Introduction
Despite advances in the treatment of esophageal
squamous cell carcinoma (ESCC), the overall mortality rate for this
disease remains high (1). The
frequency of late-stage diagnosis, high incidence of postsurgical
local-regional recurrence and occurrence of distant metastasis
contributes to this high mortality (2,3). At
present, the therapeutic strategies for ESCC include surgery,
chemotherapy regimens and radiotherapy (4). These treatment methods are unable to
eradicate all malignant cells, and are associated with frequent
side effects (5–7). Therefore, numerous investigations have
focused on developing alternative interventions, including
tumor-specific replicating viruses (4).
The well-characterized modified adenovirus (Ads),
H101 oncolytic Ads, varies from wild-type Ads in that the E1B55 kDs
gene and the E3 region are deleted (8,9). This
approach is able to produce viral agents with the ability to
selectively enter and replicate in tumor cells, consequently
leading to cancer cell lysis with minimal damage to surrounding
normal cells (10). It is
hypothesized that the infecting oncolytic virus (OV) may spread
through a solid tumor and eliminate it through the release of
progeny virions and activation of the antitumoral immune response
(11–14). However, clinical trials in patients
with head and neck cancer have revealed that the efficacy of this
treatment is limited when it is utilized as a single modality,
potentially due to inefficient intratumoral viral dispersal and the
barriers imposed by the tumor microenvironment (15). Therefore, oncolytic Ads H101 requires
combination with another modality to improve its antitumor activity
(9,14,16–18).
During transcription, histone
acetylation/deacetylation is a major regulator of chromatin
structural dynamics (19). Histone
deacetylase inhibitors (HDACIs) block the activity of histone
deacetylases, leading to the increased acetylation of histones and
causing non-histone proteins to form a compact and
transcriptionally repressed chromatin structure (20–22).
HDACIs have been reported to inhibit the ability of tumor cells to
mount a productive antiviral response (23–25). At
present, trichostatin A (TSA) is considered the most promising
HDCAI for tumor treatment, functioning as a potent inhibitor of
cyclin D1 with the ability to arrest cell-cycle progression
(26).
In the present study, the ability of TSA to augment
the oncolytic activity of H101 was evaluated. The results suggested
that the HDACI TSA potently and selectively enhanced the
replication of H101 virions in ESCC in vitro and in
vivo. Furthermore, the mechanism underlying TSA-mediated
enhancement of the oncolytic activity of H101 was examined.
Materials and methods
Cell culture
The EC1 human esophageal carcinoma cell line was
provided by The Department of Cell Biology, Hong Kong University
(Hong Kong, China). This cell line has been demonstrated to be
poorly-differentiated squamous cell carcinoma (27). EC1 cells were propagated in monolayer
culture in RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) supplemented with 10% inactivated fetal bovine
serum (56°C; 30 min; Hyclone Laboratories, Logan, UT, USA),
1×105 U/l penicillin and 100 µg/l streptomycin in a
humidified atmosphere with 5% CO2 at 37°C.
Reagents and treatments
TSA was purchased from Sigma-Aldrich (Merck
Millipore, Darmstadt, Germany) and dissolved in dimethyl sulfoxide
(DMSO) to produce a 5 mM stock solution, which was stored at −20°C.
Control cells were treated with DMSO in parallel during each
experiment.
Cell viability assay
EC1 cell lines were seeded at a density of
5×105 cells/well in 96-well microtiter plates. The cells
were incubated at 37°C with 5% CO2 for 24 h and then
were treated with TSA at various concentrations (0.1, 0.3 and 0.5
µM; prepared from a stock solution dissolved in DMSO) for 24, 48
and 72 h. Cells treated with identical concentrations of DMSO were
used as controls. A total of 4 h prior to absorbance evaluation, 10
µl Cell Counting Kit-8 (CCK-8; Dojindo Molecular Technologies,
Inc., Kumamoto, Japan) solution was added to each well and
incubated for 24 h at 37°C. Absorbance was determined at a
wavelength of 450 nm for each well using an enzyme-labeling
instrument (Multiskan G0; Thermo Fisher Scientific, Inc.). All
experiments were performed independently in triplicate.
Apoptosis assay
Following incubation with or without TSA at various
concentrations (0.1, 0.3 and 0.5 µM) for 48 h, EC1 cells were
harvested using 2.5 g/l trypsin and washed twice with PBS. A total
of 1×105 cells were stained with fluorescein
isothiocyanate (FITC)-Annexin V/propidium iodide (PI) using an
Annexin V-FITC kit (Beckman Coulter, Inc., Brea, CA, USA),
according to the manufacturer's protocol. Subsequently, the
apoptosis of 1.5×104 stained cells was quantified using
flow cytometry (BD Biosciences, Franklin Lakes, NJ, USA). Each
experiment was performed in triplicate. BD CellQuest™ software
version 3.0 (BD Biosciences) was used to calculate the proportion
of apoptotic cells. Negative staining for Annexin V and PI
indicated viable cells; early apoptotic cells were positive for
Annexin V and negative for PI, whereas late apoptotic cells were
positive for Annexin V and PI.
In vitro H101 oncolytic Ads
replication assay
EC1 cells were cultured on 6-well plates
(5×105 cells/well) at 37°C for 24 h prior to infection
with H101 Ads at a multiplicity of infection (MOI) of 100, and in
the presence or absence of 0.3 µM TSA. The cells and the
supernatants were harvested 24, 48 and 72 h following infection,
freeze-thawed 3 times and serially diluted. HEK293 cells (Shanghai
Institute of Biochemistry and Cell Biology, Chinese Academy of
Science, Shanghai, China) were seeded at a density of
1×105 (100 µl) cells/well with 2% DMEM in 96-well
microtiter plates. Each sample (cells and supernatants) that was
diluted serially 10 times with 2% DMEM was added to 96-well
microtiter plates at 37°C for 10 days. Each titer was repeated 10
times. The same volume of 2% DMEM was added as a control. Viral
titers were calculated by infecting serially diluted virus
particles in HEK293 and determined using the limiting dilution
method (4) (determination of the 50%
infective dose in tissue culture using HEK293 cells).
