Introduction
Lung cancer is the leading cause of cancer mortality
globally; 85–90% of cases of lung cancer were non-small-cell lung
cancer (NSCLC) between 1975 and 2012 (1,2). During
this time, ~80% of NSCLC cases were locally advanced (stage IIIA/B)
or metastatic (stage IV), with poor prognosis (2). For advanced NSCLC, platinum-based
doublet chemotherapy, the standard treatment, has reached a plateau
(3). The median survival time of
NSCLC from 2001 to 2004 for stages IIIB/IV was <1 year and the
3- and 5-year survival rates were 4.3 and 2.8%, respectively
(4). Precision medicine and
individualized therapy are the emerging fields in cancer research,
and multiple established and potential targets, including epidermal
growth factor receptor (EGFR) and anaplastic lymphoma kinase genes,
are the foundation of this therapy.
Multiple clinical trials indicate that, in
comparison with chemotherapy, treatment with the tyrosine kinase
inhibitor (TKI) erlotinib, which targets the EGFR, results in an
improved response rate (RR) for advanced or metastatic NSCLC and
can prolong progression-free survival (PFS), representing a valid
treatment option (5,6). However, despite its benefits, erlotinib
induces hepatotoxicity that can pose substantial harm to patients.
The EURTAC study (6) revealed that in
Western countries, the incidence rates of all-grade and grade-3
liver enzyme elevation were 6 and 2%, respectively, among
erlotinib-treated patients with advanced NSCLC with EGFR mutations.
However, the incidence of hepatotoxicity is higher in Eastern
countries. The OPTIMAL study (5)
indicated that in Eastern countries, the incidence rates of
all-grade, and grade-3/4 alanine transaminase (ALT) elevation were
37 and 4%, respectively, among erlotinib-treated patients with
NSCLC with EGFR mutations. With such occurrence and occasionally
serious severity, hepatotoxicity as a side effect of erlotinib is
