Introduction
In China, >60% of gastric cancer patients are
initially diagnosed with locally advanced (or metastatic) gastric
cancer (AGC) (1), and
fluorouracil-based combination chemotherapy is considered a
first-line treatment (2). However,
clinical outcomes for AGC patients are unsatisfactory, with
treatment offering modest responses and poor prognoses (3). Trastuzumab added to this therapy has
been documented to prolong the survival of human epidermal growth
factor receptor 2 (HER2)-positive AGC patients, but HER2 expression
is only reported to occur in 15–20% of all gastric cancers
(4). Thus, novel gastric cancer
therapies are required.
In the past, tyrosine kinase inhibitors (TKIs),
including imatinib (5), gefitinib
(6) and erlotinib (7), have been successfully used to treat
other tumors (8), but these drugs
have not been used for gastric cancer, despite animal data
suggesting their potential efficacy (9). Famitinib (SHR1020) is a novel
multi-targeted receptor TKI that targets vascular endothelial
growth factor receptor 2 (VEGFR2), c-Kit and platelet-derived
growth factor receptor β, and is inhibitory at 4.7±2.9, 2.3±2.6 and
6.6±1.1 nM, respectively (10).
Famitinib is a structural analog of sunitinib with improved cell
inhibitory activity (Xie et al, unpublished data). Due to
its anti-angiogenic effect, famitinib was effective against
metastatic renal cell carcinoma and metastatic breast cancer
(11–14). Clinical trials of famitinib against
other solid tumors such as advanced colorectal cancer and
gastroenteropancreatic neuroendocrine tumors are underway
(https://clinicaltrials.gov/). The
present study demonstrates the potential antitumor activity of
famitinib against human gastric cancer cells in vitro and
in vivo.
Materials and methods
Drugs
Famitinib (SHR1020) was a gift from Jiangsu Hengrui
Medicine Co., Ltd. (Lianyungang, China), and its appearance was a
yellow powder. The drug was stored at 4°C in the dark. For in
vitro studies, famitinib was dissolved in dimethylsulfoxide at
20 mmol/l and stored at −20°C until use. For in vivo animal
experiments, famitinib was formulated in physiological saline as a
homogeneous suspension (10 mg/ml) and stored at 4°C protected from
light. Injectable 5-florouracil (5-FU, 250 mg/10 ml) was purchased
from Tianjin Jinyao Amino Acid Co., Ltd. (Tianjin, China).
Cisplatin lyophilized powder (DDP, 10 mg) was purchased from Qilu
Pharmaceutical Co., Ltd. (Jinan, China) and was formulated in
physiological saline. Paclitaxel (PTX, 30 mg/5 ml) injection was
purchased from Hainan Haiyao Co., Ltd. (Hainan, China) and was
formulated in physiological saline.
Cell lines and cell culture
Human gastric cancer cells BGC-823 and MGC-803 were
provided by Professor Youyong Lv (Peking University Cancer Hospital
and Institute, Beijing, China). Both cell lines were cultured in
RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) supplemented with 10% fetal bovine serum (Gibco; Thermo
Fisher Scientific, Inc.) and incubated in a humidified 37°C
incubator with 5% CO2.
3-(4,5-dimethylthiazol
−2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
(MTS) cell proliferation assay
Both cell lines were seeded at ~3,000–5,000
cells/well in a 96-well plate and incubated overnight in complete
medium, followed by treatment with different concentrations of
famitinib for 24, 48 and 72 h. Cell viability was measured using
MTS tetrazolium substrate (CellTiter 96® AQueous One
Solution Cell Proliferation Assay; Promega Corporation, Madison,
WI, USA) according to the manufacturer's protocol. The absorbance
was measured at 490 nm using a spectrophotometer. All experiments
were repeated three times with at least triplicates for each
concentration.
Cell cycle analysis
Cell were treated with famitinib for 48 h, followed
by harvesting and fixing in 70% cold ethanol for ≥12 h at 4°C.
Cells were stained with 50 µg/ml propidium iodide (BD Biosciences,
Franklin Lakes, NJ, USA) at room temperature for 30 min in the
dark, and the cell cycle was analyzed using a FACSAria or a
FACSCalibur (BD Biosciences). Data were analyzed by ModFit 3.0
software (BD Biosciences). All experiments were performed in
triplicate.
Terminal deoxynucleotidyl transferase
dUTP nick end labeling (TUNEL) assay
Cell apoptosis was measured via TUNEL assay (catalog
no., C1086; Beyotime Institute of Biotechnology, Haimen, China)
according to the manufacturer's protocol. Upon treatment of the
cells with famitinib for 48 h, cells were washed with
phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde
for 10 min at room temperature. Cells were then stained with the
corresponding reagents provided in the TUNEL assay kit. Upon
overlaying the coverslips, slides were imaged under fluorescence
microscopy (TCS SP5; Leica Microsystems GmbH, Wetzlar, Germany).