Co-treatment of EC1 cells with TSA and
H101 oncolytic Ads
EC1 cells were seeded at a density of
5×105 cells/well at 37°C in 96-well microtiter plates.
Following culture at 37°C for 24 h, cells were incubated with H101
Ads at an MOI of 100 in the presence or absence of 0.3 µM TSA for
24, 48 and 72 h. Cell viability was evaluated using CCK-8. A total
of 10 µl of the CCK-8 solution was added to each well and incubated
at 37°C for 2 h. Absorbance was determined at a wavelength of 450
nm for each well using an enzyme-labeling instrument (Multiskan
G0). All experiments were performed independently in
triplicate.
Animal treatments
Nu/nu athymic female mice, 4–6 weeks old and
weighing 18–22 g, were obtained from Shanghai Laboratory Animal Co.
Ltd. (Shanghai, China). All animals were housed in specific
pathogen free laminar airflow boxes at a temperature of 25–26°C,
with a humidity of 50%, and administered sterile food and water
ad libitum. The mice were treated in accordance with the
Guide for the Care and Use of Laboratory Animals of Henan Province,
China, and experimental procedures were approved by the Medical
Ethics Committee of Zhengzhou University (Zhengzhou, China). To
obtain xenograft tumors, a 4×106 EC1 cell resuspension
(200 µl) was injected subcutaneously into the dorsal right flank of
the athymic mice. The animals were monitored for tumor growth every
other day. Upon reaching the required mean tumor volume of ~100
mm3 (volume = length × width2 × 0.5), a total
of 24 mice were randomly assigned to the following 4 groups (6 mice
per group): The TSA alone group, the H101 alone group, the TSA and
H101 combination treatment group, and the control group. The
treatment protocol comprised of TSA (0.3 µmol/l, 200 µl TSA)
administered as intratumoral injections 3 days prior to H101
injection. The H101 treatment protocol comprised of 100 µl H101
(1×108 plaque-forming units) administered as
intratumoral injections on days 2, 7, 11, 15 and 19. The control
group received five injections of 100 µl PBS on days 2, 7, 11, 15
and 19. Tumor size was measured every 7 days using Vernier
calipers. The mice from each group were sacrificed by cervical
dislocation on day 21.
Immunohistochemical analysis
Tissue sections preserved in 2.5%
glutaraldehyde-polyoxymethylene solution at room temperature for 24
h, were dehydrated and embedded in paraffin following routine
methods, and sectioned to 4-µm thick. The sections were
deparaffinized in xylene followed by treatment with a graded series
of ethanol and distilled water, and thorough rinsing with PBS.
Following microwave treatment in citrate buffer (pH 6.0), the
container was placed in boiled water for 20 min. Endogenous
peroxidase activity was blocked with 3% hydrogen peroxide in
methanol at room temperature for 10 min. The tissue samples were
incubated with a rabbit anti-coxsackie and adenovirus receptor
(CAR) monoclonal antibody (dilution, 1:200; catalog no. sc-50462;
Santa Cruz Biotechnology, Inc., Dallas, TX, USA) overnight at 4°C.
Following washing three times with PBS, the tissue samples were
incubated for 30 min with the goat anti-rabbit IgG-horseradish
peroxidase secondary antibody (dilution, 1:2,000; catalog no.
sc-2004; Santa Cruz Biotechnology, Inc.). Antibody binding was
subsequently detected using 0.5% 3,3′-diaminobenzidine
hydrochloride (DAB; Sigma-Aldrich; Merck Millipore) at room
temperature without light for 3 min. The sections were then washed
three times with PBS, counterstained with hematoxylin for 15 sec
and dehydrated at room temperature. The images were analyzed by
Image-Pro Plus 6.0 (Media Cybernetics, Inc., Rockville, MD,
USA).
Western blot analysis
Tumor tissues from xenografts of the aforementioned
mice were lysed in lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM
NaCl, 1% Triton X-100, 100 µg/ml phenylmethylsulfonyl fluoride) for
tissue homogenization. After 20 min on ice, the lysates were
centrifuged at 20,430 × g at 4°C for 10 min. The supernatants were
used as whole cell extracts. Cell lysates (50 µg) were separated
using 10% SDS-PAGE and transferred to polyvinylidene fluoride
membranes. The membranes were incubated with 5% non-fat dried milk
dissolved in Tris-buffered saline containing 0.1% Tween-20 for 1 h
at room temperature. The membranes were then incubated with a
rabbit CAR monoclonal antibody (dilution, 1:200; Santa Cruz
Biotechnology, Inc.) at 4°C overnight. After washing three times
with 0.1% TBS-T, the tissue samples were incubated with the
aforementioned horseradish peroxidase-conjugated IgG secondary
antibody for 2 h at room temperature. A Pierce™ enhanced
chemiluminescence detection kit (Thermo Fisher Scientific, Inc.)
was used to detect the target proteins. The bands were subjected to
densitometry for quantitation using the Bio-Rad Quantity
One® software (version 4.6.2; Bio-Rad Laboratories,
Inc., Hercules, CA, USA).
Statistical analysis
Quantitative data were expressed as the mean ±
standard deviation. One-way analysis of variance was used to
compare significant differences amongst the groups. Two-tailed
Student's t-tests were used for comparisons between two groups.
Data analyses were performed using SPSS 13.0 software for Windows
(SPSS Inc., Chicago, IL, USA). P<0.05 was considered to indicate
a statistically significant difference.