positively associated with the efficacy of erlotinib, and the
survival of patients. Therefore, considering the requirement of
long-term administration of EGFR-TKIs, including erlotinib, there
is a requirement for studies on erlotinib-induced
hepatotoxicity.
The mechanism of drug-induced liver injury (DILI)
has not previously been completely elucidated. Mitochondrial injury
has been proposed and acknowledged as one possible mechanism for
DILI (7). Therefore, in the present
study, the human hepatocyte L-02 cell line was used as an in
vitro model to investigate whether the mitochondrial pathway of
apoptosis was involved in erlotinib-induced hepatotoxicity.
Materials and methods
Drugs and chemicals
Erlotinib was obtained from Roche Diagnostics
(Basel, Switzerland) and dissolved in DMSO (50 mmol/l stock
solution). Erlotinib was stored at −40°C as frozen aliquots and the
solution was thawed directly prior to the experiments. The chemical
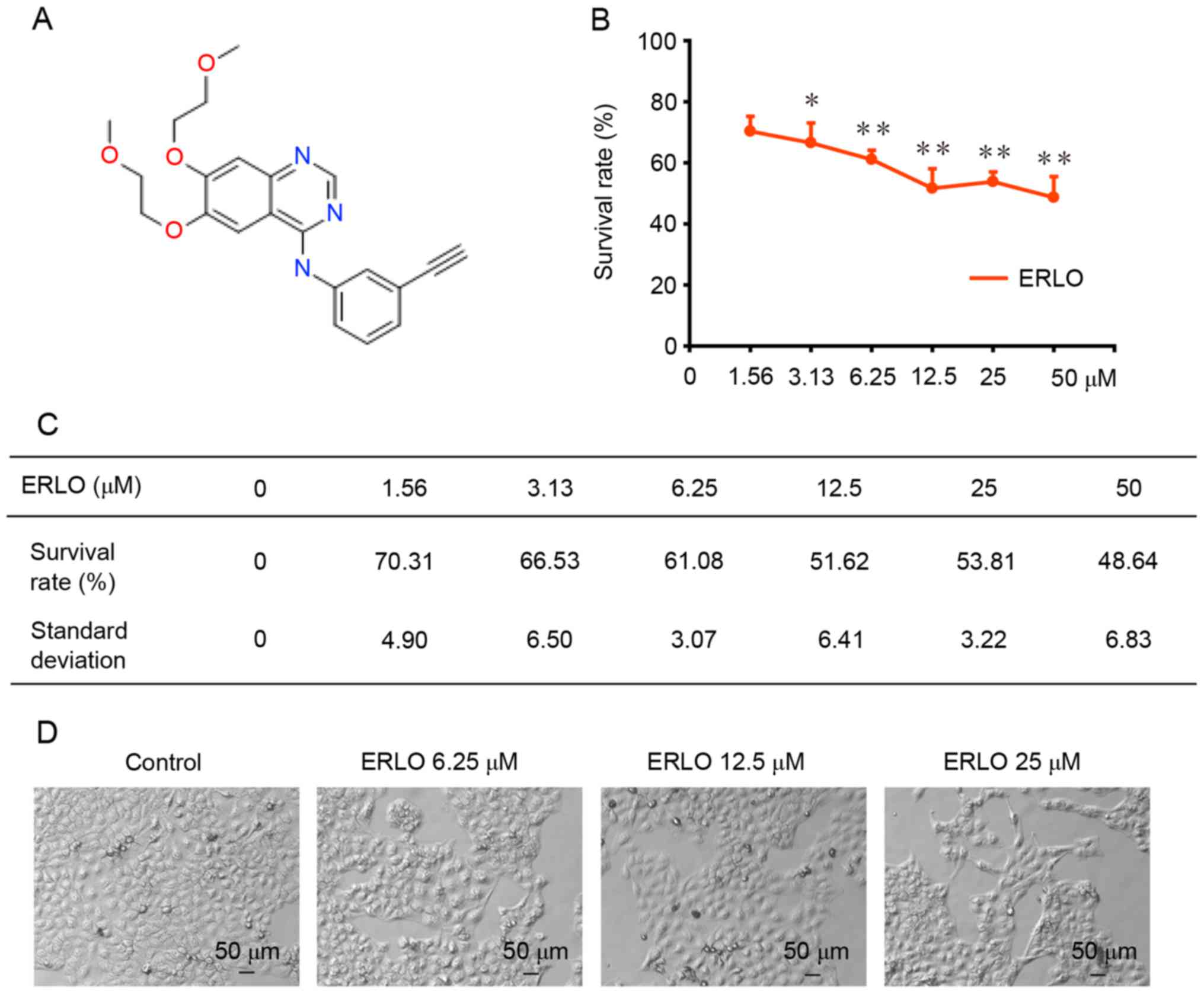
structure of erlotinib is presented in Fig. 1A.
Cell line and cell culture
L-02 cells, human hepatocytes, were purchased from
Shanghai Institute of Biochemistry and Cell Biology (Shanghai,
China). L-02 cells were maintained in complete RPMI-1640 media
(Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) at 37°C
under 5% CO2 with 10% heat-inactivated fetal bovine
serum (FBS; Invitrogen; Thermo Fisher Scientific, Inc.), containing
100 U/ml penicillin (Invitrogen; Thermo Fisher Scientific, Inc.)
and 100 µg/ml streptomycin (Invitrogen; Thermo Fisher Scientific,
Inc.).
Cell proliferation assay
The sulforhodamine B (SRB) colorimetric assay
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was used to
evaluate cell proliferation. L-02 cells (3×103
cells/well) were cultured in 96-well plates and exposed to
erlotinib (0, 1.56, 3.13, 6.25, 12.50, 25.00 or 50.00 µM) for 72 h.
