Introduction
Kasabach-Merritt syndrome (KMS) is a rare,
aggressive type of vascular tumor associated with thrombocytopenia
and consumptive coagulopathy. KMS was first reported by Kasabach
and Merritt in 1940, in a boy presenting with enlarging hemangioma
with thrombocytopenia on the left thigh (1). KMS has been associated with Kaposiform
hemangioendothelioma or tufted angioma, but not with the more
common infantile hemangioma (2). Le
Nouail et al (3) estimated the
mortality rate to be between 13% and 24%, depending on the series.
Numerous regimens have been employed for the management of KMS,
without, however, a consistent response, since patients exhibit a
variable and unpredictable response to traditional pharmacological
agents, including steroids, vincristine and interferon (IFN).
Recently, excellent response rates and prompt results have been
achieved by combining antiplatelet therapy with vincristine,
without the use of steroids. Sirolimus, which is directed against
the phosphatidylinositol-4,5-bisphosphate 3-kinase/AKT/mechanistic
target of rapamycin downstream signalling pathway and is involved
in lymphangiogenesis, has also shown promising results (4). No standard guidelines for the treatment
of the disease have been established so far. In certain cases,
multimodal therapy is necessary. The main therapeutic options
include systemic corticosteroid therapy, IFNα2a or 2b, vincristine
and platelet aggregation inhibitors, possibly in combination with
acetylsalicylic acid or ticlopidine (3).
Case report
A 1-year-old pediatric patient was admitted to the
Department of Reconstructive Plastic Surgery, The Children's
Hospital of Zhejiang University School of Medicine (Hangzhou,
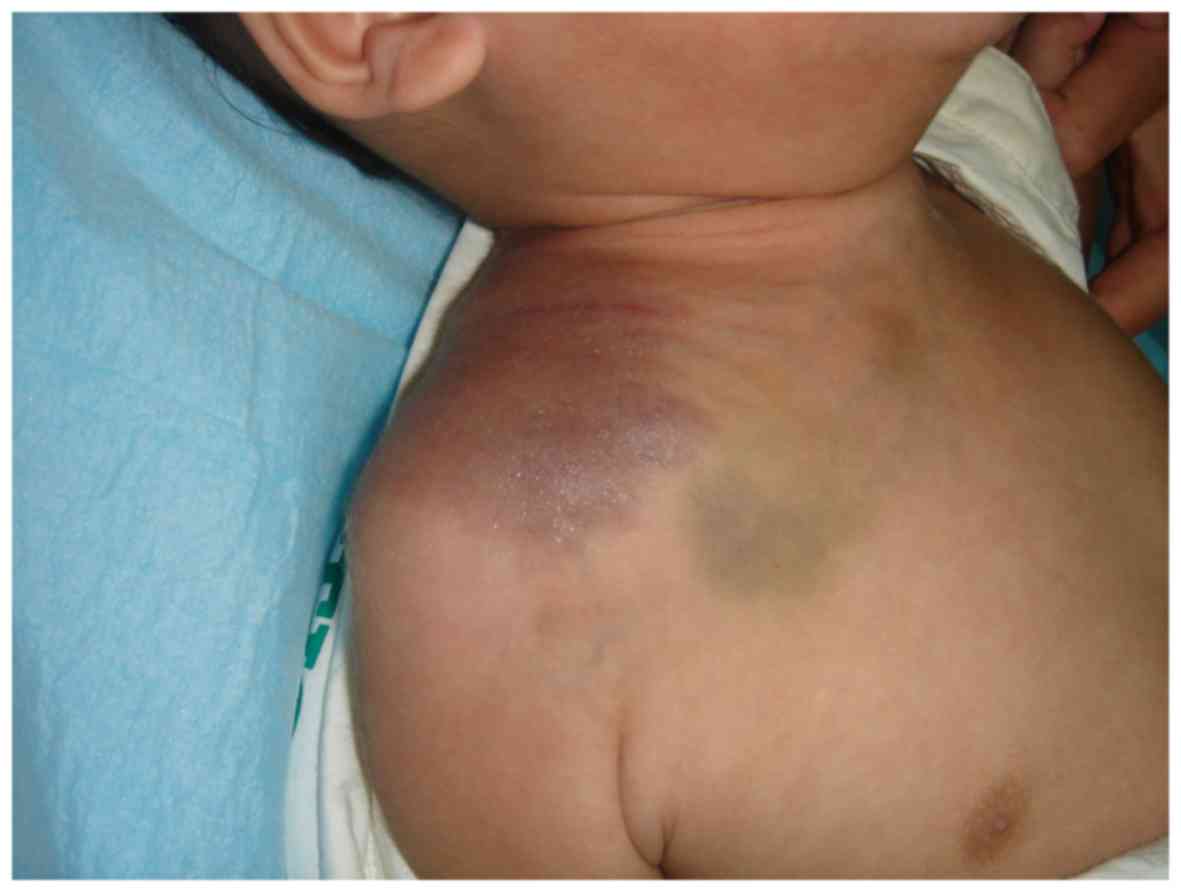
China) for the management of an erythematous and swollen process on
his right shoulder in July 2010. The lesion was a
reddish-violaceous plaque measuring 8×6 cm, irregularly shaped,
poorly demarcated and showed signs of swelling and inflammation