Results
Effect of TSA on the growth of EC1
cells
TSA, an established class I and II HDACI, has been
reported to exert numerous antitumor effects by inhibiting cell
proliferation and inducing cell apoptosis (24). The present study aimed to examine the
ability of TSA to promote the antitumor effects of oncolytic H101,
but not to affect cell viability. Therefore, EC1 cells were treated
with various concentrations of TSA, and EC1 cell viability and
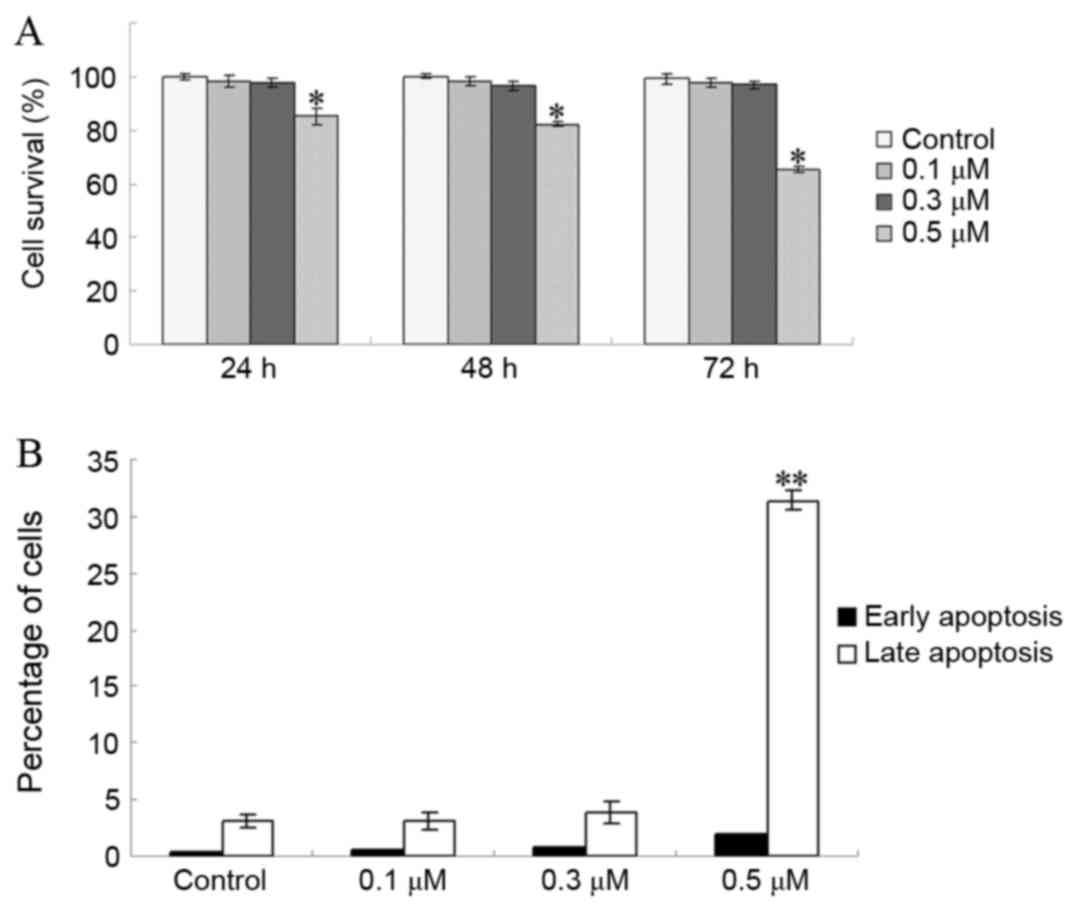
apoptosis were evaluated. As presented in Fig. 1A, the viability of EC1 cells was not
significantly inhibited by TSA at doses of 0.1 and 0.3 µM after 72
h of treatment (P=0.542 for 0.1 µM vs. control; P=0.218 for 0.3 µM
vs. control). However, the viability of EC1 cells was significantly
inhibited at doses >0.5 µM (P<0.001; Fig. 1A). In addition, the proportion of EC1
cells in early apoptosis was not markedly increased at TSA doses
<0.3 µM (P=0.773 for 0.1 µM vs. control; P=0.350 for 0.3 µM vs.
control; Fig. 1B). These results
indicated that doses of ≤0.3 µM TSA did not significantly alter EC1
cell viability.
Increased H101 replication and cell
cytotoxicity is mediated by TSA and H101 in combination
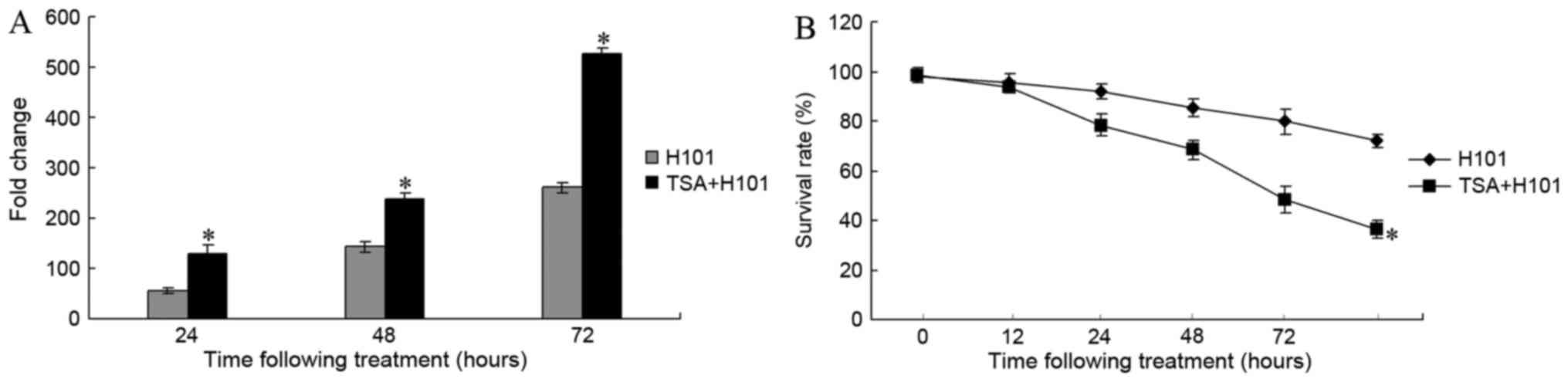
To examine whether TSA is able to impact H101
replication, end point dilution titrations were performed on HEK293
cells. Following treatment with 0.3 µM TSA for 24, 48 and 72 h,
viral titers increased 55.82-fold, 238.84-fold and 527.46-fold in
EC1 cells, respectively (Fig. 2A).
