM EDTA, 300 mM NaOH; pH 13) for 25 min to allow DNA
unwinding. Electrophoresis was subsequently performed at 20 V with
a starting current of 300 mA for 20 min. Subsequently, slides were
neutralized with 0.4 M Tris (pH 7.5) 3 times, stained with 1X SYBR
Gold solution (Thermo Fisher Scientific, Inc.), and viewed under an
epifluorescence microscope (Axio Imager M2; Zeiss AG, Oberkochen,
Germany). Data was analyzed and presented in terms of comet
occurrence (% cells containing comets) and tail extent moment (tail
length × tail intensity). Comets were scored visually without using
analysis software, classifying them as belonging to one of five
classes according to the tail intensity. Each comet class was given
a value between 0 and 4: A score of 0 indicated undamaged cells
(i.e., no comet) while a score of 4 indicated maximum damage. The
parameter ‘comet occurrence’ was calculated from this
classification and measured in arbitrary units. The comet
occurrence was calculated using the following equation: [(% of
cells in class 0) × 0 + (% of cells in class 1 × 1 + (% of cells in
class 2) × 2 + (% of cells in class 3) × 3 + (% of cells in class
4) × 4] / total cell number (26).
Tail length and tail intensity were evaluated by OpenComet version
1.3 software (www.cometbio.org). A total of 100
cells per sample were selected from random fields and counted for
comets. A single investigator analyzed all slides to minimize
scoring variation.
Statistical analysis
Two-tailed Student's t-tests were performed in
Microsoft Excel 97–2003 (Microsoft Inc., Redmond, WA, USA) to
calculate P-values. P<0.05 was considered to indicate a
statistically significant difference.
Results
STN1 suppression enhances the
cytotoxicity of commonly used chemotherapeutic reagents in various
cancer cells
The CST complex has a significant role in telomere
length control as well as in promoting efficient replication of
difficult-to-replicate genomic sequences (8–10,15). Previously the present authors and
others have demonstrated that suppression of CST in human cells
results in increased staining of the phosphorylated form of histone
H2A, member X in telomeric and non-telomeric regions in the absence
of exogenous damage (8,9), suggesting that CST deficiency may lead
to genome instability. However, the DNA damage level induced by CST
deficiency is insufficient to elicit marked growth defects in
common cancer cell lines (9,10), suggesting that cancer cells may be
able to tolerate low levels of DNA damage induced by CST
deficiency. We hypothesize that, in the presence of exogenous DNA
damaging agents, CST deficiency may augment DNA damage levels,
therefore sensitizing cancer cells to damage-inducing agents. To
test this, the present study investigated the differences in the
sensitivity between wild-type and STN1-deficient cells using the
MTT assay. The present study focused on STN1, as a previous study
indicated that STN1 is required for formation of the CST complex
(27). In agreement with this, the
present study also observed that CTC1 expression was reduced when
STN1 was absent (data not shown). Using shRNA, the present study
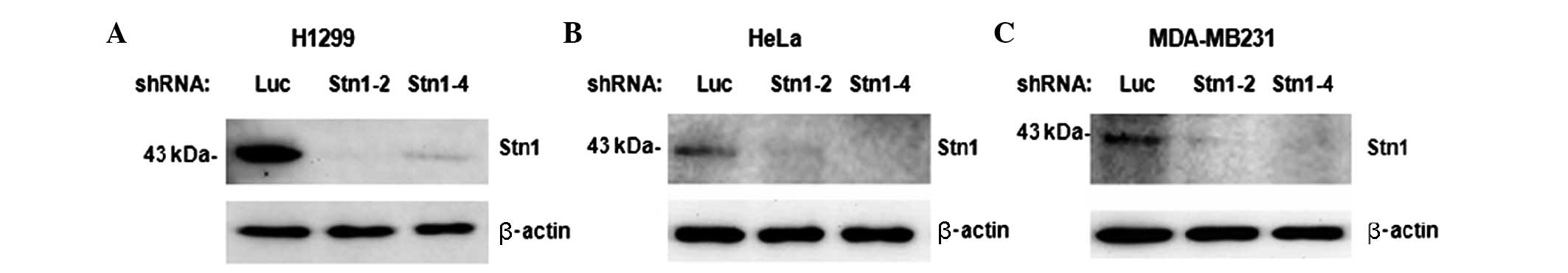
initially established cell lines with stable STN1 depletion
(Fig. 1). Cancer cell lines from
various origins, including the H1299 human non-small lung carcinoma
cell line, HeLa human cervical epithelial adenocarcinoma cell line
and human breast adenocarcinoma cell line MDA-MB231, were infected
with retroviruses expressing two distinct STN1 shRNAs (shStn1-2 and
shStn1-4). Following selection, puromycin-resistant cells were
collected and immediately treated with chemotherapeutic agents that
are commonly used in clinical treatment. STN1 depletion was
confirmed by western blotting (Fig.
1). A total of 4 chemotherapeutic agents were used:
Camptothecin (CPT), etoposide, bleomycin and TMPyP4. Among them,
CPT, bleomycin and etoposide are potent DNA damage inducers
(28–33). TMPyP4 is a cationic porphyrin compound
that binds to and stabilizes a particular DNA secondary structure
known as G-quadruplexes, which are formed by guanine residues
through Hoogsteen hydrogen bonding (34). Such stabilization creates prominent
barriers to DNA replication that stall replication forks, which may
eventually collapse, leading to DNA damage (35,36).