The cells were then fixed with 10% trichloroacetic acid
(Sigma-Aldrich; Merck KGaA) for 1 h at 4°C. Once the cells had been
stained with SRB for 30 min at room temperature and bound SRB had
been dissolved with 10 mmol/l Tris-base, a multi-well
spectrophotometer was used to measure the absorbance at 510 nm.
Calculation of the cell survival rate was according to the
following formula: A510 treated cells/A510 control cells ×100.
DAPI staining assay
L-02 cells (3×104 cells/well) cultured
exponentially in 6-well plates were exposed to erlotinib (0, 6.25,
12.50 or 25.00 µM) for 48 h. To study cell morphology, the cells
were rinsed in PBS followed by fixation with 0.1% Triton X-100 for
15 min. Next, DAPI (2.0 µg/ml; Sigma-Aldrich; Merck KGaA) was used
to stain the cells for 15 min. All steps were performed at room
temperature. The morphology of cell nuclei was examined by
fluorescence microscopy (magnification, ×100).
Apoptosis detection by flow
cytometry
An Annexin V-fluorescein isothiocyanate
(FITC)/propidium iodide (PI) apoptosis detection kit (Invitrogen;
Thermo Fisher Scientific, Inc.) was used to measure apoptosis rate.
L-02 cells (3×104 cells/well) were harvested 48 h after
treatment with erlotinib (0, 6.25, 12.50, 25.00 µM). L-02 cells
were washed twice with cold PBS, 1×106 cells/ml were
resuspended in 1X binding buffer of the kit and 100 µl cell
suspension was transferred to a 5-ml culture tube. In the dark, the
transferred suspension was stained with 5 µl Annexin V-FITC and 5
µl PI at 25°C for 15 min. Cell apoptosis was detected using a
FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ,
USA) with BD FACSDiva Software (version 8.0.1) within 1 h.
Mitochondrial membrane potential
(ΔΨm) evaluation by JC-1 stain
L-02 cells were inoculated into 6-well plates
(1.5×104 cells/well) for 24 h at 37°C. Following
treatment with erlotinib (0, 6.25, 12.50, 25.00 µM) for 48 h, L-02
cell suspension was prepared in PBS (500 µl) and stained with 2.5
µl JC-1 (20 µg/ml) for 30 min at 37°C under 5% CO2. As a
cationic dye, once in the mitochondria, JC-1 shifts its
fluorescence emission from green to red, from 525±10 to 610±10 nm,
as it accumulates owing to the high membrane potential. The cell
suspension was sampled as 1×104 cells/sample. A
FACSCalibur flow cytometer was used to analyze the fluorescence
emission. The fluorescence intensity ratio of red/green decreases
when mitochondrial depolarization occurs.
Western blot analysis
Briefly, protein was extracted from L-02 cells in
lysis buffer (50 mmol/l Tris-HCl, 150.0 mmol/l NaCl, 1 mmol/l EDTA,
1 mmol/l phenylmethylsulfonyl fluoride, 1% NP-40, 0.1% SDS, 0.5%
deoxycholic acid, 2.0 µg/ml aprotinin, 0.02% NaN3).
Lysates were centrifuged (1×104 × g) at 4°C for 15 min.
Protein expression (20 µg/lane) was detected by SDS-PAGE (8–15%
gel), and proteins were electroblotted onto polyvinylidene fluoride
membranes (EMD Millipore, Billerica, MA, USA). Primary and
horseradish peroxidase (HRP)-conjugated secondary antibodies
(Southern Biotech, Birmingham, AL, USA) were used to probe
proteins. The primary antibodies used were: B-cell lymphoma 2
(Bcl-2; cat. no. 2872) and cleaved caspase-3 (cat. no. 9961)
purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA);
cleaved poly (ADP-ribose) polymerase (PARP; cat. no. sc-56196),
Bcl-associated X (Bax; cat. no. sc-4239) and β-actin (cat. no.
sc-1615) purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX,
USA). HRP-labeled secondary antibodies (GAR007, GAM007, RAG007)
were purchased from MultiSciences Biotech Co., Ltd. (Hangzhou,
China). The primary and secondary antibodies were diluted into
1:1,000 and 1:5,000, respectively. Incubation occurred for 2 or 0.5
h at room temperature and washing for 5 min/wash for three washes.