(Fig. 1). The lesion first appeared
as a small skin lesion the size of a peanut. Six months later, it
had increased in size and become slightly tender and warm upon
palpation. Treatment with antibiotics (cefmenoxime, 80 mg/kg) for 1
month failed. Laboratory tests revealed the following: Platelet
count, 77×109/l (normal range, 100–400×109
cells/l); hemoglobin, 115 g/l (normal range,110–140 g/l); leukocyte
count, 9.49×109 cells/l (normal range,
4.00–12.00×109 cells/l); D-dimer, 302 µg/l (normal
range, <500 µg/l). Doppler ultrasound (IU Elite; Philips
Healthcare, Amsterdam, The Netherlands) showed a fluid collection
in the subcutaneous tissue and muscle plane of the patient's right
shoulder and computed tomography scan (Somatom Emotion 16; Siemens
AG, Munich, Germany) demonstrated a vascular tumor extending to the
right clavicle. Based on thrombocytopenia, consumptive coagulopathy
and purpura associated with a huge vascular tumor, the patient was
diagnosed as KMS. Informed consent was obtained from the patient's
parents prior to treatment and the present study was approved by
the Ethical Committee of Zhejiang University.
Treatment with methylprednisolone was administered
intravenously at a dose of 10 mg/kg/day for 3 days and tapered off
over 2 weeks. Subsequently, 5 intralesional injections of compound
betamethasone (diprospan) were administered at a dose of 1 ml (7
mg) twice a week, which was equivalent to 10 mg/kg/day
methylprednisolone. Despite the stabilization of the platelet count
following treatment, the lesion continued to enlarge. A weekly
intravenous injection of 1.5 mg/m2 vincristine was
therefore added upon termination of the treatment. Following 6
injections of vincristine at a dose 0.68 mg (according his body
surface area), the platelet count was found to have increased to
264×109/l, and the mass in the right shoulder to have
shrank. The patient was discharged and received weekly blood tests
to ensure that the platelet count was stable.