H101 replication was significantly increased in EC1 cells treated
with TSA, compared with the untreated control cells (P=0.002 for
TSA 24 h vs. control; P<0.001 for TSA 48 h vs. control;
P<0.001 for TSA 72 h vs. control). Subsequently, the antitumor
effects of TSA on H101 in EC1 cells were examined. A CCK-8 assay
was used to measure the EC1 cell survival rate at 12, 24, 48, 72
and 96 h following treatment. As compared with the H101 monotherapy
group, the cell survival rate in the TSA and H101 combination group
exhibited a significant decrease at 24, 48 and 72 h (P<0.001;
Fig. 2B).
Effect of TSA and H101 combination
treatment on the EC1 xenograft model
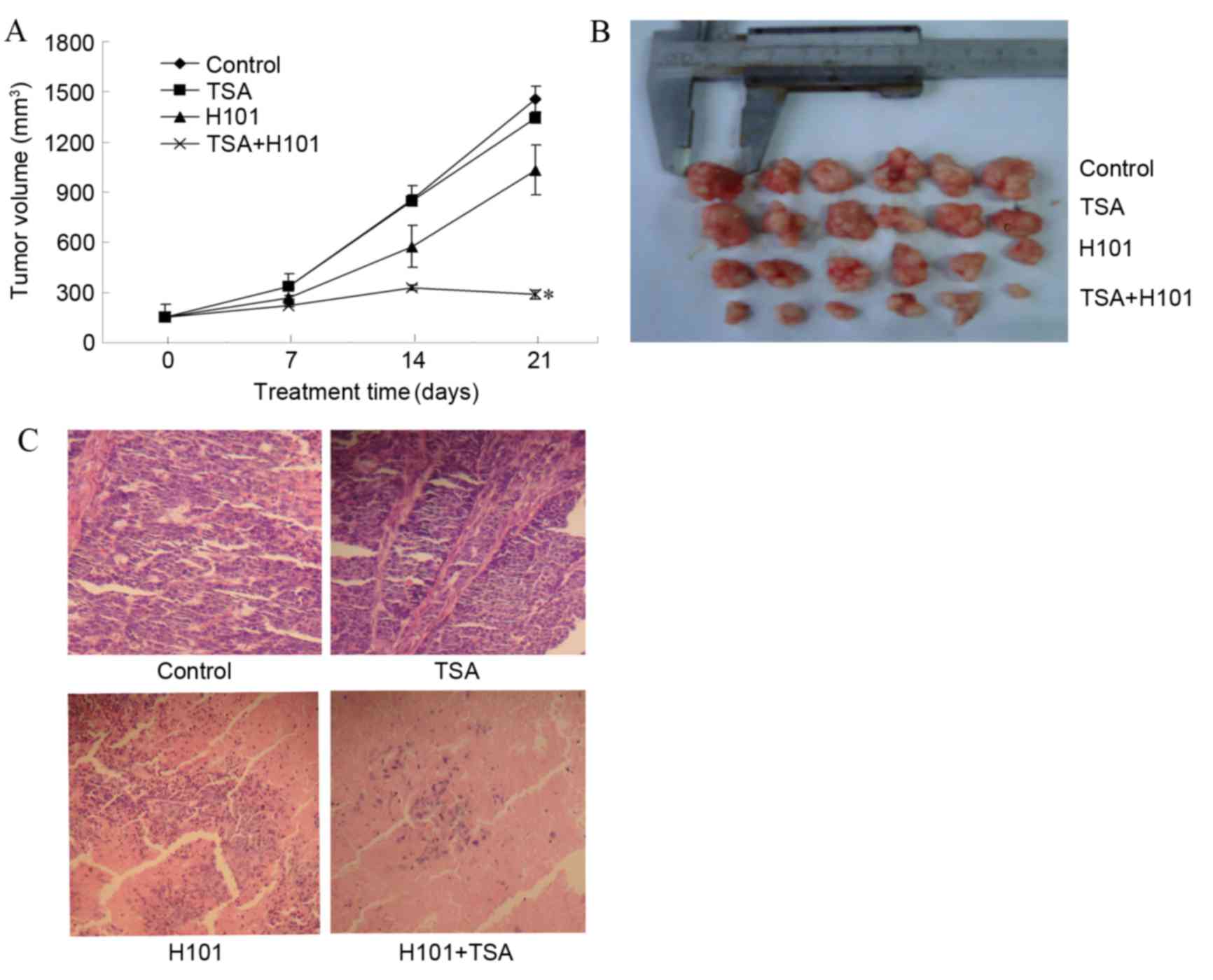
TSA and H101 in combination enhanced tumor cell
cytotoxicity in vitro. Therefore, in order to examine
whether this treatment is able to inhibit EC1 tumor growth in
vivo, tumor bearing mice were divided into various treatment
groups as described in material and methods. As compared with the
PBS control group, mice treated with TSA alone did not exhibit
tumor regression and there was no significant difference in the
tumor volume at the end of treatment (P=0.148). In the TSA and H101
combination treatment group, a significant decrease in tumor volume
(286.53±28.99 mm3) was observed, as compared with the
untreated controls (1459.79±76.81 mm3) (P<0.001).