An enhanced chemiluminescence detection system (Biological
Industries, Beit Haemek, Israel) was used for protein
detection.
Statistical analysis
SPSS version 18.0 (SPSS, Inc., Chicago, IL, USA) was
used for statistical analyses. Survival rates were presented as
mean ± standard deviation and analyzed using two-way analysis of
variance. P<0.05 (two-way) was considered to indicate a
statistically significant difference.
Results
Erlotinib induces cytotoxicity in L-02
cells
Cell viability was assessed using a SRB colorimetric
assay following treatment of L-02 cells with erlotinib (0, 1.56,
3.13, 6.25, 12.50, 25.00, 50.00 µM) for 72 h. As presented in
Fig. 1B and C, a dose-dependent
inhibition of L-02 cell proliferation was detected in L-02 cells
following exposure to erlotinib for 72 h (P<0.05). The
percentage of cells that survived (compared with untreated control
cells) was 70.31±4.90 and 48.64±6.83% at the lowest (P=0.03), and
highest (P=0.003) concentration tested, respectively. The growth of
L-02 cells following the 72-h treatment was analyzed using light
microscopy. Cellular swelling, a cloudy cytoplasm, slow cell growth
and impaired cell adherence were observed (Fig. 1D).
Erlotinib induces apoptosis in L-02
cells
Following treatment of L-02 cells with 6.25, 12.50
and 25.00 µM erlotinib for 48 h, DAPI staining was used to detect
variations in the nucleus morphology caused by apoptosis. Upon
examination by fluorescence microscopy, L-02 cells treated with
12.50 µM erlotinib exhibited chromatin condensation and
karyopyknosis, which are typical apoptotic phenomena. Further
marked nuclear pyknosis was observed at a concentration of 25.00
µM, indicating the dose-dependent effect of erlotinib on the
apoptosis of L-02 cells (Fig. 2A).
Furthermore, detection of apoptotic protein expression was
performed by western blot analysis. In concordance with the
morphological changes, the levels of cleaved caspase-3 and cleaved
PARP increased in a dose-dependent manner in treated cells,
indicating the upregulation of apoptotic proteins (Fig. 2B). Subsequently, measurement by flow
cytometry of the apoptotic rate of cells stained with Annexin
V-FITC/PI was performed. Compared with the control, the late
apoptotic rate in erlotinib-treated cells increased significantly
and dose-dependently (P<0.05; Fig.
3). These results revealed that apoptosis contributes to
erlotinib-induced hepatotoxicity.
 | Figure 3.Erlotinib induces apoptosis in L-02
cells. Following treatment of cells with increasing doses of
erlotinib for 48 h, flow cytometry analysis determined the degree
of apoptosis induction in cells stained with Annexin V-FITC and PI.
(A) FACS analysis of Annexin V-FITC/PI staining for apoptotic
cells. upper left, upper right, lower left, and lower right
quadrants indicate necrotic cells (PI-positive), late apoptotic
cells (Annexin V-FITC- and PI-positive), viable cells, and early
apoptotic cells (Annexin V-FITC-positive), respectively. Data are
representative of three independent experiments. (B) Quantification
of necrotic, late apoptotic, early apoptotic and viable cells in
(A). Data are presented as the mean of three independent
experiments. *P<0.05, **P<0.01 vs. control (0 µM). ERLO,
erlotinib; FITC, fluorescein isothiocyanate; PI, propidium iodide;
FACS, fluorescence-activated cell sorting. |
Mitochondrial pathway is involved in
erlotinib-induced apoptosis
During drug-induced apoptosis, mitochondrial changes
are the key events. Therefore, the function of mitochondria in
erlotinib-induced apoptosis was investigated. JC-1 staining and
western blot analysis was used to examined the variation of
ΔΨm, and associated protein expression in L-02 cells
treated with erlotinib (0, 6.25, 12.50 and 25.00 µM) for 48 h.