A month later, the patient was readmitted to the
Department of Reconstructive Plastic Surgery, The Children's
Hospital of Zhejiang University School of Medicine, presenting with
a cough and a decreased platelet count of 78×109/l. A
posterior-anterior view of the chest was obtained using X-rays,
which demonstrated pneumonia. Several antibiotics were altered for
dealing with pneumonia and the weekly injection of vincristine was
reinitiated for 2 weeks. No further improvement in the platelet
count was observed. Laboratory test results showed the following:
Platelet count, 86×109 cells/l (normal range,
100–400×109 cells/l); white blood cell count,
5.96×109 cells/l (normal range,
4.00–12.00×109 cells/l); hemoglobin, 97 g/l (normal
range, 110–140 g/l); C-reactive protein, <1 mg/l (normal range,
<1 mg/l); D-dimer, 853 µg/l (normal range, <500 µg/l);
prothrombin time, 11.6 sec (normal range, 9.0–14.0 sec); activated
partial thromboplastin time, 23.0 sec (normal range, 23.0–28.0
sec); thrombin time, 18.8 sec (normal range, 15.0–22.0 sec). An
emergency surgery was scheduled. Intraoperatively, completely
resecting the tumor and stopping the bleeding around the right
clavicle was challenging. In the end, partial resection (>90%)
was performed and the wound was repaired by skin grafting. The
patient was intraoperatively supplied with 3 units platelets
(60×109 cells), 1 packed unit red blood cells (20 g
hemoglobin) and 200 ml plasma transfusions. On day 3 following
surgery, the platelet count was 170×109 cells/l, and on
day 10 it was 233×109 cells/l. The surgical specimen was
fixed with 10% neutral formaldehyde (Shanghai Ling Feng Chemical
Reagent Co., Ltd., Changshu, China) for >24 h and paraffin
embedded. The tissue slices (thickness, 4 µm) were stained with
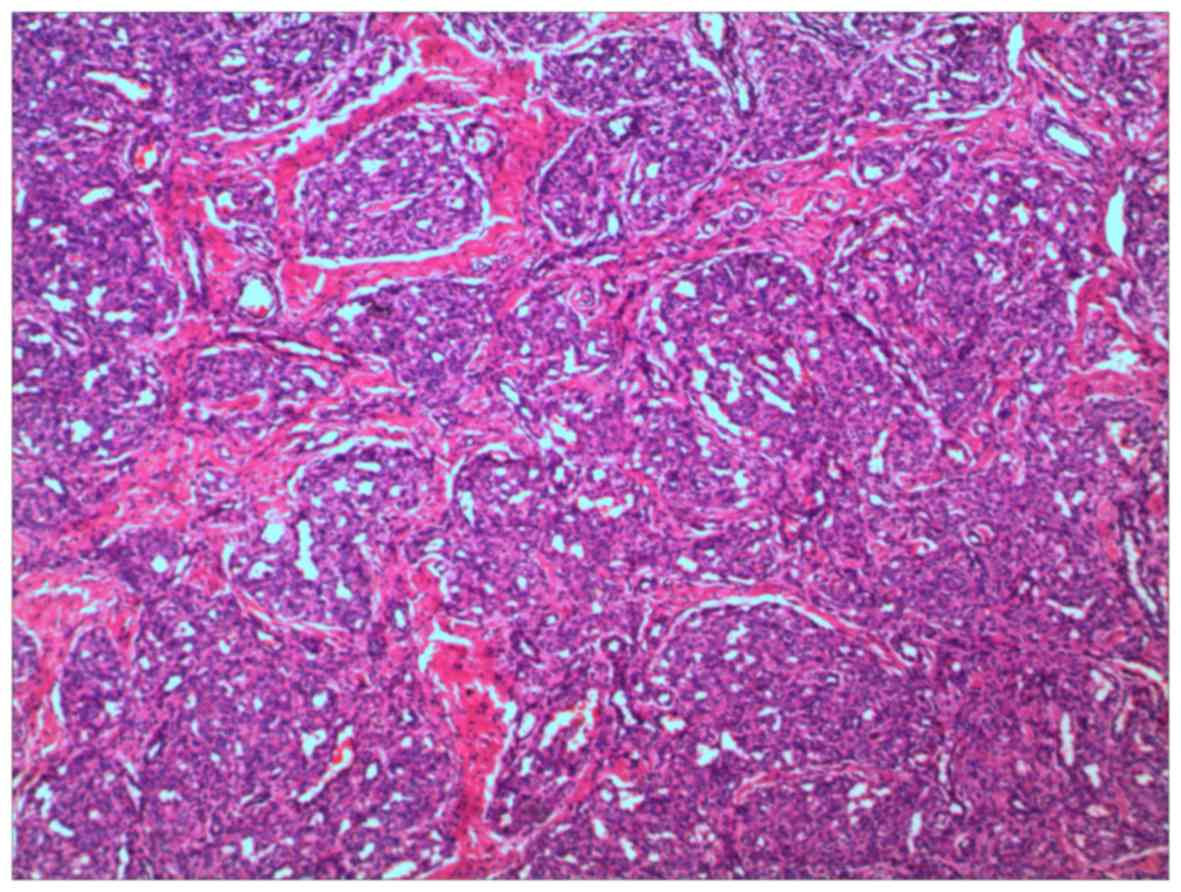
hematoxylin and eosin. Histopathology of the KMS revealed tufted
angioma (TA). A tumor-like distribution of visible clustered
capillary plexus in the dermis and subcutaneous tissue was
observed. Low-magnification microscopy (DMLB2; Leica Microsystems
GmbH, Wetzlar, Germany) showed a cannon-like appearance of the
lesion (Fig. 2). No atypia was
identified. Immunohistochemical staining revealed the following:
Cluster of differentiation (CD)31+ (mouse; 1:100
dilution; GA610; DakoCytomation, Glostrup, Denmark),
CD34+ (mouse; 1:100 dilution; IR632; DakoCytomation),
smooth muscle actin+ (mouse; 1:100 dilution; IR611;
DakoCytomation), vascular endothelial growth factor+
