Introduction
Rhabdomyosarcoma (RMS) is a highly malignant type of
soft tissue tumor with skeletal muscle differentiation. The
incidence of RMS is ~43 cases per 10 million each year for
individuals under the age of 20 (1).
In patients with localized disease, the relapse-free survival rate
has improved to 70–80% (2). However,
the prognosis for patients with metastases is relatively poor, with
a 5-year survival rate of only 30% (3). Diagnostic methods for RMS include
clinical and laboratory examination, imaging analysis, pathological
diagnosis and immunohistological examination (4). RMS has been divided into 3 main
subtypes: Embryonal, alveolar and pleomorphic RMS (PRMS). The most
common subtypes are the embryonal and alveolar subtypes (5). Primary PRMS is relatively rare and
primarily affects adults, with a peak incidence in the fifth decade
of life (6–9). It most commonly arises in the deep soft
tissues of the extremities. Due to the similarities in clinical
manifestations and imaging features between PRMS and other soft
tissue tumors, PRMS is often misdiagnosed (10). The present study presents a case of
PRMS that was misdiagnosed as schwannoma. This misdiagnosis
resulted in the progression of the mass, and only following
fine-needle aspiration and histological and immunohistochemical
analysis was the tumor origin confirmed to be the skeletal muscle,
and a final diagnosis of PRMS of the right thigh was provided. The
present study was approved by the Ethics Review Committee of The
First Affiliated Hospital of Nanchang University (Nanchang, China),
and written informed consent was obtained from the patient.
Case report
In August 2014, a 28-year-old male patient presented
to the Orthopedic Clinic of Taihe County People's Hospital (Ji'an,
China) with a chief complaint of swelling in his right thigh for 1
month. According to the local district general hospital
radiologist, magnetic resonance imaging (MRI) revealed the presence
of a schwannoma and the patient was advised to undergo regular
follow-up. However, 6 months later in December 2014, a gradual
increase in the size of the mass and pain in the right thigh were
noted. The patient was referred to the Department of Orthopedics of
The First Affiliated Hospital of Nanchang University for further