Following 5 days of exposure, the growth of H1299
shStn1-2 and shStn1-4 cells was remarkably inhibited by all four
agents, with IC50 values two to three times lower
compared with the shLuc control cells (Table I). When the HeLa and MDA-MB231 cell
lines were investigated, a similar degree of chemosensitivity was
observed (Tables II and III). Therefore, STN1 knockdown reduced the
survival of tumor cells following exposure to various
damage-inducing anti-tumor agents, independent of cell type.
 | Table I.IC50 values of
camptothecin, etoposide, bleomycin and TMPyP4 in H1299 cells with
either STN1 knockdown or control. |
Table I.
IC50 values of
camptothecin, etoposide, bleomycin and TMPyP4 in H1299 cells with
either STN1 knockdown or control.
|
| IC50
value, µM |
|---|
|
|
|
|---|
| Cell type | Camptothecin | Etoposide | Bleomycin | TMPyP4 |
|---|
| shLuc | 0.57±0.01 | 0.43±0.29 | 7.10±1.60 | 8.65±0.44 |
| shStn1-2 | 0.20±0.03 | 0.13±0.10 | 2.36±0.94 | 5.76±1.76 |
| shStn1-4 | 0.18±0.17 | 0.11±0.06 | 3.07±0.92 | 5.14±1.39 |
| Table II.IC50 values of
camptothecin, etoposide, bleomycin and TMPyP4 in HeLa cells with
either STN1 knockdown or control. |
Table II.
IC50 values of
camptothecin, etoposide, bleomycin and TMPyP4 in HeLa cells with
either STN1 knockdown or control.
|
| IC50
value, µM |
|---|
|
|
|
|---|
| Cell type | Camptothecin | Etoposide | Bleomycin | TMPyP4 |
|---|
| shLuc | 0.51±0.06 | 0.60±0.13 | 2.78±0.56 | 7.41±0.95 |
| shStn1-2 | 0.15±0.04 | 0.24±0.09 | 1.12±0.17 | 3.54±0.72 |
| shStn1-4 | 0.23±0.07 | 0.22±0.05 | 1.29±0.39 | 3.11±1.04 |
| Table III.IC50 values of
camptothecin, etoposide, bleomycin and TMPyP4 in MDA-MB231 cells
with either STN1 knockdown or control. |
Table III.
IC50 values of
camptothecin, etoposide, bleomycin and TMPyP4 in MDA-MB231 cells
with either STN1 knockdown or control.
|
| IC50
value, µM |
|---|
|
|
|
|---|
| Cell type | Camptothecin | Etoposide | Bleomycin | TMPyP4 |
|---|
| shLuc | 0.25±0.08 | 0.53±0.12 | 9.33±0.76 | 8.85±1.12 |
| shStn1-2 | 0.12±0.13 | 0.27±0.08 | 7.78±1.02 | 5.54±0.88 |
| shStn1-4 | 0.16±0.11 | 0.28±0.10 | 6.59±0.85 | 6.07±1.08 |
STN1 suppression diminishes
colony-formation ability and anchorage-independent growth ability
in the presence of anticancer agents