(mouse; 1:100 dilution; M7273; DakoCytomation),
vimentin+ (mouse; 1:100 dilution; IR630; DakoCytomation)
and glucose transporter 1− (polyclonal rabbit; 1:250
dilution; ab652; Abcam, Cambridge, MA, USA). Upon changing the
dressing, the wound was found to be clear and fresh. Two weeks
later, however, wound dehiscence and skin graft necrosis were
reported, while the platelet count was 187×109/l, white
blood cell count 7.33×109/l and C-reactive protein <1
mg/l. Unluckily, a decrease in the platelet count
(79×109/l) was again recorded 3 weeks after the surgery,
so vincristine injections were repeated postoperatively. Following
4 injections over 1 month, the patient was discharged with a
platelet count of 166×109/l and a healing wound.
Half a year later, the patient was readmitted to the
Department of Reconstructive Plastic Surgery, The Children's
Hospital of Zhejiang University School of Medicine presenting with
a severely low platelet count of 36×109/l. During
preparation for platelet transfusion, a 63.08% positivity for
platelet antibodies was detected (normal value, <30%). Platelet
transfusion was delayed and propranolol was introduced to the
parents. Due to refractoriness to previous treatments, the parents
agreed to oral administration of propranolol for the treatment of
KMS. First, the dose of propranolol was increased from 6 mg/day to
24 mg/day (split into 3 equal doses) for three days. The dose was
maintained at 24 mg/day in 3 equal doses (2 mg/kg/day) for 3 weeks
until the platelet count increased to 134×109/l. During
that period, a bone marrow puncture was made to confirm the origin
of platelet was normal. Following discharge from the hospital, oral
propranolol at the dose of 30 mg/day in 3 equal doses was
administered for 5 months and subsequently tapered off. Meanwhile,
vincristine was administered intravenously once a month, which
resulted in a continued response for 5 months.
At the time of writing the present study, the
patient was healthy without evidence of relapse for 2 years.
Discussion
KMS is a rare, aggressive type of vascular tumor
that has been associated with thrombocytopenia and consumptive
coagulopathy, which can result in a high risk of bleeding (5). KMS was first noted by Kasabach and
Merritt in 1940 (1). Enjolras et
al (2) suggested that KMS was
caused by either hemangioendothelioma (KHE), TA or a combination of
the two. KHE and TA probably belong to the same neoplastic spectrum
and histological continuum (6).