Positive cells exhibited green fluorescence and were counted from
three random microscopic fields.
Western blotting
Total proteins prior and subsequent to famitinib
treatment were extracted from BGC-823 and MGC-803 cell pellets
using CytoBuster Protein Extraction Reagent (Merck Millipore,
Darmstadt, Germany). Proteins were quantified with a Pierce BCA
Protein Assay kit (Thermo Fisher Scientific, Inc.), and ~20 µg of
protein was separated on 15% sodium dodecyl sulfate-polyacrylamide
gel electrophoresis, and proteins were then transferred to a
nitrocellulose membrane (GE Healthcare Life Sciences, Chalfont,
UK), which was subsequently incubated with anti-cyclin B1
(dilution, 1:1,000; catalog no., AJ1208a; Abgent Inc., San Diego,
CA, USA), rabbit polyclonal anti-B-cell lymphoma 2 (BCL2; dilution,
1:1,000; catalog no., 2872; Cell Signaling Technology, Inc.,
Danvers, MA, USA) and mouse monoclonal anti-β-actin (dilution,
1:3,000; catalog no., A5441; Sigma-Aldrich, St. Louis, MO, USA)
antibodies at 4°C overnight. Secondary anti-rabbit and anti-mouse
horseradish peroxidase-conjugated IgG antibodies (dilution,
1:3,000; catalog nos., 7074 and 7076, respectively; Cell Signaling
Technology, Inc.) were applied and allowed to incubate at room
temperature for 1 h. Proteins were visualized with ECL Plus Western
Blotting Detection Reagent (GE Healthcare Life Sciences).
In vivo xenograft model
experiments
BGC-823 cells were suspended in PBS
(1×107 cells/ml), and 100 µl of the cell suspension was
subcutaneously injected into the right axillary area of 18-20-g
female BALB/c athymic nu/nu mice (n=40; age, 6–8
weeks; Vital River Laboratories Co., Ltd., Beijing, China). The
temperature of the housing conditions was maintained at 23–25°C
with a humidity of 50–60% and a 10/14 h light/dark cycle. Food and
water were changed 3 times a week. When the tumor volume reached
~100 mm3, mice were randomized into treatment groups.
Tumors and animal weights were measured twice weekly, and tumor
volume was calculated using the following formula:
V=LxW2x1/2 (where V represents tumor volume, L is the
length of the tumor and W is the width of the tumor).
To measure famitinib, three groups were randomized
(n=5 mice/group) as follows: Control group (gavage, physiological
saline, once daily for 3 weeks); low-dose famitinib group (gavage,
50 mg/kg, once daily for 3 weeks); and high-dose famitinib group
(gavage, 100 mg/kg, once for 3 weeks). A dose of 50 mg/kg was used
for the following experiments.
To compare famitinib with other drugs, animals were
randomized (n=5 mice/group) as follows: Control group (gavage,
physiological saline, once daily for 3 weeks); famitinib group
(gavage, 50 mg/kg, once daily for 3 weeks); 5-FU group [10 mg/kg,
intraperitoneal (ip), once every 2 days for 3 weeks]; DDP group (3
mg/kg, ip, once weekly for 3 weeks); and PTX group (10 mg/kg, ip,
once a week for 3 weeks). Then, tumors and weight were quantified.
All animal experiments were approved by the Institutional Ethics
and Animal Care Committee and performed in accordance with the
animal experimental guidelines of Peking University Cancer Hospital
(Beijing, China).
Hematoxylin and eosin (H&E) and
immunohistochemical staining
Once the mice were sacrificed, xenografts were
isolated, and formalin-fixed, paraffin-embedded (FFPE) tissue
blocks were obtained. FFPE tumor sections (4-µm thick) were
deparaffinized in xylene and hydrated in graded alcohol. Then,
tumor sections were either stained using a H&E staining kit
(catalog no., C0105; Beyotime Institute of Biotechnology),
according to the manufacturer's protocol, or incubated with rabbit
polyclonal anti-cluster of differentiation (CD)34 antibody
(dilution, 1:2,500; catalog no., ab81289; Abcam, Cambridge, UK)
upon antigen retrieval and endogenous peroxidase treatment. Signals
were visualized using an immunoglobulin G-horseradish peroxidase
polymer (Beijing CoWin Biotech Co., Ltd., Beijing, China) and
3,3′-diaminobenzidine substrate. Sections were scored by two
independent pathologists as previously described (15).
Statistical analysis
SPSS 18.0 software (SPSS, Inc., Chicago, IL, USA)
was used to perform the statistical analysis. One-way analysis of
variance was used for the in vivo experiments. P<0.05 was
considered to indicate a statistically significant difference.