treatment. No personal or family history of injury or illness was
recorded. A general physical examination demonstrated that the
passive and active range of motion of the right knee joint was
normal, with the exception of paresthesia in the right lower
extremity. No fever or respiratory embarrassment accompanied the
mass, and no history of weight loss or exposure to tuberculosis was
reported by the patient.
Additional physical examination revealed a
poorly-defined, tender and firm soft tissue mass over the right
inner thigh, but no palpable head, neck, supraclavicular, axillary
or epitrochlear lymph nodes were identified. Inflammatory markers,
including erythrocyte sedimentation rate and C-reactive protein,
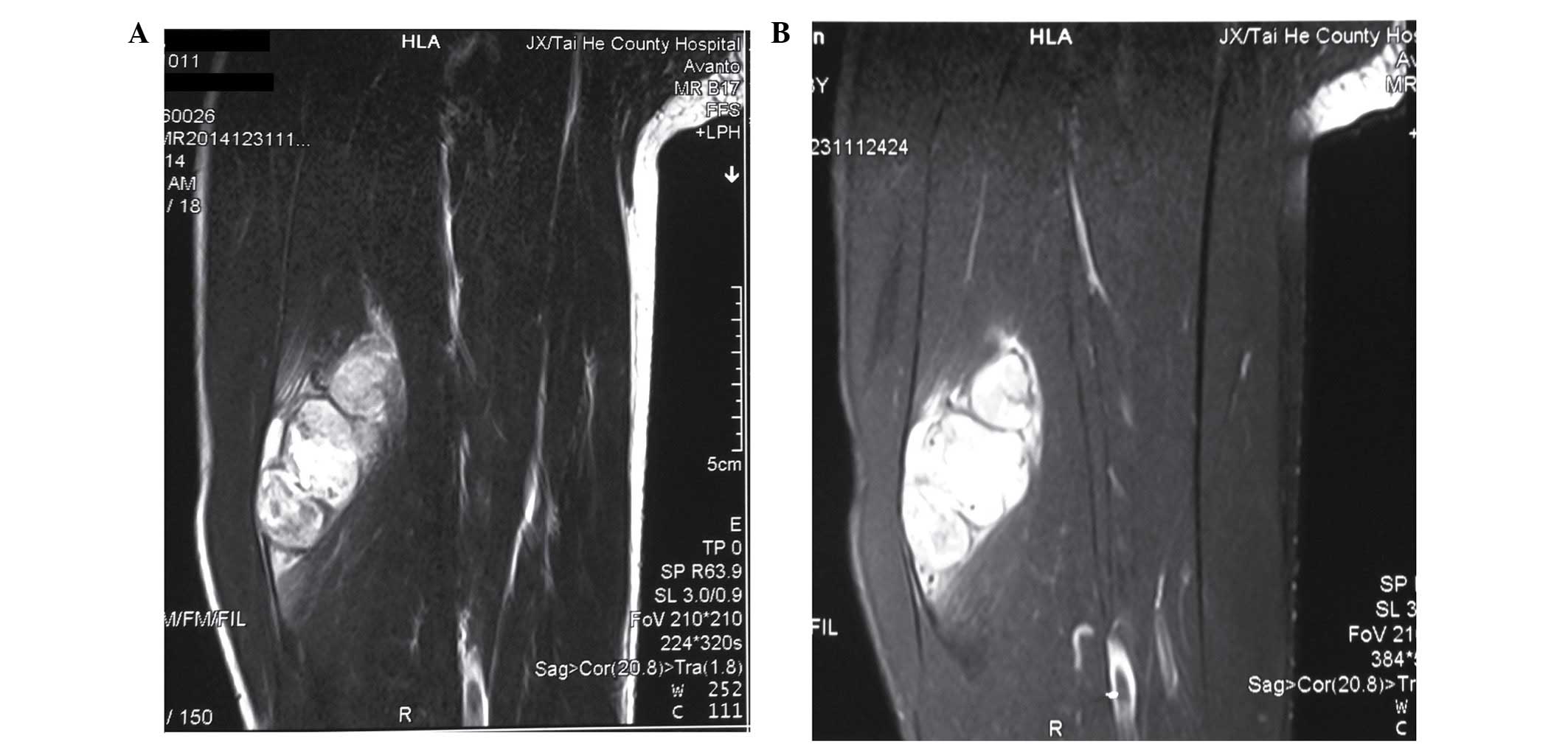
were within the normal ranges. MRI was performed for the evaluation
of the mass. Axial T2-weighted images revealed multiple cystic
lesions of varying sizes of high-signal intensity. Fat-suppressed
T2-weighted MRI also exhibited high-signal intensity (Fig. 1A and B). In addition, fine-needle
aspiration was performed to assess the mass, and cytological
diagnosis was consistent with malignant neoplasm; pleomorphic
spindle cell neoplasm with marked nuclear atypia and prominent
mitotic activity was observed.
Based on the exclusion of surgical
contraindications, surgeons affiliated with the Department of
Orthopedics, The First Affiliated Hospital of Nanchang University,
who specialize in treating bone tumors, performed the surgery. The
patient was placed in the supine position, and following the
success of epidural anesthesia, sterile drapes were disinfected and
paved routinely in the right lower extremity to expose the
operative field. First, a medial thigh incision was performed, ~17
cm in length. Next, the subcutaneous tissue, superficial and deep
fascia, and vastus intermedius muscle were resected layer by layer,
until the femoral artery and vein were observed. The vessels above
were intact. The tumor was dark brown and located in the vastus
intermedius muscle. The tumor was excised completely with negative
margins. Intraoperative tissue samples were extracted for
pathological examination. A wound drainage tube was placed and each
layer of tissue was sutured strictly following complete hemostasis.