To additionally investigate the effectiveness of
STN1 suppression on the survival and proliferation of cancer cells,
the present study evaluated whether STN1 suppression affected the
abilities of cancer cells to form colonies and to grow
independently of anchorage using clonogenic and soft agar assays.
In both assays, the same enhanced growth inhibitory effect was
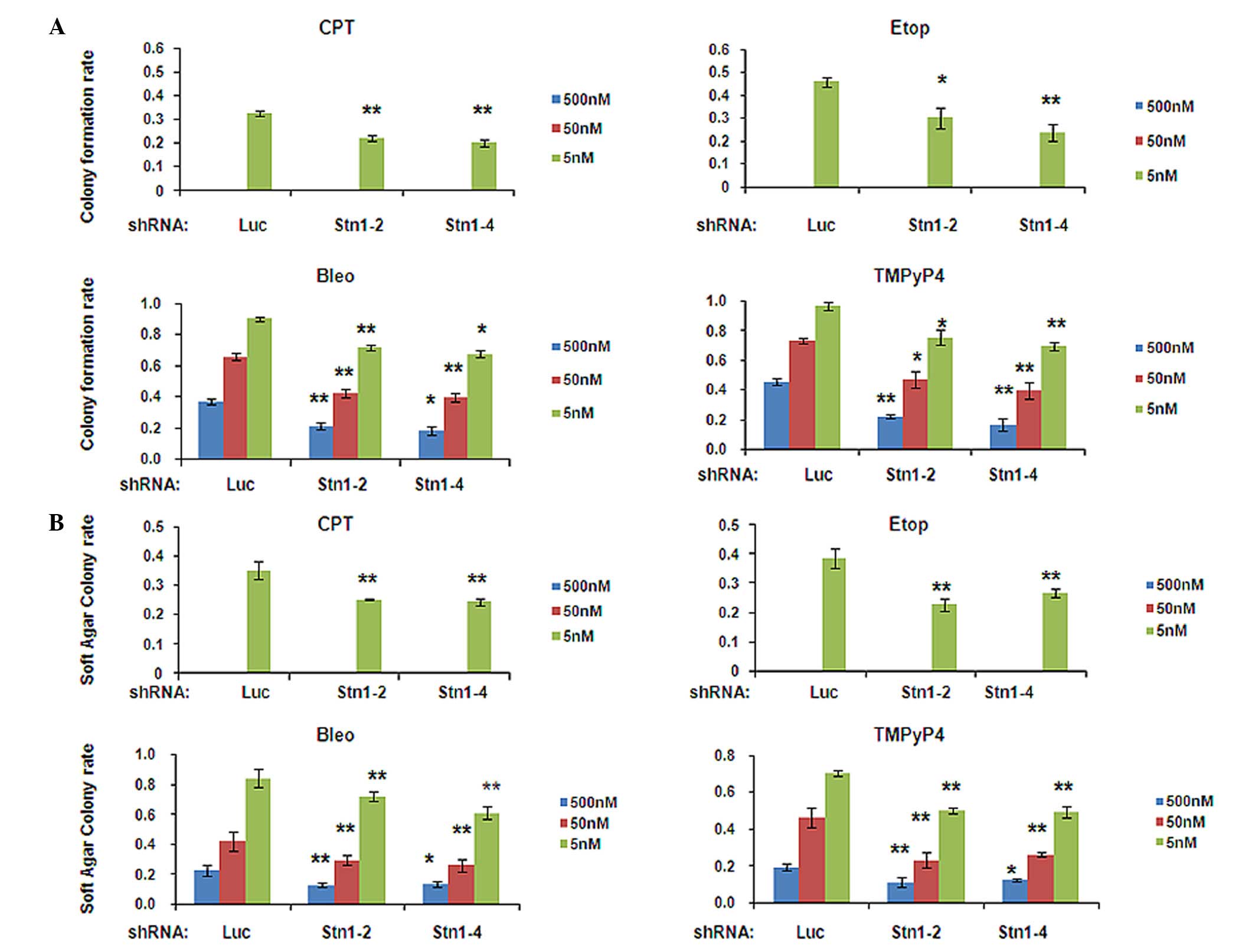
observed in H1299 (Fig. 2A and B) and
HeLa (Fig. 3A and B) cells with STN1
suppression following drug treatment, compared with shLuc cells. In
the clonogenic assay, cells were treated with a gradient
concentration of 5, 50 and 500 nM of each drug for 10 days. In the
absence of drug treatment, no significant growth defects were
observed in H1299 or HeLa cells with STN1 knockdown compared with
the shLuc control (data not shown). Colony formation was largely
inhibited by drug treatment in all cells (Figs. 2A and 3A). Notably, this inhibition was
significantly escalated in STN1 knockdown H1299 cells (Fig. 2A). Under all three drug
concentrations, colony numbers of drug-treated shStn1-2 and
shStn1-4 H1299 cells were greatly diminished compared with the
drug-treated shLuc control. The most significant changes were
observed in cells treated with 500 nM of bleomycin (shStn1-2,
P=0.005; shStn1-4, P=0.022) or TMPyP4 (shStn1-2, P<0.001;
shStn1-4, P<0.001; Fig. 2A). In
HeLa cells, the long-term synergetic effects of STN1 knockdown and
drug treatment were additionally observed. The colony growth in
shStn1-2 and shStn1-4 cells was reduced in all drug-treated groups,
in contrast to their shLuc controls (Fig.
3A).
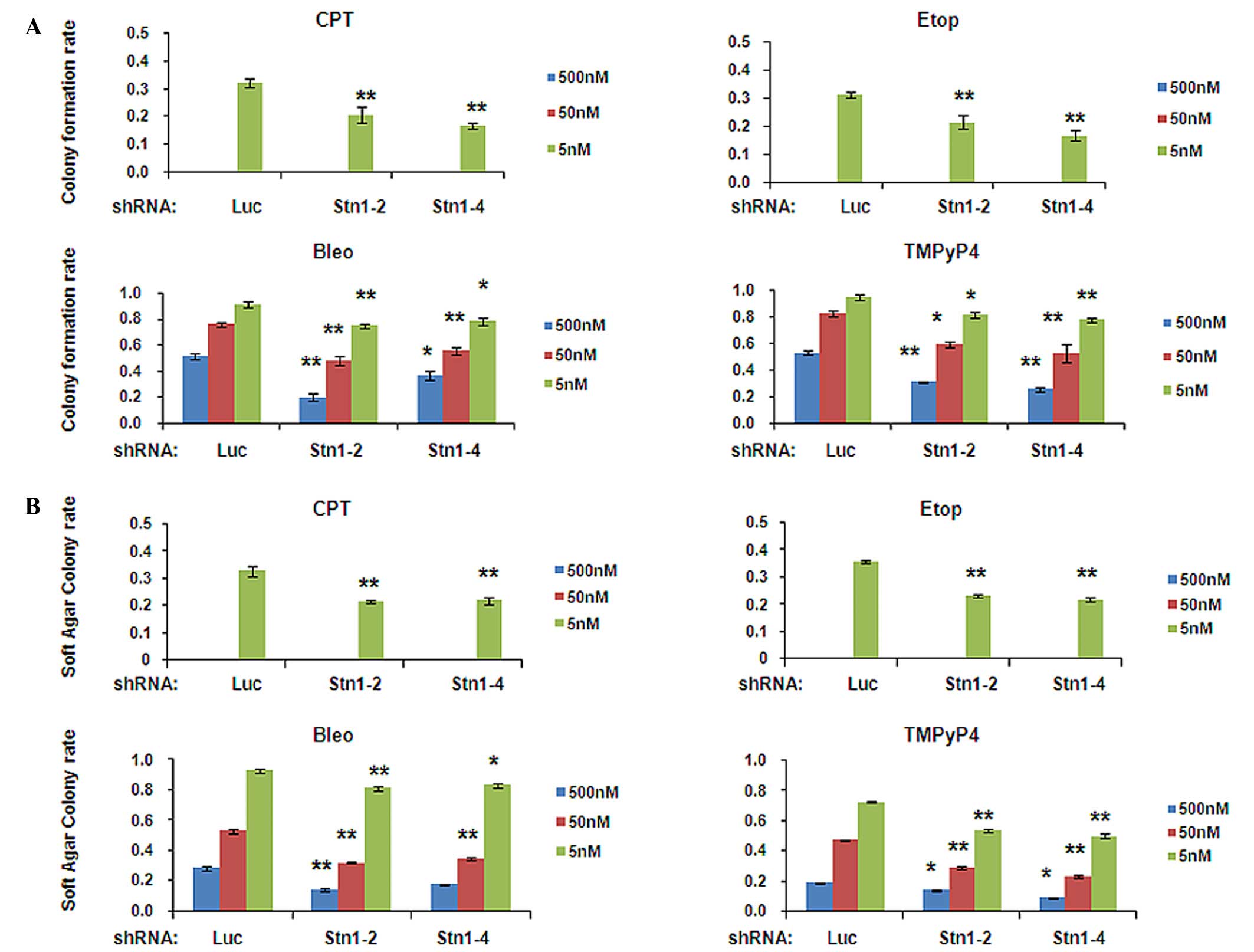 | Figure 2.STN1 suppression enhances growth
inhibition by various anticancer agents in the H1299 cell line. (A)
Decreased colony formation of STN1 knockdown cells following
exposure to CPT, Etop, Bleo and TMPyP4. The colony formation rate
was calculated by dividing the number of colonies formed by
drug-treated cells by the number of colonies formed by untreated
cells. Each drug was tested at 5, 50 and 500 nM. High
concentrations of CPT or Etop (50 and 500 nM) caused severe cell
death and no colonies were formed. The results are representative
of three independent experiments. Data are presented as the mean ±