Using JC-1 staining, treated cells exhibited a decreased red/green
fluorescence intensity ratio in line with the increasing
concentration of erlotinib (P<0.05), which indicated that
erlotinib treatment induced a dose-dependent loss of ΔΨm
and mitochondrial depolarization (Fig.
4). Since the mitochondrial pathway of apoptosis is regulated
by proteins of the Bcl-2 family (8),
the levels of the pro-apoptotic Bax and anti-apoptotic Bcl-2
proteins were analyzed. The levels of Bcl-2 protein decreased
whereas those of Bax increased in L-02 cells 24 h after erlotinib
treatment (6.25, 12.50, 25.00 µmol/l), in a dose-dependent manner
(Fig. 5). The present study indicated
that the mitochondrial pathway may be involved in apoptosis induced
by erlotinib.
Discussion
The mechanism of drug-induced liver injury (DILI)
has not been completely elucidated. Russmann et al (9) suggested that the mechanism may include
specific injury ‘upstream’ and unspecific ‘downstream’ events. The
upstream events of DILI are initial hepatocyte injury caused by
sophisticated interactions between hereditary and environmental
risk factors, whereas the downstream events involve the equilibrium
between the processes of injury and protection in mitochondrion. To
study the mechanism of DILI, a three-step working model was
proposed (9). First, drugs associated
with DILI and their reactive metabolites directly cause cellular
stress, inhibit mitochondrial functions and activate specific
immune responses. Secondly, mitochondrial permeability transition
(MPT) caused by initial injury may proceed through an intrinsic
pathway by activating cascades of intracellular stressors and an
extrinsic pathway modulated by cytokines, and immune reactions.
Finally, depending on ATP availability, MPT may result in necrosis
or apoptosis.
Liver injury is a frequent side effect in patients
undergoing long-term erlotinib treatment. Hepatotoxicity can
influence the efficacy of erlotinib and survival (10). Thus, the purpose of the present study
was to investigate the potential mechanism of erotinib-induced
hepatotoxicity via in vitro experiments with L-02 cells. The
present study revealed that, in L-02 cells, cell viability
decreased dose-dependently and cell morphology was altered 72 h
after erlotinib treatment. As a form of programmed cell death,
apoptosis is associated with DILI. In the present study, DAPI
staining revealed chromatin condensation and karyopyknosis in the
nucleus, and increased apoptosis in a dose-dependent manner in L-02
cells following erlotinib treatment. Similar results have been
reported in hepatocytes upon treatment with other drugs, including
17-demethoxy-reblastatin, schisandrin B and saikosaponin D and
(11–13). PARP is a DNA repair enzyme whose
cleaved form contributes to apoptosis owing to drug toxicity.
Caspase-3 is responsible for PARP cleavage during the early stage
of apoptosis (14). Therefore,
increased levels of cleaved caspase-3 and cleaved PARP lead to
apoptosis. In the present study, cleaved caspase-3 and cleaved PARP
protein expression levels increased following erlotinib treatment.
These data demonstrated that erlotinib-induced hepatotoxicity
occurs through the process of apoptosis.
Generally, the biological mechanism of apoptosis is
composed of an extrinsic pathway that is death-receptor-dependent
and an intrinsic pathway that is mitochondrial-dependent (15,16). The
two pathways are involved in drug-induced apoptosis (13,17). A
collapse of the ΔΨm was observed alongside an alteration
of Bax and Bcl-2 protein expression during the apoptosis of
hepatocytes induced by saikosaponin D (13). Data from the present study
demonstrated that erlotinib-treated hepatocytes exhibit
significantly decreased ΔΨm and Bcl-2 protein levels,
but increased Bax expression, all in a dose-dependent manner. These
results indicated that erlotinib-induced apoptosis may proceed
through the mitochondrial-dependent pathway.