Results
Famitinib inhibits gastric cancer cell
growth in a dose-dependent manner
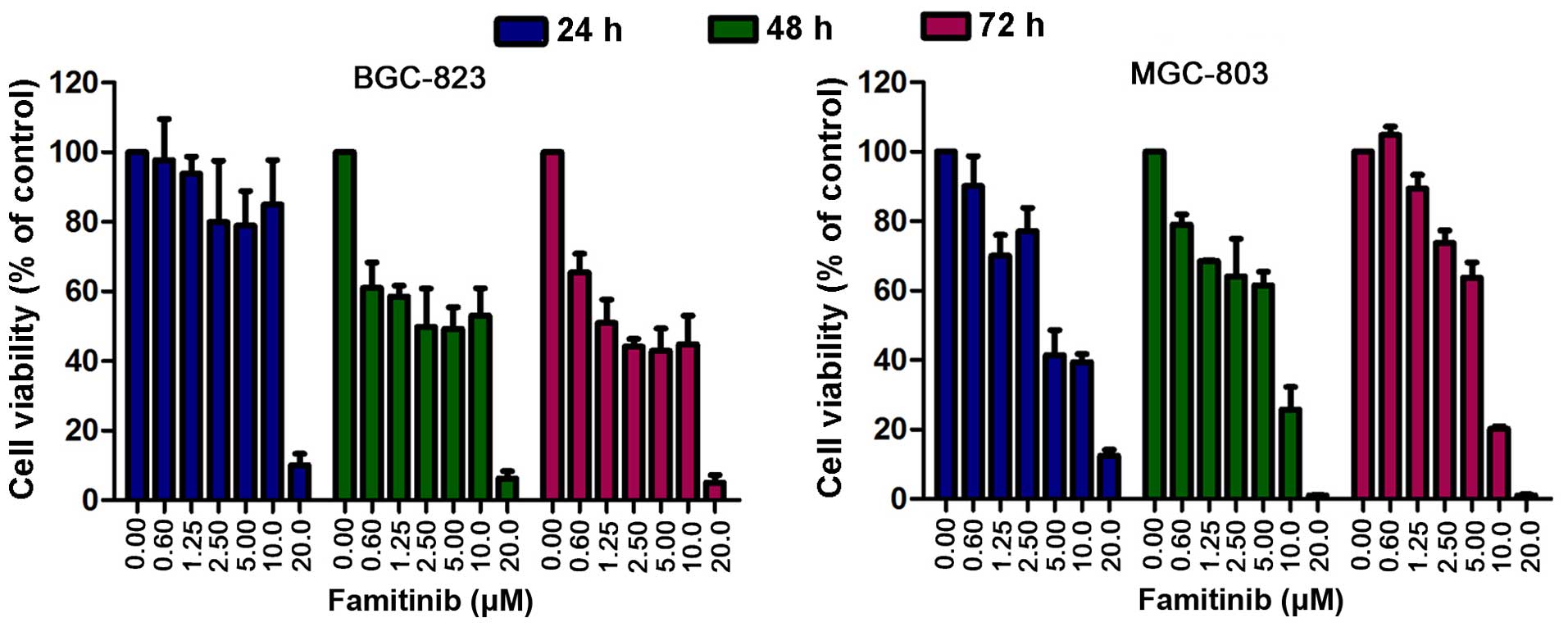
BGC-823 and MGC-803 cells were treated with
famitinib (0, 0.6, 1.25, 2.5, 5.0, 10.0 and 20.0 µM) for 24, 48 and
72 h, followed by MTS assay. Famitinib inhibited cell growth in a
dose-dependent manner (Fig. 1). The
half maximal inhibitory concentration (IC50) values of
famitinib in BGC-823 and MGC-803 cells were 3.6 and 3.1 µM,
respectively. Based on these results, the IC50 and 1/2
IC50 were used for in vitro experiments.