standard error. **P<0.01, *P<0.05. (B) Reduced
anchorage-independent growth ability of STN1 knockdown cells
assessed by soft agar colony formation assay. Each drug was tested
at 5, 50 and 500 nM. The results are representative of three
independent experiments. Data are presented as the mean ± standard
error. **P<0.01, *P<0.05. CPT, camptothecin; Etop, etoposide;
Bleo, bleomycin; TMPyP4, meso-Tetra (N-methyl-4-pyridyl) porphine
tetra tosylate; shRNA, small hairpin RNA; shLuc, shRNA targeting
luciferase. |
 | Figure 3.STN1 suppression causes increased
growth-inhibitory effects by various anticancer agents in the HeLa
cell line. (A) Decreased colony formation of STN1 knockdown cells
following exposure to CPT, Etop, Bleo and TMPyP4. The colony
formation rate was normalized to untreated cells. Each drug was
tested at 5, 50 and 500 nM. The results are representative of three
independent experiments. Data are presented as the mean ± standard
error. **P<0.01, *P<0.05. (B) Reduced anchorage-independent
growth ability of STN1 knockdown HeLa cells assessed by soft agar
colony formation assay. Each drug was tested at 5, 50 and 500 nM.
The results are representative of three independent experiments.
Data are presented as the mean ± standard error. **P<0.01,
*P<0.05. CPT, camptothecin; Etop, etoposide; Bleo, bleomycin;
TMPyP4, meso-Tetra (N-methyl-4-pyridyl) porphine tetra tosylate;
shRNA, small hairpin RNA; shLuc, shRNA targeting luciferase. |
Anchorage-independent growth is one of the hallmarks
of cancerous cell transformation (37,38). In
general, there is a positive correlation between in vitro
transformation and in vivo carcinogenesis (39–41). Soft
agar assay is considered the most accurate and stringent in
vitro assay for detection of malignant transformation of cells
(37,38). The present study used soft agar assay
to determine the effect of STN1 knockdown on anchorage-independent
growth in H1299 and HeLa cells. Following 10 days of treatment with
drugs, STN1 knockdown enhanced growth inhibition induced by all
four drugs in soft agar (Figs. 2B and
3B). The results of the present study
additionally support the conclusion that STN1 suppression enhanced
the growth inhibitory effect of tested anticancer agents.