The estimated blood loss was 100 ml and no blood transfusion was
required during the procedure.
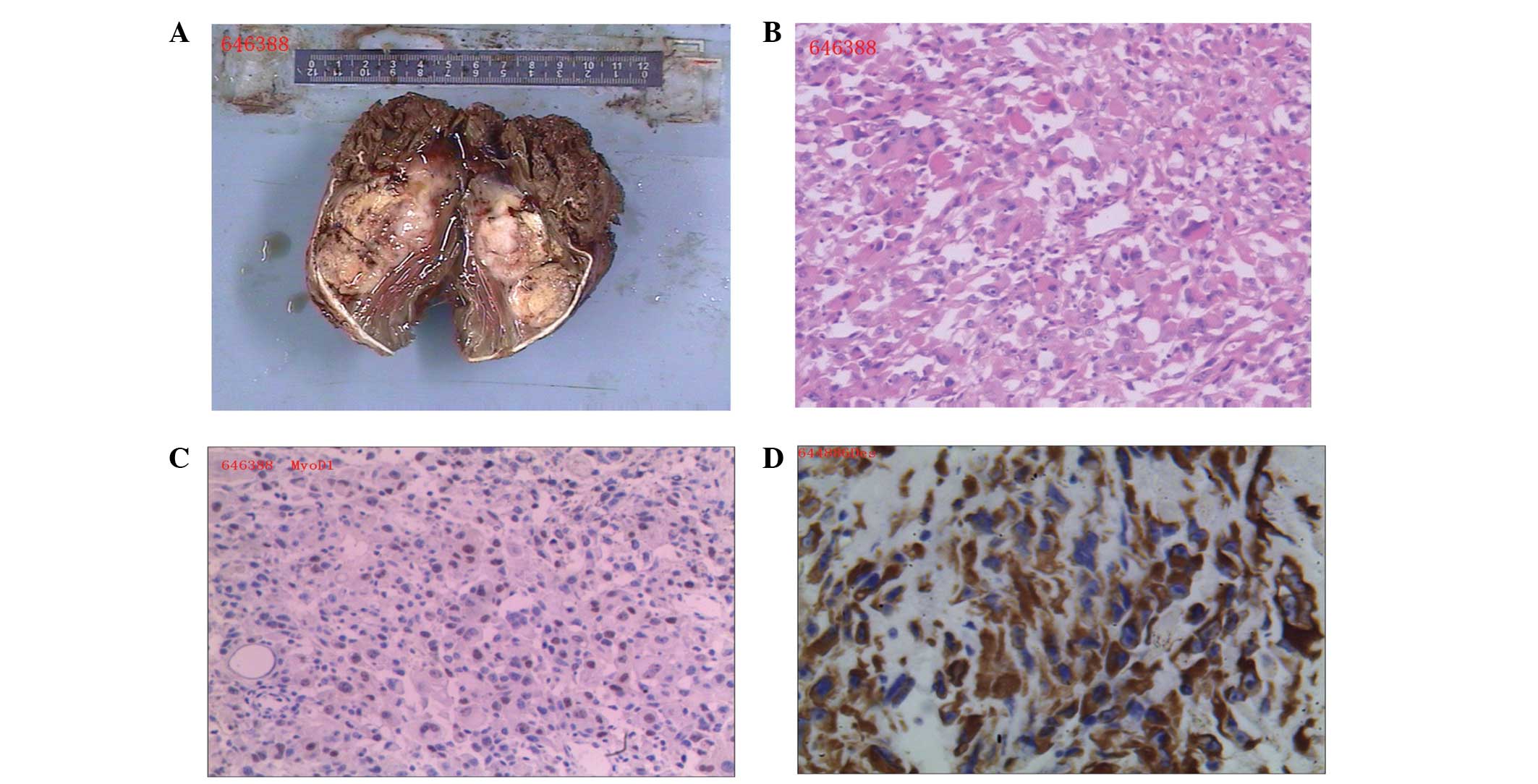
The tumor was large, smooth-surfaced and dark brown
upon gross examination. The irregular tissue mass measured ~11×9×5
cm3 in size (Fig. 2A). The
resected tumor tissue was fixed in 10% formalin, embedded in
paraffin and cut into 5-µm sections using a microtome. The sections
were subsequently stained with hematoxylin and eosin and visualized
under a microscope. Microscopic examination revealed a tumor
composed of interconnected bundles of atypical, spindled,
pleomorphic and giant cells with high-grade atypical nuclear
features. Numerous abnormal and multinucleated giant cells were
observed, and the majority of cells exhibited prominent nucleoli
and abundant eosinophilic cytoplasms (Fig. 2B). For immunohistochemistry, tissue
sections were incubated at 25°C for 60 min with monoclonal mouse
antibodies against desmin (catalog no., kit-0023) and myogenic
differentiation 1 (MyoD1; catalog no., MAB-0119) (dilution,
1:1,000; Fuzhou Maixin Biotech Co., Ltd., Fuzhou, China).
Immunohistochemical analysis revealed that the cells were positive
for actin, MyoD1 and desmin, and negative for human melanoma black
45, calponin and melan-A (Fig. 2C and
D). Based on these findings, a diagnosis of PRMS of the right
thigh was provided.
The patient was discharged without any complications
1 week following surgery. The patient was administered 6 cycles of
chemotherapy as follows: Doxorubicin, 90 mg/day for 3 days; 14 days
off followed by ifosfamide 3.8 g/day for 5 days, followed by 14
days off prior to the next treatment cycle. At the 3-month follow
up, which consisted of plain radiography and MRI, the patient was
symptom-free and able to return to work. At present, the patient is
currently alive and well. However, in cases like the present one,
it is necessary for patients to be closely monitored, due to the