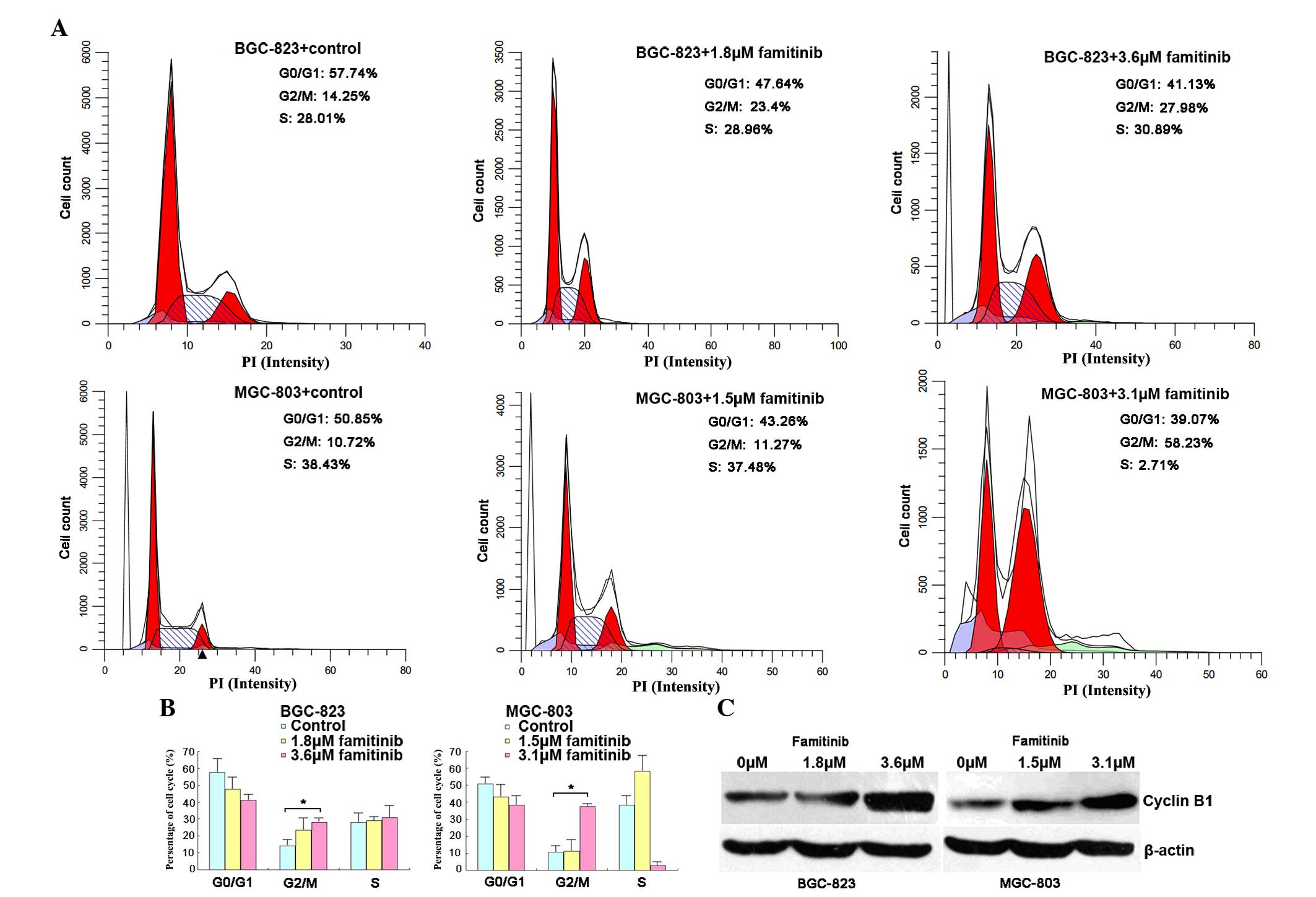
Famitinib induces cell cycle arrest at
the G2/M phase
The cell cycle was next analyzed following famitinib
treatment, and it was observed that the number of G2/M-phase cells
increased in BGC-823 (27.98 vs. 14.25%) and MGC-803 (58.23 vs.
10.72%) cells compared with the control (P=0.04 and P=0.02,
respectively; Fig. 2A and B). Cell
cycle arrest at the G2/M phase was confirmed by increased
expression of the cell metaphase-specific protein cyclin B1
(Fig. 2C).
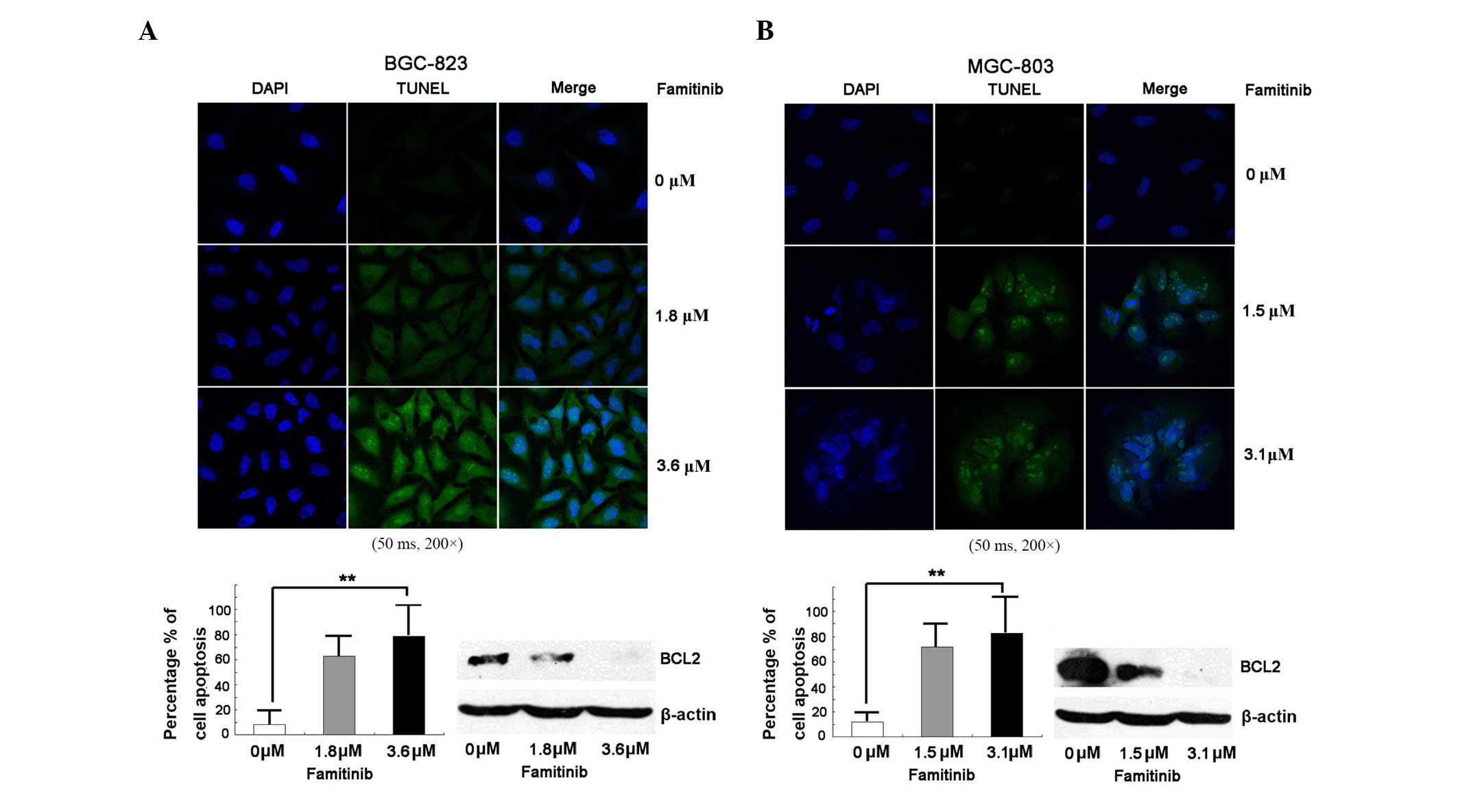
Famitinib triggers apoptosis
Cell apoptosis is an important mechanism of cell
growth inhibition (16); therefore,
apoptosis was measured via TUNEL assay. Fig. 3 indicates that, compared with the
control, famitinib increased apoptosis in BGC-823 and MGC-803 cell
lines significantly (P<0.01), and downregulated BCL2.
Famitinib reduces xenograft growth in
vivo via inhibition of angiogenesis
To identify the optimal famitinib dose for the in
vivo study, two doses were used, 50 and 100 mg/kg. Both doses
exerted a similar inhibitory power, but greater toxicity was
observed with the highest dose (data not shown).
Mice were sacrificed 21 days after treatment, and
tumors were isolated. Famitinib inhibited BGC-823 xenograft growth