STN1 suppression augments DNA damage
in cancer cells treated with various DNA damage inducers
Given the role of CST in maintaining genome
stability and countering replication stress, it was suspected that
STN1 deficiency may elevate DNA damage levels caused by damage
inducers, eliciting rapid growth inhibition. To investigate this
hypothesis, the present study employed the widely-used comet assay
to detect DNA lesions in STN1 knockdown cells. The principle of
comet assay is based on the ability of denatured broken DNA
fragments to migrate out of the nucleoid under the influence of an
electric field, whereas undamaged DNA migrates more slowly and
remains within the confines of the nucleoid when a current is
applied. Evaluation of the DNA ‘comet’ tail shape and migration
pattern allows for assessment of DNA damage (26,42). Comet
assays were performed on H1299 and HeLa control cells and the
corresponding STN1 knockdown cells following treatment with each
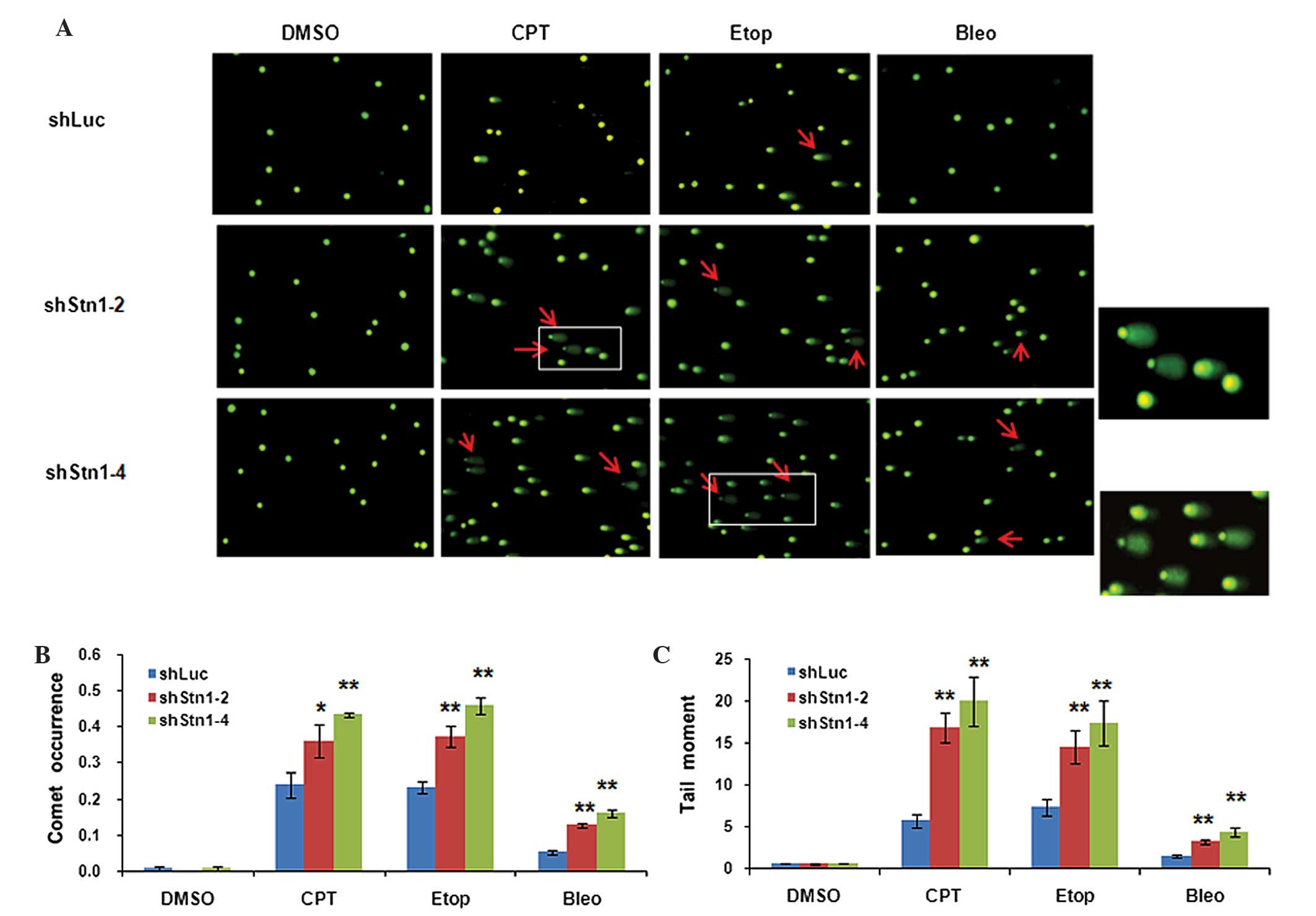
chemotherapeutic agent. As shown in Fig.
4A, untreated H1299 cells in all three groups exhibited a
normal organized of nucleus without tails, indicative of undamaged
DNA. As expected, treating H1299 cells with damage-inducing agent
(CPT, etoposide and bleomycin) induced comet tails in all groups
(Fig. 4A). Notably, quantification of
comet occurrence and tail moment revealed that an increased number
of comets as well as longer tails were present in shStn1-2 and
shStn1-4 cells compared with control shLuc cells (Fig. 4B and C). In CPT-treated cells, STN1
knockdown increased comet occurrence by 1.5- (shStn1-2, P=0.011)
and 1.8-fold (shStn1-4, P=0.005). In etoposide-treated cells,
shStn1-2 and shStn1-4 increased comet occurrence by 1.6- (P=0.002)
and 2-fold (P=0.015). In bleomycin-treated cells, comet occurrence
in H1299 cells expressing shStn1-2 and shStn1-4 was 2- (P=0.001)
and 3-fold (P=0.001) higher compared with control cells (Fig. 4B). Similarly, STN1 knockdown also
significantly increased the tail moment by 2- to 3-fold in cells
treated with CPT (shStn1-2, P=0.004; shStn1-4, P=0.0004) etoposide
(shStn1-2, P=0.002; shStn1-4, P=0.003;) and bleomycin (shStn1-2,
P=0.0005; shStn1-4, P=0.0003; Fig.
4C).