Furthermore, a significant decrease in the tumor volume was
detected in the TSA and H101 combination group, as compared with
the two treatments administered individually (both P<0.001). The
results indicate that TSA and H101 in combination produce an
enhanced antitumor effect, compared with the two treatments
administered individually (Fig. 3A).
In addition, marked variations were identified in the degree of
inflammation and necrosis observed in the tumor specimens in the
TSA and H101 combination group, compared with the groups in which
TSA and H101 were administered individually (both P<0.001;
Fig. 3A and B).
TSA alone, or combined with H101,
upregulates the expression of CAR in an EC1 cell- xenograft
model
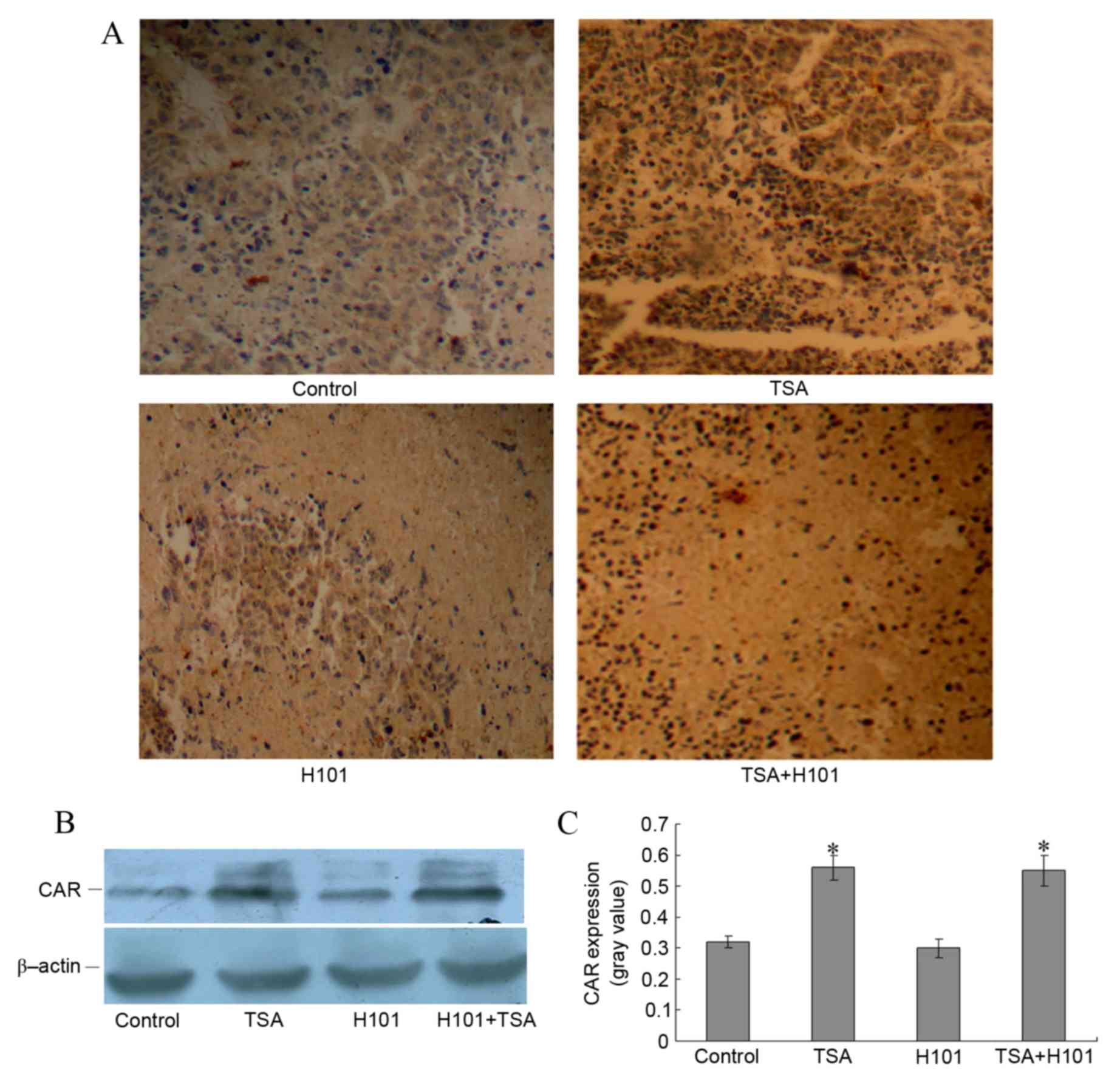
To investigate whether the enhanced antitumor
effects of TSA and H101 combined in vivo were achieved via
the upregulation of CAR, the expression levels of CAR in the
xenograft tumor tissues were detected using immunohistochemistry.
An increase in the expression levels of CAR in xenograft tumors was
observed in the TSA group and in the TSA and H101 combination
group, as compared with the control group and the H101 group
(Fig. 4A). Western blot analysis also
demonstrated that the CAR protein levels were increased in the TSA
group and the TSA in combination with H101 group (both P<0.001;
Fig. 4B and C). These results
indicated that TSA intratumoral injections may result in increased
levels of CAR expression in xenograft tumors in mice.
Discussion
The tumor suppressor gene tumor protein p53 is
considered an attractive target for cancer gene therapy (28–30). The
human p53 gene is known as the ‘guardian of the genome’ for its
roles in regulating the cell cycle, apoptosis and cellular
senescence, as well as inducing a variety of activities to maintain
genomic stability (31,32). Mutant p53 has been demonstrated to be
overexpressed in the tumor tissues of patients with ESCC and its
expression levels are correlated with tumor progression (33). Therefore, Ads-mediated p53 cancer gene
therapy constitutes a promising treatment approach for patients
with ESCC (34). H101 is a
recombinant human type 5 Ads with a total deletion of the E1B 55 K
gene, which is able to proliferate effectively in p53 mutant cells,
but not in p53 wild-type cells (35).