(tumor volume, 395.2 vs. 2,690.5 mm3, P<0.01;
Fig. 4A), and animal weights were
similar between groups (21.6 vs. 18.7 g, P=0.17; Fig. 4B).
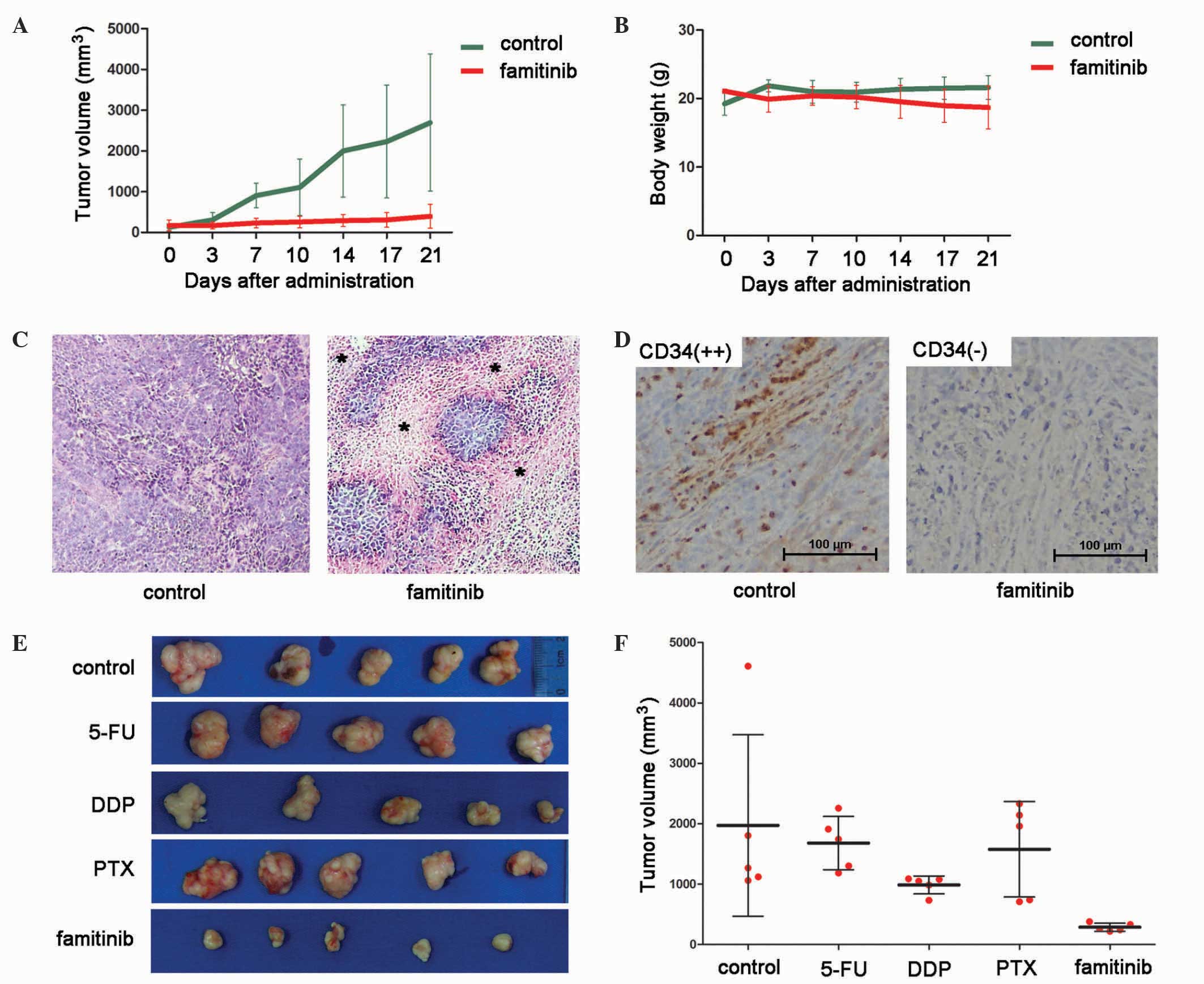 | Figure 4.Inhibitory effect of famitinib alone
compared with 5-fluorouracil, cisplatin or paclitaxel alone on in
BGC-823 xenografts. Growth curves of (A) xenografts and (B) animal
weight treated with famitinib (50 mg/kg) or control (n=5
mice/group, mean ± SD). (C) H&E staining revealed areas of
tissue necrosis following famitinib treatment (*) (magnification,
×200). (D) Tumor vascularization was significantly suppressed by
famitinib according to CD34 expression (magnification, ×200). (E)
Upon treatment, tumor xenografts were isolated and photographed.
(F) Tumor volumes from each group were measured (n=5 mice/group,
mean ± SD). CD, cluster of differentiation; 5-FU, 5-fluorouracil;
DDP, cisplatin; PTX, paclitaxel; SD, standard deviation. |
Famitinib is considered to inhibit angiogenesis;
therefore, microvessel density was measured with CD34 staining.
Upon famitinib treatment, xenografts had greater tissue necrosis
(Fig. 4C) and exhibited significantly
weaker CD34 staining than the controls (Fig. 4D). Thus, famitinib inhibits tumor
vascularization.
Famitinib has greater tumor inhibitory