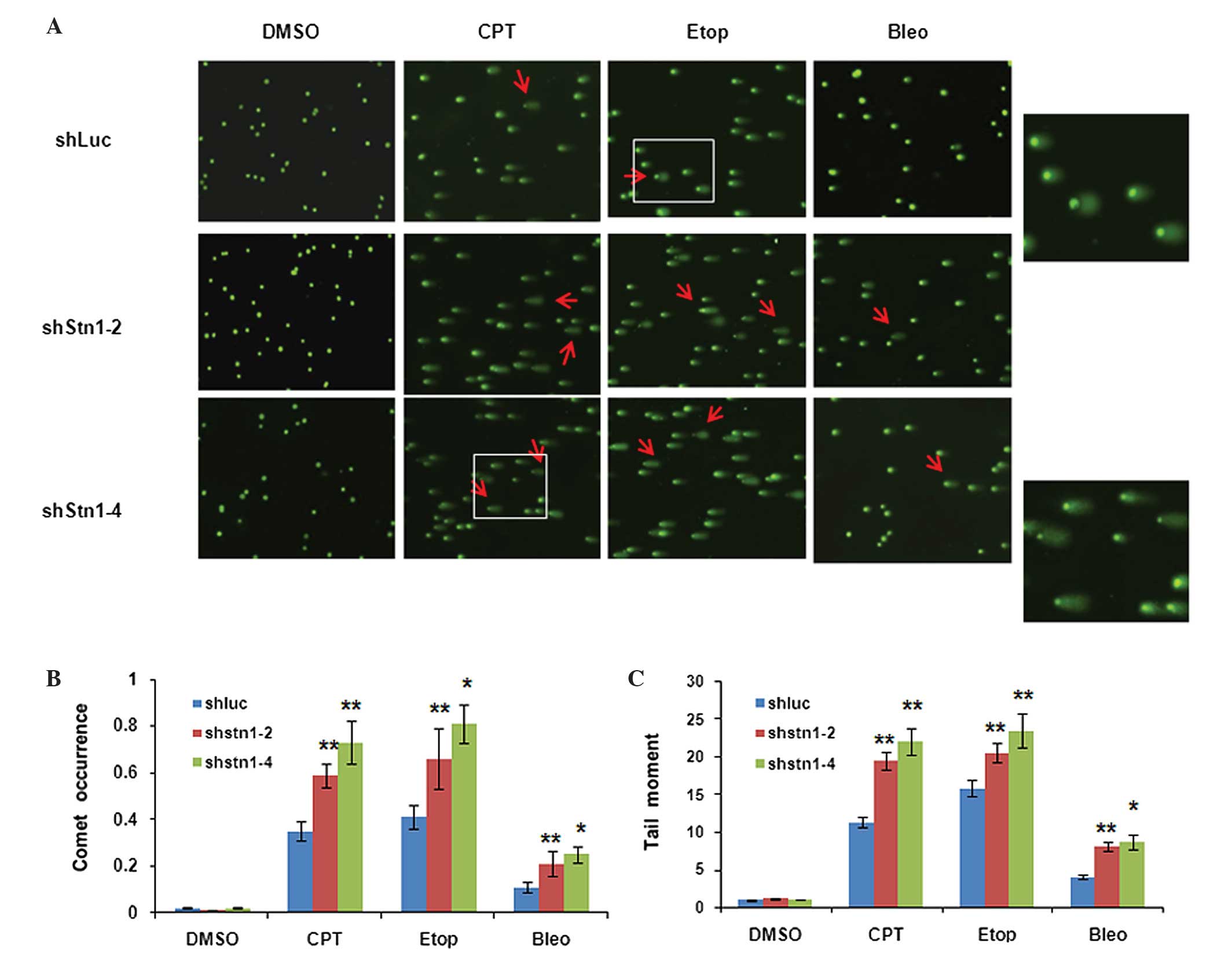
In HeLa cells, a significant fraction of shLuc cells
showed comet tails following treatment with DNA-damaging agents.
However, in all drug-treated groups, shStn1-2 and shStn1-4 cells
had increased numbers of comets and longer tail moments compared
with shLuc controls (Fig. 5A). Among
the three DNA damage inducers, etoposide gave rise to the most
marked increase in terms of comet occurrence, while CPT treatment
led to the largest increase in tail moments in shStn1-2 and
shStn1-4 cells, in contrast to shLuc controls (Fig. 5B and C). These results suggest that
STN1 knockdown cells are more susceptible to DNA damage. The
results of the present study suggest that the chemosensitivity
induced by STN1 suppression is at least in part due to elevated
levels of DNA damage in STN1 knockdown cells.
Discussion
DNA-damaging agents have been commonly used in
clinical studies to eliminate cancer cells and reduce tumor growth
(1,2,43).
Sensitization of cancer cells to chemotherapeutic agents reduces
side effects, as well as balances the efficacy and toxicity of
chemotherapy (44). Given the
significant role of the human CST complex in maintaining genomic
stability and telomere integrity, the authors of the present study
reasoned that suppression of CST expression may augment DNA damage
levels, therefore sensitizing cancer cells to chemotherapeutic
agents. The results of the present study revealed that suppression
of STN1 increases the sensitivity of various cancer cells to
several commonly used DNA damage inducing agents. Stable knockdown
of STN1 using two independent shRNA sequences enhances the
cytotoxicity of DNA damage inducers (camptothecin, etoposide and
bleomycin) in common cancer cell lines. These results are in
agreement with a previous study demonstrating that overexpression
of CST proteins facilitate the recovery from replication
stress-induced DNA damage (45).
Mechanistically, the present study demonstrates that STN1 knockdown
elevates the levels of DNA lesions induced by DNA damage inducers,
implying that the enhanced cytotoxicity may be at least in part due
to inefficiency in repairing DNA damage or induction of increased
DNA damage by STN1 deficiency. The results of the present study
provide proof of principle that combining inhibition of CST with
chemotherapeutic regimens may enhance their effectiveness against
malignant cell proliferation.
The results of the present study additionally
suggest that the CST complex may be a novel player in the DNA
repair pathway. Prior to the present study, it had only been
demonstrated that the CST complex is important for countering
replication stress, likely by promoting genome-wide replication
restart following replication fork stalling (9,10). To the
best of our knowledge, no previous studies have linked CST with
protecting cells from DNA damage. In fact, the results of the
present comet assay reveal that there is no significant increase in
DNA lesions in STN1 knockdown cells without exposure to drugs,
indicating that STN1 deficiency only induces moderate DNA damage
that is tolerated by cancer cells or is easily repaired. This is in
agreement with previously published work demonstrating that
downregulation of STN1 in cancer cells does not affect cell growth
(9,10,15). By
contrast, following exposure to various DNA-damaging agents, DNA
damage levels are markedly elevated in STN1 knockdown cells
compared with control cells. The authors of the present study
propose that the CST complex may have an unidentified yet
significant role in repairing DNA double-strand breaks. This
important function of CST remains to be elucidated in additional
studies.