However, H101 has limited potential for the eradication of tumors
when used as a monotherapy due to its low infection efficiency
(10). Therefore, a high degree of
viral transduction within the tumor is key to the success of gene
therapy approaches. H101 is often used in combination with
traditional modalities, including chemotherapy (36). In the present study, the antitumor
efficacy of H101 in combination with the HDACI TSA was
evaluated.
The H101 OV enters malignant cells through a
receptor-mediated endocytosis mechanism (37). CAR is necessary for adenoviral entry
into the cell; however, this receptor is frequently downregulated
in malignant cells, rendering them less vulnerable to viral attack
(38). It has been reported that
HDACIs are able to enhance transgene expression, making them
suitable for use in conjunction with adenoviral vector-based
therapies due to their ability to increase CAR expression levels
(39).
Although TSA was one of the first HDACIs to be
identified, its suboptimal in vivo stability limits its use
as a widely administered cancer treatment (40). Furthermore, TSA is more effective at
promoting vaccinia virus spread in vitro, compared with TSA
derivatives, by increasing the expression levels of CAR in
malignant cells, and leading to more efficient cell killing
compared with other HDACIs, including vorinistat (40). In the present study, TSA was selected
to enhance the antitumor efficacy of H101 with the purpose of
evaluating the ability of TSA to enhance H101 viral oncolysis
without altering cell viability. Following the treatment of EC1
cells with various concentrations of TSA and the subsequent
evaluation of cell viability, the data indicated that a dose of 0.3
µM TSA delivered to EC1 cells was well-tolerated and did not induce
apoptosis. However, the treatment of EC1 cells with 0.3 µM TSA
increased H101 replication and cell cytotoxicity. These results
indicate that TSA is able to enhance the antitumor efficacy of H101
in vitro.
Previous studies have indicated that a number of
factors may lead to a discrepancy between the efficacy data
obtained from cell culture in vitro, and the in vivo
data (15,41). In cell culture monolayer, all
infectable cells are easily accessed by viruses. By contrast,
aspects of the tumor architecture, including fibrotic septa and
necrotic areas in tumor tissue, prevent the virus from spreading
in vivo (9). This leads to an
inconsistency between the efficacy obtained from cell culture
experiments in vitro and in clinical trials. Therefore,
their efficacy of viral oncolysis need to be improved (42–45). The
present study examined whether the enhanced in vitro tumor
cytotoxicity, mediated by TSA and H101 in combination, was also
exhibited during EC1 cell tumor growth in vivo. In
comparison with the TSA group and the H101 group, a significant
decrease in tumor volume in the TSA and H101 combination group was
observed. This result indicates that the use of TSA and H101 in
combination produced an enhanced antitumor effect in
vivo.
Subsequently, the mechanisms underlying the ability
of TSA to enhance the antitumor effects of H101 were investigated.
Lower expression levels of CAR protein have been reported in ESCC
cells (46). In addition, Wei Lu
et al proved that an intratumoral injection of chemotherapy
in combination with H101 exhibits better antitumor activity to
refractory malignant tumors than H101 alone (46). It has become apparent that a major
determinant of Ads-mediated gene transfer efficacy is the
expression of its primary receptor, CAR, on target cells (47). In order to infect tumor cells
efficiently, H101 requires CAR for attachment and αv
integrin for internalization (48).
In the present study, the expression levels of CAR in the mouse
xenograft tumor tissue were increased in the TSA group, and in the
TSA and H101 combination group. These results suggest that TSA
intratumoral injections may enhance the H101 antitumor effect by
increasing CAR expression levels in vivo.
In conclusion, the HDACI TSA is able to enhance the
antitumor effect of the OV H101 on ESCC cells in vitro and
in vivo. HDACIs combined with OVs may therefore be able to
overcome the obstacle of the low infection efficiency of H101 when
used as monotherapy.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 81372269), the Science
Foundation of Henan Education Department (grant no. 13A310622) and
the Key project on Science and Technology provided by Henan
Province, China (grant nos. 152102410067, 162102310142 and
201403035).
References
|
1
|
Fujiwara Y, Yoshikawa R, Kamikonya N,
Nakayama T, Kitani K, Tsujie M, Yukawa M, Inoue M and Yamamura T:
Trimodality therapy of esophagectomy plus neoadjuvant
chemoradiotherapy improves the survival of clinical stage II/III
esophageal squamous cell carcinoma patients. Oncol Rep. 28:446–452.
2012.PubMed/NCBI
|
|
2
|
Jemal A, Center MM, DeSantis C and Ward
EM: Global patterns of cancer incidence and mortality rates and
trends. Cancer Epidemiol Biomarkers Prev. 19:1893–1907. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bonavina L, Incarbone R, Saino G, Clesi P
and Peracchia A: Clinical outcome and survival after esophagectomy
for carcinoma in elderly patients. Dis Esophagus. 16:90–93. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang Y, Thorne S, Hannock J, Francis J, Au
T, Reid T, Lemoine N, Kirn D and Halldén G: A novel assay to assess
primary human cancer infectibility by replication-selective
oncolytic adenoviruses. Clin Cancer Res. 11:351–360.