The histopathology of the present case revealed that
KMS was associated with TA, which is an infrequently observed
benign vascular tumor that was first described by Nakagawa in 1949
under the name angioblastoma (7). The
term TA was introduced by Wilson-Jones and Orkin in 1976, based on
the characteristic histology of the lesion (8). TA is a solitary tumor or infiltrated
plaque believed to have more of an inflammatory appearance than a
vascular abnormality (5). The lesion
has been described as small, cannonball-like, circumscribed
angiomatous tufts and nodules in the dermis and subcutaneous tissue
with characteristic lymphangioma-like vessels (9).
The pathogenesis of KMS remains unknown. Platelet
trapping by an abnormally proliferating endothelium within the
hemangioma may lead to the platelet activation, followed by
secondary activation of coagulation cascades, eventually resulting
in the consumption of various clotting factors (3,10).
Immunohistochemical analysis, using monoclonal antibodies against
platelet marker CD61, and isotope analysis, using
111indium- and 51Cr-labeled platelets, have
supported the importance of platelet trapping for the development
of KMS (10). In addition, excessive
blood flow and sheer stress, secondary to arteriovenous shunts
within the tumors, could lead to further platelet activation
(11).
The management of KMS is challenging due to its
rarity and the lack of well-established systematic treatment
strategies (11). Numerous
therapeutic modalities have been employed for KMS, with no clear
evidence that any type of treatment is superior over others. It has
been suggested that multidisciplinary treatment is required for KMS
(5). Several treatment regimens for
KMS have been reported, including topical or systemic
corticosteroid, IFN, chemotherapy, radiation, laser, propranolol,
sirolimus and surgery (5,11–19; Table
I).
 | Table I.Clinical profile of patients with
tufted angioma with Kasabach-Merritt syndrome arising from tufted
angioma. |
Table I.
Clinical profile of patients with
tufted angioma with Kasabach-Merritt syndrome arising from tufted
angioma.
| Author, year | Age | Gender | Site | Treatment | Outcome | Follow-up | Year of
treatment | Country | Ref. |
|---|
| Ferrandiz-Pulido
et al, 2010 | 1 m | Female | Chin | Predinisone + aspirin
+ VCR + VAC combination + IFN α-2a + Megadose
methylprednisolone | Partial response | 5 y | 2005 | Spain | (5) |
| Kim et al,
2010 | 2 m | Male | Left pubis | Systemic
costicosteroid (DXM + predinisone) | Complete
response | 1 y | 2008 | Korea | (11) |
| Rodriguez et
al, 2009 | 6 w | Female | Left elbow | High-dose
methylprednisolone (30 mg/kg/day) + VCR | Near-complete
response | 4 y | 2002 | USA | (16) |
| Wang et al,
2014 | 2 m | Male | Left knee | Predinisolone +
propranolol + VCR + surgery | Complete
response | 3 y | 2010 | China | (17) |
| Chiu et al,
2012 | 2 d | Female | Right thigh | Propranolol (3
mg/kg/day) | Complete
response | 6 m | 2011 | USA | (18) |
| Choi et al,
2013 | 15 d | Male | Left cheek | IFN α-2b +
predinisolone + propranolol + VCR + VAC combination | No response | 6 m | 2011 | Korea | (19) |
In the present study, multimodal treatment with
steroids, vincristine, surgery and propranolol was selected. An
intralesional injection of compound betamethasone (diprospan) was
administered at a dose of 1 ml (7 mg) twice a week, equivalent to
10 mg/kg/day of methylprednisolone. As a megadose of a normal
intralesional injection, compound betamethasone (diprospan)
provided fast-acting and slow-acting treatment to ensure the drug
concentration in the lesion was stable.