effect than 5-FU, DDP or PTX
In clinical practice, 5-FU, DDP and PTX are the most
commonly used chemotherapeutic drugs for gastric cancer. Thus, the
activity of famitinib alone was compared with that of these
compounds, and it was observed that famitinib exerted better tumor
inhibition than 5-FU, DDP and PTX (Fig.
4E and F). The mean tumor volumes of the control, 5-FU, DDP,
PTX and famitinib groups were 1,973.0, 1,680.3, 987.3, 1,577.6 and
287.6 mm3, respectively; and the corresponding tumor
inhibitory ratios were 0.0, 14.9, 49.9, 20.0 and 85.4%,
respectively.
Discussion
According to the data from phase I studies of
famitinib against advanced solid tumors (11), one third of patients with AGC
responded to famitinib and had a stabilized disease without further
evaluation. Thus, the present data offer additional evidence for
future trials of famitinib against gastric cancer. The current
study confirmed that famitinib alone inhibited the growth of
BGC-823 and MGC-803 gastric cancer cells in a dose-dependent manner
in vitro (Fig. 1). From the
in vivo results, it was concluded that the inhibitory effect
of famitinib in mice xenografts was greater than that of 5-FU, DDP
or PTX alone (Fig. 4E and F). Thus,
famitinib has promising antitumor activity against gastric cancer.