Acetaminophen (APAP) is one of the most common
causes of DILI owing to its wide use as an analgesic (18). The mechanisms of APAP-induced
hepatotoxicity include the damage of reactive drug
intermediates/binding/adducts, mitochondrial dysfunction, nuclear
DNA damage, karyorrhexis and inflammation (19). Additionally, hepatoprotective factors,
including heat shock protein exhibit a protective function in
hepatotoxicity (20). Certain studies
(21,22) have provided evidence that
mitochondrial dysfunction and damage is involved in APAP
hepatotoxicity in humans, and inflammation is necessary for
recovery and regeneration, whereas apoptosis has only a minor
function. By contrast, the present study demonstrated that
erlotinib-induced hepatotoxicity involved mitochondrial damage
accompanied with apoptotic phenomena, including chromatin
condensation, nuclear fragmentation, and caspase expression and
cleavage.
Nowadays, TKIs are increasingly applied in clinical
practice and TKI-induced hepatotoxicity is common (23). However, studies of TKI-induced
hepatotoxicity are relatively rare and the mechanisms involved are
poorly understood. Xue et al (24) reported that mitochondria-mediated cell
death pathway serves an essential function via oxidative stress,
and activation of nuclear factor erythroid 2-related factor 2 and
the mitogen-activated protein kinase pathway in dasatinib-induced
hepatotoxicity. Researchers from the same group also identified
that autophagy protects against dasatinib-induced hepatotoxicity by
activating p38 signaling in liver tissues and primary cultured rat
hepatocytes (25). The current study
demonstrated that erlotinib induces apoptosis in hepatocytes with a
correspondent decrease in the ΔΨm and alterations to
protein expression typical of the mitochondrial apoptotic pathway.
These results indicated that the potential mechanism of
erlotinib-induced hepatotoxicity may involve the mitochondrial
apoptosis pathway.
The evidence provided by the present study expands
the understanding of the fundamental mechanism of TKI-induced
hepatotoxicity. Further in vivo research is; however,
required to support these observations.
Acknowledgements
The present study was supported by the Zhejiang
Provincial Foundation of National Science (grant nos. LZ13H160001
and LY14H160006) and the Health Foundation of Hangzhou City
Zhejiang Province (grant no. 2015Z003).
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Howlader N, Noone AM, Krapcho M, Garshell
J, Miller D, Altekruse SF, Kosary CL, Yu M, Ruhl J, Tatalovich Z,
et al: SEER cancer statistics review 1975–2012. Natl Cancer Inst.
Nov 18–2015.
|
|
3
|
Iranzo V, Bremnes RM, Almendros P, Gavilá
J, Blasco A, Sirera R and Camps C: Induction chemotherapy followed
by concurrent chemoradiation for patients with non-operable stage
III non-small-cell lung cancer. Lung Cancer. 63:63–67. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ozkaya S, Findik S, Dirican A and Atici
AG: Long-term survival rates of patients with stage IIIB and IV
non-small cell lung cancer treated with cisplatin plus vinorelbine
or gemcitabine. Exp Ther Med. 4:1035–1038. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhou C, Wu YL, Chen G, Feng J, Liu XQ,
Wang C, Zhang S, Wang J, Zhou S, Ren S, et al: Erlotinib versus
chemotherapy as first-line treatment for patients with advanced
EGFR mutation-positive non-small-cell lung cancer (OPTIMAL,
CTONG-0802): A multicentre, open-label, randomized, phase 3 study.