The reported adverse effects of steroid treatment
include hypertension, cushingoid appearance and opportunistic
infections (10). Vincristine is
considered to be an effective treatment option for TA/KHE; it has
been associated with a low incidence of side-effects and should,
therefore, be used as first-line treatment (12). Vincristine is a vinca alkaloid
antimitotic agent able to block the formation of microtubules in
cells (20). Toxicities associated
with myelosuppression and neurotoxicity require relevant
precautions. In particular, vincristine has been reported to cause
loss of deep tendon reflexes, peripheral neuropathy and abdominal
autonomic disturbance, such as constipation (21). The effects of angiography and
embolization have been shown to be temporary and are used primarily
as an adjunct to surgery, in order to minimize bleeding during a
planned resection (22). In certain
refractory cases, in which the patient cannot be tapered off of the
corticosteroids, chemotherapy is used. Therefore, the most common
chemotherapy used in such cases is cyclophosphamide, either alone
or combined with agents such as vincristine and actinomycin-D
(23). Propranolol is a non-selective
β-adrenergic antagonist, widely used in the treatment of infantile
hemangioma; KMP-associated tumors have been shown to have a
variable response to propranolol, and therefore the optimal dose
has not yet been established (13,18).
Sirolimus, a mammalian target of rapamycin inhibitor, was
previously tested in a prospective clinical trial for complicated
vascular anomalies (15). However,
due to the refractoriness of the treatment, the study did not
conclude the best way to deal with KMS.
In a previous study, no platelet antibodies or
evidence of an anti-immune process were found to be responsible for
the destruction of platelets, which suggests that their destruction
may have been due to another mechanism (24). In the present case, however, a
positivity of 63.08% for platelet antibodies was detected (normal
value, <30%). Prothrombin time and activated partial
thromboplastin time have been shown to be either normal or slightly
elevated in KMS (16). There were no
signs of bleeding and platelet transfusion may have been
unnecessary in the present study.
The present study reports a case of KMS as a result
of TA, which was managed with multimodal treatment. Although the
present patient obtained an optimal objective long-term response
with disease control, well-designed, large-scale studies are
required in order to clearly determine the benefits and risks of
multidisciplinary treatment for KMS. However, accumulating
sufficient patients for such studies may be challenging.
Acknowledgements
The authors would like to thank Mr. Grahay Rester
(Zhejiang Ivy International High School, Hangzhou, China) for
reviewing the present manuscript.
References
|
1
|
Kasabach HH and Merritt KK: Capillary
hemangioma with extensive purpura: report of a case. Am J Dis
Child. 59:1063–1070. 1940. View Article : Google Scholar
|
|
2
|
Enjolras O, Mulliken JB, Wassef M, Frieden
IJ, Rieu PN, Burrows PE, Salhi A, Léauté-Labreze C and Kozakewich
HP: Residual lesions after Kasabach-Merritt phenomenon in 41
patients. J Am Acad Dermatol. 42:225–235. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Le Nouail P, Viseux V and Enjolras O:
Groupe de Recherche Clinique en Dermatologie Pédiatrique:
Kasabach–Merritt phenomenon. Ann Dermatol Venereol. 134:580–586.
2007.(In French). View Article : Google Scholar : PubMed/NCBI
|
|
4
|
O'Rafferty C, O'Regan GM, Irvine AD and
Smith OP: Recent advances in the pathobiology and management of
Kasabach-Merritt phenomenon. Br J Haematol. 171:38–51. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ferrandiz-Pulido C, Mollet J, Sabado C,
Ferrer B and Garcia-Patos V: Tufted angioma associated with
Kasabach-Merritt phenomenon: A therapeutic challenge. Act Derm
Venereol. 90:535–537. 2010.
|
|
6
|
Chu CY, Hsiao CH and Chiu HC:
Transformation between Kaposiform hemangioendothelioma and tufted
angioma. Dermatology. 206:334–337. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nakagawa K: Case report of angioblastoma
of the skin. Jpn J Dermatol. 59:92–94. 1949.