2005.PubMed/NCBI
|
|
5
|
Zhu H, Huo X, Chen L, Wang H and Yu H:
Clinical experience with radio-, chemo- and hyperthermotherapy
combined trimodality on locally advanced esophageal cancer. Mol
Clin Oncol. 1:1009–1012. 2013.PubMed/NCBI
|
|
6
|
Fujiwara Y, Yoshikawa R, Kamikonya N,
Nakayama T, Kitani K, Tsujie M, Yukawa M, Hara J, Yamamura T and
Inoue M: Neoadjuvant chemoradiotherapy followed by esophagectomy
vs. surgery alone in the treatment of resectable esophageal
squamous cell carcinoma. Mol Clin Oncol. 1:773–779. 2013.PubMed/NCBI
|
|
7
|
Minsky BD, Pajak TF, Ginsberg RJ, Pisansky
TM, Martenson J, Komaki R, Okawara G, Rosenthal SA and Kelsen DP:
INT 0123 (Radiation Therapy Oncology Group 94–05) phase III trial
of combined-modality therapy for esophageal cancer: High-dose
versus standard-dose radiation therapy. J Clin Oncol. 20:1167–1174.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Garber K: China approves world's first
oncolytic virus therapy for cancer treatment. J Natl Cancer Inst.
98:298–300. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lu W, Zheng S, Li XF, Huang JJ, Zheng X
and Li Z: Intra-tumor injection of H101, a recombinant adenovirus,
in combination with chemotherapy in patients with advanced cancers:
A pilot phase II clinical trial. World J Gastroenterol.
10:3634–3638. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Toth K and Wold WS: Increasing the
efficacy of oncolytic adenovirus vectors. Viruses. 2:1844–1866.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Thorne SH, Hermiston T and Kirn D:
Oncolytic virotherapy: Approaches to tumor targeting and enhancing
antitumor effects. Semin Oncol. 32:537–548. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lichtenstein DL and Wold WS: Experimental
infections of humans with wild-type adenoviruses and with
replication-competent adenovirus vectors: Replication, safety and,
transmission. Cancer Gene Ther. 11:819–829. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nettelbeck DM, Jérôme V and Müller R: Gene
therapy: Designer promoters for tumour targeting. Trends Genet.
16:174–181. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tysome JR, Lemoine NR and Wang Y:
Combination of anti-angiogenic therapy and virotherapy: Arming
oncolytic viruses with anti-angiogenic genes. Curr Opin Mol Ther.
11:664–669. 2009.PubMed/NCBI
|
|
15
|
Nguyen TL, Wilson MG and Hiscott J:
Oncolytic viruses and histone deacetylase inhibitors-a
multi-pronged strategy to target tumor cells. Cytokine Growth
Factor Rev. 21:153–159. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Reid TR, Freeman S, Post L, McCormick F
and Sze DY: Effects of Onyx-015 among metastatic colorectal cancer
patients that have failed prior treatment with 5-FU/leucovorin.
Cancer Gene Ther. 12:673–681. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kumar S, Gao L, Yeagy B and Reid T: Virus
combinations and chemotherapy for the treatment of human cancers.
Curr Opin Mol Ther. 10:371–379. 2008.PubMed/NCBI
|
|
18
|
Bhattacharyya M, Francis J, Eddouadi A,
Lemoine NR and Halldén G: An oncolytic adenovirus defective in
pRb-binding (dl922-947) can efficiently eliminate pancreatic cancer
cells and tumors in vivo in combination with 5-FU or gemcitabine.
Cancer Gene Ther. 18:734–743. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Barbetti V, Gozzini A, Cheloni G, Marzi I,
Fabiani E, Santini V, Sbarba P Dello and Rovida E: Time- and
residue-specific differences in histone acetylation induced by VPA
and SAHA in AML1/ETO-positive leukemia cells. Epigenetics.
8:210–219. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ellis L, Atadja PW and Johnstone RW:
Epigenetics in cancer: Targeting chromatin modifications. Mol
Cancer Ther. 8:1409–1420. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Meng F, Sun G, Zhong M, Yu Y and Brewer
MA: Inhibition of DNA methyltransferases, histone deacetylases and
lysine-specific demethylase-1 suppresses the tumorigenicity of the
ovarian cancer ascites cell line SKOV3. Int J Oncol. 43:495–502.
2013.PubMed/NCBI
|
|
22
|
Wilson PM, Labonte MJ, Martin SC, Kuwahara
ST, El-Khoueiry A, Lenz HJ and Ladner RD: Sustained inhibition of
deacetylases is required for the antitumor activity of the histone
deactylase inhibitors panobinostat and vorinostat in models of
colorectal cancer. Invest New Drugs. 31:845–857. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Minucci S and Pelicci PG: Histone
deacetylase inhibitors and the promise of epigenetic (and more)
treatments for cancer. Nat Rev Cancer. 6:38–51. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Stankov MV, El Khatib M, Thakur B Kumar,
Heitmann K, Panayotova-Dimitrova D, Schoening J, Bourquin JP,
Schweitzer N, Leverkus M, Welte K, et al: Histone deacetylase
inhibitors induce apoptosis in myeloid leukemia by suppressing
autophagy. Leukemia. 28:577–588. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ye RR, Ke ZF, Tan CP, He L, Ji LN and Mao
ZW: Histone-deacetylase-targeted fluorescent ruthenium(II)
polypyridyl complexes as potent anticancer agents. Chemistry.
19:10160–10169. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen X, Xiao W, Chen W, Luo L, Ye S and
Liu Y: The epigenetic modifier trichostatin A, a histone
deacetylase inhibitor, suppresses proliferation and
epithelial-mesenchymal transition of lens epithelial cells. Cell
Death Dis. 4:e8842013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yen CC, Chen YJ, Chen JT, Hsia JY, Chen
PM, Liu JH, Fan FS, Chiou TJ, Wang WS and Lin CH: Comparative
genomic hybridization of esophageal squamous cell carcinoma:
Correlations between chromosomal aberrations and disease
progression/prognosis. Cancer. 92:2769–2777. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li LX, Zhang YL, Zhou L, Ke ML, Chen JM,
Fu X, Ye CL, Wu JX, Liu RY and Huang W: Antitumor efficacy of a
recombinant adenovirus encoding endostatin combined with an
E1B55KD-deficient adenovirus in gastric cancer cells. J Transl Med.