high rate of recurrence and metastasis associated with misdiagnosed
PRMS.
Discussion
RMS is a highly malignant type of soft tissue tumor
that arises from striated muscle cells, exhibits skeletal muscle
differentiation and is associated with early and wide-spread
metastasis (11,12). RMS is divided into 3 main subtypes:
Embryonal and alveolar RMS and PRMS, according to the 2002 World
Health Organization Classification of Soft Tissue and Bone
Neoplasms (5). The embryonal and
alveolar subtypes are the most common, and the most frequent sites
of origin of RMS include the head and neck, extremities and soft
tissues. The disease has a male predominance, with a male-to-female
ratio of 1.3:1 (13).
PRMS was first described by Stout in 1946 (14). Primary PRMS is relatively rare and
primarily affects adults in the fifth decade of life (15). Its occurrence in young adults, as in
the present case, is extremely rare. Notably, PRMS is often
misdiagnosed or missed entirely, since its clinical manifestations
and imaging features are similar to those of other soft tissue
tumors, including fibrous histiocytoma and schwannoma (9). In the present study, the patient was
misdiagnosed with schwannoma, which led to the progression of the
disease for half a year; therefore, fine-needle aspiration is
crucial to the diagnosis of soft tissue masses. The histological
manifestations of RMS widely vary, and the histopathological
diagnosis is based on morphological, immunohistochemical and
ultrastructural findings that reveal a skeletal muscle phenotype
(16,17). PRMS is histologically distinguished
from the two more common subtypes (embryonal and alveolar) by the
haphazard arrangement of cells that are composed of large,
pleomorphic nuclei and eosinophilic cytoplasms. PRMS cells may also
be arranged in fascicles, which resembles the cell pattern observed
in leiomyosarcoma (18). Histological
subtyping is crucial, due to the various prognoses and therapeutic
approaches used in PRMS as opposed to other soft tissue tumors.