In conclusion, the present study demonstrates the
synergistic effect of STN1 depletion and established
chemotherapeutic agents. The results of the present study
contribute to the knowledge concerning the function of the CST
complex in cancer cell proliferation, and provide proof of
principle that CST may potentially be a novel molecular target for
anticancer therapy.
Acknowledgements
The present study was supported in part by the
National Institute of Health (grant nos., R01GM112864 and
R15GM099008 and the CONCERN Now Award to Professor Weihang
Chai.
Glossary
Abbreviations
Abbreviations:
|
CPT
|
camptothecin
|
|
DMSO
|
dimethylsulfoxide
|
|
OBFC1
|
oligonucleotide/oligosaccharide
binding fold containing protein 1
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
|
References
|
1
|
Cheung-Ong K, Giaever G and Nislow C:
DNA-damaging agents in cancer chemotherapy: Serendipity and
chemical biology. Chem Biol. 20:648–659. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lord CJ and Ashworth A: The DNA damage
response and cancer therapy. Nature. 481:287–294. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bouwman P and Jonkers J: The effects of
deregulated DNA damage signaling on cancer chemotherapy response
and resistance. Nat Rev Cancer. 12:587–598. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Luo Y and Leverson JD: New opportunities
in chemosensitization and radiosensitization: Modulating the
DNA-damage response. Expert Rev Anticancer Ther. 5:333–342. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Holohan C, Van Schaeybroeck S, Longley DB
and Johnston PG: Cancer drug resistance: An evolving paradigm. Nat
Rev Cancer. 13:714–727. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Coley HM: Mechanisms and strategies to
overcome chemotherapy resistance in metastatic breast cancer.
Cancer Treat Rev. 34:378–390. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhou BB and Bartek J: Targeting the
checkpoint kinases: Chemosensitization versus chemoprotection. Nat
Rev Cancer. 4:216–225. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gu P, Min JN, Wang Y, Huang C, Peng T,
Chai W and Chang S: CTC1 deletion results in defective telomere
replication, leading to catastrophic telomere loss and stem cell
exhaustion. EMBO J. 31:2309–2321. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huang C, Dai X and Chai W: Human Stn1
protects telomere integrity by promoting efficient lagging-strand
synthesis at telomeres and mediating C-strand fill-in. Cell Res.
22:1681–1695. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Stewart JA, Wang F, Chaiken MF, Kasbek C,
Chastain PD II, Wright WE and Price CM: Human CST promotes telomere
duplex replication and general replication restart after fork
stalling. EMBO J. 31:3537–3549. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Casteel DE, Zhuang S, Zeng Y, Perrino FW,
Boss GR, Goulian M and Pilz RB: A DNA polymerase-{alpha}{middle
dot}primase cofactor with homology to replication protein A-32
regulates DNA replication in mammalian cells. J Biol Chem.
284:5807–5818. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wan M, Qin J, Songyang Z and Liu D:
OB-fold containing protein 1 (OBFC1), a human homolog of yeast
Stn1, associates with TPP1 and is implicated in telomere length
regulation. J Biol Chem. 284:26725–26731. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen LY, Majerská J and Lingner J:
Molecular basis of telomere syndrome caused by CTC1 mutations.
Genes Dev. 27:2099–2108. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Anderson BH, Kasher PR, Mayer J,
Szynkiewicz M, Jenkinson EM, Bhaskar SS, Urquhart JE, Daly SB,
Dickerson JE, O'Sullivan J, et al: Mutations in CTC1, encoding
conserved telomere maintenance component 1, cause Coats plus. Nat
Genet. 44:338–342. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen LY, Redon S and Lingner J: The human
CST complex is a terminator of telomerase activity. Nature.
488:540–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang F, Stewart JA, Kasbek C, Zhao Y,
Wright WE and Price CM: Human CST has independent functions during
telomere duplex replication and C-strand fill-in. Cell Rep.
2:1096–1103. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu P, Takai H and de Lange T: Telomeric 3′
overhangs derive from resection by Exo1 and Apollo and fill-in by
POT1b-associated CST. Cell. 150:39–52. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shay JW and Wright WE: Telomerase
therapeutics for cancer: Challenges and new directions. Nat Rev
Drug Discov. 5:577–584. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
De Cian A, Lacroix L, Douarre C,
Temime-Smaali N, Trentesaux C, Riou JF and Mergny JL: Targeting
telomeres and telomerase. Biochimie. 90:131–155. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mondello C and Scovassi IA: Telomeres,
telomerase, and apoptosis. Biochem Cell Biol. 82:498–507. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chan SR and Blackburn EH: Telomeres and
telomerase. Philos Trans R Soc Lond B Biol Sci. 359:109–121. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu Y, Bohr VA and Lansdorp P: Telomere,
telomerase and aging. Mech Ageing Dev. 129:1–2. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen M and McLeskey SW: Telomere-based
cancer treatment: emerging targeted therapies. Clin J Oncol
Nursing. 14:720–727. 2010. View Article : Google Scholar
|
|
24
|
Herbert BS, Gellert GC, Hochreiter A,
Pongracz K, Wright WE, Zielinska D, Chin AC, Harley CB, Shay JW and
Gryaznov SM: Lipid modification of GRN163, an N3′-P5′
thio-phosphoramidate oligonucleotide, enhances the potency of
telomerase inhibition. Oncogene. 24:5262–5268. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dai X, Huang C, Bhusari A, Sampathi S,
Schubert K and Chai W: Molecular steps of G-overhang generation at
human telomeres and its function in chromosome end protection. EMBO
J. 29:2788–2801. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Collins AR: The comet assay. Principles,
applications, and limitations. Methods Mol Biol. 203:163–177.