11:2572013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Huang H, Xiao T, He L, Ji H and Liu XY:
Interferon-β-armed oncolytic adenovirus induces both apoptosis and
necroptosis in cancer cells. Acta Biochim Biophys Sin (Shanghai).
44:737–745. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Essmann F and Schulze-Osthoff K:
Translational approaches targeting the p53 pathway for anti-cancer
therapy. Br J Pharmacol. 165:328–344. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Krell J, Frampton AE, Colombo T, Gall TM,
de Giorgio A, Harding V, Stebbing J and Castellano L: The p53 miRNA
interactome and its potential role in the cancer clinic.
Epigenomics. 5:417–428. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee JY, Kim HJ, Yoon NA, Lee WH, Min YJ,
Ko BK, Lee BJ, Lee A, Cha HJ, Cho WJ and Park JW: Tumor suppressor
p53 plays a key role in induction of both tristetraprolin and let-7
in human cancer cells. Nucleic Acids Res. 41:5614–5625. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Huang K, Chen L, Zhang J, Wu Z, Lan L,
Wang L, Lu B and Liu Y: Elevated p53 expression levels correlate
with tumor progression and poor prognosis in patients exhibiting
esophageal squamous cell carcinoma. Oncol Lett. 8:1441–1446.
2014.PubMed/NCBI
|
|
34
|
D'Assoro AB, Leontovich A, Amato A,
Ayers-Ringler JR, Quatraro C, Hafner K, Jenkins RB, Libra M, Ingle
J, Stivala F, et al: Abrogation of p53 function leads to metastatic
transcriptome networks that typify tumor progression in human
breast cancer xenografts. Int J Oncol. 37:1167–1176.
2010.PubMed/NCBI
|
|
35
|
Song X, Zhou Y, Jia R, Xu X, Wang H, Hu J,
Ge S and Fan X: Inhibition of retinoblastoma in vitro and in vivo
with conditionally replicating oncolytic adenovirus H101. Invest
Ophthalmol Vis Sci. 51:2626–2635. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Alvarez-Breckenridge C, Kaur B and Chiocca
EA: Pharmacologic and chemical adjuvants in tumor virotherapy. Chem
Rev. 109:3125–3140. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Walters RW, Freimuth P, Moninger TO,
Ganske I, Zabner J and Welsh MJ: Adenovirus fiber disrupts
CAR-mediated intercellular adhesion allowing virus escape. Cell.
110:789–799. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kothari V, Joshi G, Nama S, Somasundaram K
and Mulherkar R: HDAC inhibitor valproic acid enhances tumor cell
kill in adenovirus-HSVtk mediated suicide gene therapy in HNSCC
xenograft mouse model. Int J Cancer. 126:733–742. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Auer D, Reimer D, Porto V, Fleischer M,
Roessler J, Wiedemair A, Marth C, Müller-Holzner E, Daxenbichler G
and Zeimet AG: Expression of coxsackie-adenovirus receptor is
related to estrogen sensitivity in breast cancer. Breast Cancer Res
Treat. 116:103–111. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
MacTavish H, Diallo JS, Huang B, Stanford
M, Le Boeuf F, de Silva N, Cox J, Simmons JG, Guimond T, Falls T,
et al: Enhancement of vaccinia virus based oncolysis with histone
deacetylase inhibitors. PLoS One. 5:e144622010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sauthoff H, Hu J, Maca C, Goldman M,
Heitner S, Yee H, Pipiya T, Rom WN and Hay JG: Intratumoral spread
of wild-type adenovirus is limited after local injection of human
xenograft tumors: Virus persists and spreads systemically at late
time points. Hum Gene Ther. 14:425–433. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Eikenes L, Brekken ØS, Bruland C and Cde L
Davies: Collagenase increases the transcapillary pressure gradient
and improves the uptake and distribution of monoclonal antibodies
in human osteosarcoma xenografts. Cancer Res. 64:4768–4773. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ferreira TB, Ferreira AL, Carrondo MJ and
Alves PM: Two different serum-free media and osmolality effect upon
human 293 cell growth and adenovirus production. Biotechnol Lett.
27:1809–1813. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Henry O, Dormond E, Perrier M and Kamen A:
Insights into adenoviral vector production kinetics in acoustic
filter-based perfusion cultures. Biotechnol Bioeng. 86:765–774.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jardon M and Garnier A: PH, pCO2, and
temperature effect on R-adenovirus production. Biotechnol Prog.
19:202–208. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ma J, Zhao J, Lu J, Jiang Y, Yang H, Li P,
Zhao M, Liu K and Dong Z: Coxsackievirus and adenovirus receptor
promotes antitumor activity of oncolytic adenovirus H101 in
esophageal cancer. Int J Mol Med. 30:1403–1409. 2012.PubMed/NCBI
|
|
47
|
Chen G, Wang BB, Li FJ, Liu DY, Zhou JF,
Lu YP and Ma D: Enhancive effect of histone deacetylase inhibitor
trichostatin a on transfection efficiency of adenovirus in ovarian
carcinoma cell line A2780. Ai Zheng. 24:1196–1200. 2005.(In
Chinese). PubMed/NCBI
|
|
48
|
Pandha HS, Stockwin LH, Eaton J, Clarke
IA, Dalgleish AG, Todryk SM and Blair GE: Coxsackie B and
adenovirus receptor, integrin and major histocompatibility complex
class I expression in human prostate cancer cell lines:
Implications for gene therapy strategies. Prostate Cancer Prostatic
Dis. 6:6–11. 2003. View Article : Google Scholar : PubMed/NCBI
|