For animal experiments, 5-FU, DDP and PTX alone were dosed at 10, 3
and 10 mg/kg, respectively. According to our previous study
(15) and the results from the
present study (data not shown), although high doses of 5-FU, DDP
and PTX (20, 6 and 20 mg/kg, respectively) alone had better
antitumor activity than low doses of the same drugs, these were all
toxic at higher doses. Furthermore, based on our preliminary
results, the antitumor activity of famitinib was greater than high
doses of 5-FU, DDP and PTX.
Combination regimens of ≥2 drugs for gastric cancer
are commonly used in clinical practice (2). In the present study, famitinib was not
used with other drugs due to its impressive inhibitory activity
(>85%). Thus, we anticipate that patients who do not have
success with traditional treatment may be treated with famitinib
for AGC and advanced colorectal cancer (NCT01762293; clinicaltrials.gov/ct2/show/NCT0176229).
The present study revealed that famitinib induced
cell cycle arrest at the G2/M phase, which was validated by the
upregulation of cyclin B1 (Fig. 2C).
In addition, cell apoptosis was triggered by famitinib, according
to the results of TUNEL assay, and this was confirmed by the
downregulation of BCL2 (Fig. 3).
Anti-angiogenesis was reported to be an important mechanism of
famitinib (13), which was confirmed
by the present study. Upon famitinib treatment, tumor microvessel
density represented by CD34 staining was significantly lower than
that exhibited by the controls (Fig.
4D). Famitinib may have other targets, which may be studied in
future experiments.
Anti-angiogenic therapy has been used since it was
first proposed by Dr Judah Folkman in 1971 (17). Although the frequent failures of
anti-angiogenic drugs such as bevacizumab and sorafenib have been
documented in the treatment of gastric cancer (18–20),
ramucirumab and apatinib, which mainly block VEGFR2, do improve the
survival of patients with chemotherapy-refractory AGC compared with
placebo (21–23). Similar to ramucirumab and apatinib,
famitinib mainly targets VEGFR2, which was not investigated in the
present study. However, tumor microvessel density decreased upon
famitinib treatment compared with the controls (Fig. 4D).
In additional studies, we will futher validate the
inhibitory effect of famitinib in gastric cancer patient-derived
xenografts, as well as the synergistic effects of famitinib
combined with common chemotherapeutic drugs. In conclusion, the
present study demonstrated that famitinib induces cell cycle arrest
at the G2/M phase and causes cell apoptosis and anti-angiogenesis.
The present data can be used as a foundation for future studies to
identify drugs to treat gastric cancer.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (Beijing, China; grant nos.
81172110, 81472789 and 81301853) and the National High Technology
Research and Development Program (Beijing, China; grant no. 2012AA
02A 504). The authors would like to thank LetPub (Woburn, MA, USA)
for its linguistic assistance during the preparation of the present
manuscript.
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ajani JA, Bentrem DJ, Besh S, D'Amico TA,
Das P, Denlinger C, Fakih MG, Fuchs CS, Gerdes H, Glasgow RE, et
al: National Comprehensive Cancer Network: Gastric cancer, version
2.2013: featured updates to the NCCN Guidelines. J Natl Compr Canc
Netw. 11:531–546. 2013.PubMed/NCBI
|
|
3
|
Shen L, Shan YS, Hu HM, Price TJ, Sirohi
B, Yeh KH, Yang YH, Sano T, Yang HK, Zhang X, et al: Management of
gastric cancer in Asia: Resource-stratified guidelines. Lancet
Oncol. 14:e535–e547. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bang YJ, Van Cutsem E, Feyereislova A,
Chung HC, Shen L, Sawaki A, Lordick F, Ohtsu A, Omuro Y, Satoh T,
et al: Trastuzumab in combination with chemotherapy versus
chemotherapy alone for treatment of HER2-positive advanced gastric
or gastro-oesophageal junction cancer (ToGA): A phase 3,
open-label, randomised controlled trial. Lancet. 376:687–697. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Druker BJ, Tamura S, Buchdunger E, Ohno S,
Segal GM, Fanning S, Zimmermann J and Lydon NB: Effects of a
selective inhibitor of the Abl tyrosine kinase on the growth of
Bcr-Abl positive cells. Nat Med. 2:561–566. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Barker AJ, Gibson KH, Grundy W, Godfrey
AA, Barlow JJ, Healy MP, Woodburn JR, Ashton SE, Curry BJ, Scarlett
L, et al: Studies leading to the identification of ZD1839 (IRESSA):
An orally active, selective epidermal growth factor receptor
tyrosine kinase inhibitor targeted to the treatment of cancer.
Bioorg Med Chem Lett. 11:1911–1914. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Perez-Soler R: The role of erlotinib
(Tarceva, OSI 774) in the treatment of non-small cell lung cancer.