Immunohistochemistry is considered valuable for the diagnosis of
PRMS, as a series of markers with a range of specificity and
sensitivity is available. The primary markers of PRMS are MyoD1,
desmin, sarcomeric actin and myosin (19,20).
Surgical excision of PRMS is considered to be the
preferred treatment, since it relieves swelling and allows for the
final diagnosis to be confirmed histologically. The ideal surgical
management involves complete tumor resection with negative
microscopic margins (21). By
contrast, RMS has particular sensitivity to external beam
radiation; thus complete resection may be delayed following size
reduction by radiotherapy. Certain studies have reported that RMS
has an 85% overall response rate to chemotherapy (22,23), in
contrast to PRMS, which Ferrari et al (11) have reported a lower response rate of
6.25%. PRMS predominance in adults, as well as its resistance to
chemotherapy, has led to PRMS being often considered as a distinct
entity from other subtypes of RMS.
In conclusion, the present case describes a
28-year-old male patient who suffered from primary PRMS of the
right thigh. Fine-needle aspiration and total tumor resection was
performed, and at the 3-month follow-up, the patient had no
evidence of recurrent disease or residual side effects from
therapy. The performance of laboratory tests and imaging
examination is particularly important in the differential diagnosis
of patients presenting with soft-tissue tumors. Although the
follow-up time of the current patient was relatively short, in
consideration of the high risk of recurrence and metastasis in
misdiagnosed PRMS, long-term follow-ups are often recommended in
such cases.
Acknowledgements
The present study was supported by the Gan-Po
Talents Project 555 of Jiangxi Province, the Support Plan of the
Science and Technology Department of Jiangxi Province (grant no.
20112BBG70020) and the Natural Science Foundation of Jiangxi
Province (grant no. 20132BAB205067).
References
|
1
|
Cranmer LD, Chen CC, Morgan S, Martino G
and Ray J: Pleomorphic rhabdomyosarcoma in a patient with
hereditary nonpolyposis colorectal cancer. J Clin Oncol.
31:e108–e110. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Malempati S and Hawkins DS:
Rhabdomyosarcoma: Review of the Children's Oncology Group (COG)
Soft-Tissue Sarcoma Committee experience and rationale for current
COG studies. Pediatr Blood Cancer. 59:5–10. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Breneman JC, Lyden E, Pappo AS, Link MP,
Anderson JR, Parham DM, Qualman SJ, Wharam MD, Donaldson SS, Maurer
HM, et al: Prognostic factors and clinical outcomes in children and
adolescents with metastatic rhabdomyosarcoma - a report from the
Intergroup Rhabdomyosarcoma Study IV. J Clin Oncol. 21:78–84. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Scull C, Amar S, Feiz-Erfan I, Dave H and
Gridley D: Adult onset primary pineal rhabdomyosarcoma. J Clin
Oncol. 34:e137–e40. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li JJ, Forstner D and Henderson C:
Cutaneous pleomorphic rhabdomyosarcoma occurring on sun damaged
skin: A case report. Am J Dermatopathol. 37:653–657. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Stock N, Chibon F, Binh MB, Terrier P,
Michels JJ, Valo I, Robin YM, Guillou L, Ranchère-Vince D,
Decouvelaere AV, et al: Adult-type rhabdomyosarcoma: Analysis of 57
cases with clinicopathologic description, identification of 3
morphologic patterns and prognosis. Am J Surg Pathol. 33:1850–1859.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Moretti G, Guimarães R, Oliveira KM,
Sanjar F and Voegels RL: Rhabdomyosarcoma of the head and neck: 24
Cases and literature review. Braz J Otorhinolaryngol. 76:533–537.