|
|
8
|
Jones EW and Orkin M: Tufted angioma
(angioblastoma). A benign progressive angioma, not to be confused
with Kaposi's sarcoma or low-grade angiosarcoma. J Am Acad
Dermatol. 20:214–225. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sadeghpour M, Antaya RJ, Lazova R and Ko
CJ: Dilated lymphatic vessels in tufted angioma: A potential source
of diagnostic confusion. Am J Dermatopathol. 34:400–403. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hall GW: Kasabach-Merritt syndrome:
Pathogenesis and management. Br J Haematol. 112:851–862. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim T, Roh MR, Cho S and Chung KY:
Kasabach-merritt syndrome arising from tufted angioma successfully
treated with systemic corticosteroid. Ann Dermatol. 22:426–430.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fahrtash F, McCahon E and Arbuckle S:
Successful treatment of kaposiform hemangioendothelioma and tufted
angioma with vincristine. J Pediatr Hematol Oncol. 32:506–510.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hermans DJ, van Beynum IM, van der Vijver
RJ, Kool LJ, de Blaauw I and van der Vleuten CJ: Kaposiform
hemangioendothelioma with Kasabach-Merritt syndrome: A new
indication for propranolol treatment. J Pediatr Hematol Oncol.
33:e171–e173. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mahendran R, White SI, Clark AH and
Sheehan-Dare RA: Response of childhood tufted angioma to the
pulsed-dye laser. J Am Acad Dermatol. 47:620–622. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hammill AM, Wentzel M, Gupta A, Nelson S,
Lucky A, Elluru R, Dasgupta R, Azizkhan RG and Adams DM: Sirolimus
for the treatment of complicated vascular anomalies in children.
Pediatr Blood Cancer. 57:1018–1024. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rodriguez V, Lee A, Witman PM and Anderson
PA: Kasabach-merritt phenomenon: Case series and retrospective
review of the mayo clinic experience. J Pediatr Hematol Oncol.
31:522–526. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang Z, Li K, Dong K, Xiao X and Zheng S:
Variable response to propranolol treatment of kaposiform
hemangioendothelioma, tufted angioma and Kasabach-Merritt
phenomenon. Pediatr Blood Cancer. 61:1518–1519. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chiu YE, Drolet BA, Blei F, Carcao M,
Fangusaro J, Kelly ME, Krol A, Lofgren S, Mancini AJ, Metry DW, et
al: Variable response to propranolol treatment of kaposiform
hemangioendothelioma, tufted angioma and Kasabach-Merritt
phenomenon. Pediatric Blood Cancer. 59:934–938. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Choi JW, Na JI, Hong JS, Kwon SH, Byun SY,
Cho KH, Youn SW, Choi HS, Park KD and Park KC: Intractable tufted
angioma associated with kasabach-merritt syndrome. Ann Dermatol.
25:129–130. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Brunton L, Lazo J and Paker K: Goodman
& Gliman's The Pharmacological Basis of Therapeutics. 11th.
McGraw-Hill; New York: 2005
|
|
21
|
Haisley-Royster C, Enjolras O, Frieden IJ,
Garzon M, Lee M, Oranje A, de Laat PC, Madern GC, Gonzalez F,
Frangoul H, et al: Kasabach-merritt phenomenon: A retrospective
study of treatment with vincristine. J Pediatr Hematol Oncol.
24:459–462. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Drolet BA, Trenor CC III, Brandão LR, Chiu
YE, Chun RH, Dasgupta R, Garzon MC, Hammill AM, Johnson CM, Tlougan
B, et al: Consensus-derived practice standards plan for complicated
Kaposiform hemangioendothelioma. J Pediatr. 163:285–291. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hauer J, Graubner U, Konstantopoulous N,
Schmidt S, Pfluger T and Schmid I: Effective treatment of
kaposiform hemangioendotheliomas associated with Kasabach-Merritt
phenomenon using four-drug regimen. Pediatr Blood Cancer.
49:852–854. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
de la Hunt MN: Kasabach-Merritt syndrome:
Dangers of interferon and successful treatment with pentoxifylline.
J Pediatr Surg. 41:e29–e31. 2006. View Article : Google Scholar : PubMed/NCBI
|