Clin Cancer Res. 10:4238s–4240s. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rask-Andersen M, Zhang J, Fabbro D and
Schiöth HB: Advances in kinase targeting: Current clinical use and
clinical trials. Trends Pharmacol Sci. 35:604–620. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang W, Raufi A and Klempner SJ: Targeted
therapy for gastric cancer: Molecular pathways and ongoing
investigations. Biochim Biophys Acta. 1846:232–237. 2014.PubMed/NCBI
|
|
10
|
Lou L, Mi Y, Xu Y, Xie C and Zhao H:
Preclinical antitumor study of famitinib, an orally available
multi-targeted kinase inhibitor of VEGFR/PDGFR/c-Kit in phase I
clinical trials. Proceedings of AACR 102nd Annual Meeting (Cancer
Res, Orlando, FL). 712011.
|
|
11
|
Zhou A, Zhang W, Chang C, Chen X, Zhong D,
Qin Q, Lou D, Jiang H and Wang J: Phase I study of the safety,
pharmacokinetics and antitumor activity of famitinib. Cancer
Chemother Pharmacol. 72:1043–1053. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang W, Zhou AP, Qin Q, Chang CX, Jiang
HY, Ma JH and Wang JW: Famitinib in metastatic renal cell
carcinoma: A single center study. Chin Med J (Engl). 126:4277–4281.
2013.PubMed/NCBI
|
|
13
|
Xie C, Zhou J, Guo Z, Diao X, Gao Z, Zhong
D, Jiang H, Zhang L and Chen X: Metabolism and bioactivation of
famitinib, a novel inhibitor of receptor tyrosine kinase, in cancer
patients. Br J Pharmacol. 168:1687–1706. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cao J, Zhang J, Wang Z, Wang B, Lv F, Wang
L and Hu X: Hypothyroidism as a potential biomarker of efficacy of
famitinib, a novel VEGFR-2 inhibitor in metastatic breast cancer.
Cancer Chemother Pharmacol. 74:389–398. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
He Q, Gao J, Ge S, Wang T, Li Y, Peng Z,
Li Y and Shen L: Axitinib alone or in combination with
chemotherapeutic drugs exerts potent antitumor activity against
human gastric cancer cells in vitro and in vivo. J Cancer Res Clin
Oncol. 140:1575–1583. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shen L, Shan YS, Hu HM, Price TJ, Sirohi
B, Yeh KH, Yang YH, Sano T, Yang HK, Zhang X, et al: Management of
gastric cancer in Asia: Resource-stratified guidelines. Lancet
Oncol. 14:e535–e547. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Folkman J: Tumor angiogenesis: Therapeutic
implications. N Engl J Med. 285:1182–1186. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shen L, Li J, Xu J, Pan H, Dai G, Qin S,
Wang L, Wang J, Yang Z, Shu Y, et al: Bevacizumab plus capecitabine
and cisplatin in Chinese patients with inoperable locally advanced
or metastatic gastric or gastroesophageal junction cancer:
Randomized, double-blind, phase III study (AVATAR study). Gastric
Cancer. 18:168–176. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ohtsu A, Shah MA, Van Cutsem E, Rha SY,
Sawaki A, Park SR, Lim HY, Yamada Y, Wu J, Langer B, et al:
Bevacizumab in combination with chemotherapy as first-line therapy
in advanced gastric cancer: A randomized, double-blind,
placebo-controlled phase III study. J Clin Oncol. 29:3968–3976.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Martin-Richard M, Gallego R, Pericay C,
Foncillas Garcia J, Queralt B, Casado E, Barriuso J, Iranzo V, Juez
I, Visa L, et al: Multicenter phase II study of oxaliplatin and
sorafenib in advanced gastric adenocarcinoma after failure of
cisplatin and fluoropyrimidine treatment. A GEMCAD study. Invest
New Drugs. 31:1573–1579. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ilson DH: Angiogenesis in gastric cancer:
Hitting the target? Lancet. 383:4–6. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fuchs CS, Tomasek J, Yong CJ, Dumitru F,
Passalacqua R, Goswami C, Safran H, dos Santos LV, Aprile G, Ferry
DR, et al: Ramucirumab monotherapy for previously treated advanced
gastric or gastro-oesophageal junction adenocarcinoma (REGARD): An
international, randomised, multicentre, placebo-controlled, phase
III trial. Lancet. 383:31–39. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li J, Qin S, Xu J, Guo W, Xiong J, Bai Y,
Sun G, Yang Y, Wang L, Xu N, et al: Apatinib for
chemotherapy-refractory advanced metastatic gastric cancer: Results
from a randomized, placebo-controlled, parallel-arm, phase II
trial. J Clin Oncol. 31:3219–3225. 2013. View Article : Google Scholar : PubMed/NCBI
|