2002.PubMed/NCBI
|
|
27
|
Gu P and Chang S: Functional
characterization of human CTC1 mutations reveals novel mechanisms
responsible for the pathogenesis of the telomere disease Coats
plus. Aging Cell. 12:1100–1109. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pizarro JG, Folch J, Junyent F, Verdaguer
E, Auladell C, Beas-Zarate C, Pallàs M and Camins A: Antiapoptotic
effects of roscovitine on camptothecin-induced DNA damage in
neuroblastoma cells. Apoptosis. 16:536–550. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Berniak K, Rybak P, Bernas T, Zarębski M,
Biela E, Zhao H, Darzynkiewicz Z and Dobrucki JW: Relationship
between DNA damage response, initiated by camptothecin or oxidative
stress, and DNA replication, analyzed by quantitative 3D image
analysis. Cytometry A. 83:913–924. 2013.PubMed/NCBI
|
|
30
|
Nguyen TV, Chen JK and Murray V: Bleomycin
DNA damage: Anomalous mobility of 3′-phosphoglycolate termini in an
automated capillary DNA sequencer. J Chromatogr B Analyt Technol
Biomed Life Sci. 913–914:113–122. 2013. View Article : Google Scholar
|
|
31
|
Patel JR, Dhorajiya BD, Dholakiya BZ,
Badria FA and Ibrahim AS: In-vitro cytotoxicity, antioxidant,
bleomycin-dependent DNA damage and immunomodulatory evaluation of
1-(4-acetylphenyl)-3-aryloxypyrrolidine-2, 5-dione based
derivatives. Med Chem Res. 23:3907–3915. 2014. View Article : Google Scholar
|
|
32
|
Li X, Liu W, Wang H, Yang L, Li Y, Wen H,
Ning H, Wang J, Zhang L, Li J and Fan D: Rap1 is indispensable for
TRF2 function in etoposide-induced DNA damage response in gastric
cancer cell line. Oncogenesis. 4:e1442015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Griaud F, Williamson AJK, Taylor S, Potier
DN, Spooncer E, Pierce A and Whetton AD: BCR/ABL modulates protein
phosphorylation associated with the etoposide-induced DNA damage
response. J Proteomics. 77:14–26. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Grand CL, Han H, Muñoz RM, Weitman S, Von
Hoff DD, Hurley LH and Bearss DJ: The cationic porphyrin TMPyP4
down-regulates c-MYC and human telomerase reverse transcriptase
expression and inhibits tumor growth in vivo. Mol Cancer
Ther. 1:565–573. 2002.PubMed/NCBI
|
|
35
|
Tarsounas M and Tijsterman M: Genomes and
G-quadruplexes: For better or for worse. J Mol Biol. 425:4782–4789.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bochman ML, Paeschke K and Zakian VA: DNA
secondary structures: Stability and function of G-quadruplex
structures. Nat Rev Genet. 13:770–780. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Borowicz S, Scoyk MV, Avasarala S,
Rathinam Karuppusamy MK, Tauler J, Bikkavilli RK and Winn RA: The
soft agar colony formation assay. J Vis Exp.
92:e519982014.PubMed/NCBI
|
|
38
|
Horibata S, Vo TV, Subramanian V, Thompson
PR and Coonrod SA: Utilization of the soft agar colony formation
assay to identify inhibitors of tumorigenicity in breast cancer
cells. J Vis Exp. 99:e527272015.PubMed/NCBI
|
|
39
|
IARC/NCI/EPA Working Group: Cellular and
molecular mechanisms of cell transformation and standardization of
transformation assays of established cell lines for the prediction
of carcinogenic chemicals: Overview and recommended protocols.
Cancer Res. 45:2395–2399. 1985.
|
|
40
|
Kolber AR, Wong TK, Grant LD, DeWoskin RS
and Hughes TJ: In Vitro Toxicity Testing of Environmental Agents.
Current and Future Possibilities Part A: Survey of Test Systems.
Plenum Press. (New York). 321–322. 1979.
|
|
41
|
Pullman B, Ts'O POP and Schneider EL:
Interrelationship Among Aging, Cancer and Differentiation. The
Jerusalem Symposia on Quantum Chemistry and Biochemistry. 18:D.
Reidel Publishing Company. (Dordrecht, The Netherlands). 247–248.
1985.
|
|
42
|
Collins AR: The comet assay for DNA damage
and repair: Principles, applications, and limitations. Mol
Biotechnol. 26:249–261. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hosoya N and Miyagawa K: Targeting DNA
damage response in cancer therapy. Cancer Sci. 105:370–388. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dickerson EB, Blackburn WH, Smith MH, Kapa
LB, Lyon LA and McDonald JF: Chemosensitization of cancer cells by
siRNA using targeted nanogel delivery. BMC Cancer. 10:102010.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang F, Stewart J and Price CM: Human CST
abundance determines recovery from diverse forms of DNA damage and
replication stress. Cell Cycle. 13:3488–3498. 2014. View Article : Google Scholar : PubMed/NCBI
|