2010.(In English, Portuguese). View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kefeli M, Kandemir B, Akpolat I, Yildirim
A and Kokcu A: Rhabdomyosarcoma arising in a mature cystic teratoma
with contralateral serous carcinoma: Case report and review of the
literature. Int J Gynecol Pathol. 28:372–375. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hakozaki M, Hojo H, Kuze T, Tajino T,
Yamada H, Kikuta A, Qualman SJ, Kikuchi S and Abe M: Primary
rhabdomyosarcoma of the sacrum: A case report and review of the
literature. Skeletal Radiol. 37:683–687. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mondal SK, Mandal PK, Adhikari A and Basak
B: Primary pleomorphic rhabdomyosarcoma of breast: Report of a rare
neoplasm. J Res Med Sci. 19:1200–1202. 2014.PubMed/NCBI
|
|
11
|
Ferrari A, Dileo P, Casanova M, Bertulli
R, Meazza C, Gandola L, Navarria P, Collini P, Gronchi A, Olmi P,
et al: Rhabdomyosarcoma in adults. A retrospective analysis of 171
patients treated at a single institution. Cancer. 98:571–580. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kishore B, Khare P, Gupta RJ, Gupta C and
Khare V: A rare case of paratesticular pleomorphic rhabdomyosarcoma
diagnosed by fine needle aspiration: A case report. Diagn
Cytopathol. 38:121–126. 2010.PubMed/NCBI
|
|
13
|
Dang ND, Teh BS and Paulino AC:
Rhabdomyosarcoma arising in a previously irradiated field: An
analysis of 43 patients. Int J Radiat Oncol Biol Phys. 85:598–603.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Stout AP: Rhabdomyosarcoma of the skeletal
muscles. Ann Surg. 123:447–472. 1946. View Article : Google Scholar
|
|
15
|
Fadare O, Bonvicino A, Martel M, Renshaw
IL, Azodi M and Parkash V: Pleomorphic rhabdomyosarcoma of the
uterine corpus: A clinicopathologic study of 4 cases and a review
of the literature. Int J Gynecol Pathol. 29:122–134. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Caserto BG: A comparative review of canine
and human rhabdomyosarcoma with emphasis on classification and
pathogenesis. Vet Pathol. 50:806–826. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yao JC, Wang WC, Tseng HH and Hwang WS:
Primary rhabdomyosarcoma of the prostate: Diagnosis by needle
biopsy and immunocytochemistry. Acta Cytol. 32:509–512. 1998.
|
|
18
|
Atahan S, Aksu O and Ekinci C: Cytologic
diagnosis and subtyping of rhabdomyosarcoma. Cytopathology.
9:389–397. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Morgenstern DA, Rees H, Sebire NJ, Shipley
J and Anderson J: Rhabdomyosarcoma subtyping by immunohistochemical
assessment of myogenin: Tissue array study and review of the
literature. Pathol Oncol Res. 14:233–238. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cessna MH, Zhou H, Perkins SL, Tripp SR,
Layfield L, Daines C and Coffin CM: Are myogenin and myoD1
expression specific for rhabdomyosarcoma? A study of 150 cases,
with emphasis on spindle cell mimics. Am J Surg Pathol.
25:1150–1157. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ge X, Ma J, Dai H, Ren L, Li Q and Shi J:
Clinical research on the treatment effects of radioactive (125) I
seeds interstitial brachytherapy on children with primary orbital
rhabdomyosarcoma. Med Oncol. 31:272014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fuchs J, Dantonello TM, Blumenstock G,
Kosztyla D, Klingebiel T, Leuschner I, Schuck A, Niggli FK,
Koscielniak E and Seitz G: Treatment and outcome of patients
suffering from perineal/perianal rhabdomyosarcoma: Results from the
CWS trials-retrospective clinical study. Ann Surg. 259:1166–1172.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Petrović B, Djan I, Markov B, Petrović M,
Erak M, Teodorović M and Baucal M: Optimal postoperative
radiotherapy treatment of orbital rhabdomyosarcoma. Srp Arh Celok
Lek. 141:375–379. 2013.(In Serbian). View Article : Google Scholar : PubMed/NCBI
|