Lancet oncol. 12:735–742. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rosell R, Carcereny E, Gervais R,
Vergnenegre A, Massuti B, Felip E, Palmero R, Garcia-Gomez R,
Pallares C, Sanchez JM, et al: Erlotinib versus standard
chemotherapy as first-line treatment for European patients with
advanced EGFR mutation-positive non-small-cell lung cancer
(EURTAC): A multicentre, open-label, randomized phase 3 trial.
Lancet Oncol. 13:239–246. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Russmann S, Kullak-Ublick GA and
Grattagliano I: Current concepts of mechanisms in drug-induced
hepatotoxicity. Curr Med Chem. 16:3041–3053. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Leanza L, Henry B, Sassi N, Zoratti M,
Chandy KG, Gulbins E and Szabò I: Inhibitors of mitochondrial Kv1.
3 channels induce Bax/Bak-independent death of cancer cells. EMBO
Mol Med. 4:577–593. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Russmann S, Jetter A and Kullak-Ublick GA:
Pharmacogenetics of drug-induced liver injury. Hepatology.
52:748–761. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Teo YL, Ho HK and Chan A: Risk of tyrosine
kinase inhibitors-induced hepatotoxicity in cancer patients: A
meta-analysis. Cancer Treat Rev. 39:199–206. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhao S, Li H, Jiang C, Ma T, Wu C, Huo Q
and Liu H: 17-Demethoxy-reblastatin, an Hsp90 inhibitor, induces
mitochondria-mediated apoptosis through downregulation of Mcl-1 in
human hepatocellular carcinoma cells. J Bioenerg Biomembr.
47:373–381. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang Y, Zhou ZW, Jin H, Hu C, He ZX, Yu
ZL, Ko KM, Yang T, Zhang X, Pan SY and Zhou SF: Schisandrin B
inhibits cell growth and induces cellular apoptosis and autophagy
in mouse hepatocytes and macrophages: Implications for its
hepatotoxicity. Drug Des Devel Ther. 9:2001–2027. 2015.PubMed/NCBI
|
|
13
|
Chen L, Zhang F, Kong D, Zhu X, Chen W,
Wang A and Zheng S: Saikosaponin D disrupts platelet-derived growth
factor-β receptor/p38 pathway leading to mitochondrial apoptosis in
human LO2 hepatocyte cells: A potential mechanism of
hepatotoxicity. Chem Biol Interact. 206:76–82. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Oliver FJ, de la Rubia G, Rolli V,
Ruiz-Ruiz MC, de Murcia G and Murcia JM: Importance of
poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson
from an uncleavable mutant. J Biol Chem. 273:33533–33539. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kaiser WJ, Upton JW, Long AB,
Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T and
Mocarski ES: RIP3 mediates the embryonic lethality of
caspase-8-deficient mice. Nature. 471:368–372. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Guha M, Maity P, Choubey V, Mitra K,
Reiter RJ and Bandyopadhyay U: Melatonin inhibits free
radical-mediated mitochondrial-dependent hepatocyte apoptosis and
liver damage induced during malarial infection. J Pineal Res.
43:372–381. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang F, Chen L, Jin H, Shao J, Wu L, Lu Y
and Zheng S: Activation of Fas death receptor pathway and Bid in
hepatocytes is involved in saikosaponin D induction of
hepatotoxicity. Environ Toxicol Pharmacol. 41:8–13. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee WM: Etiologies of acute liver failure.
Semin Liver Dis. 28:142–152. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
McGill MR and Jaeschke H: Mechanistic
biomarkers in acetaminophen-induced hepatotoxicity and acute liver
failure: From preclinical models to patients. Expert Opin Drug
Metab Toxicol. 10:1005–1017. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Williams CD, McGill MR, Farhood A and
Jaeschke H: Fas receptor-deficient lpr mice are protected against
acetaminophen hepatotoxicity due to higher glutathione synthesis
and enhanced detoxification of oxidant stress. Food Chem Toxicol.
58:228–235. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
McGill MR, Sharpe MR, Williams CD, Taha M,
Curry SC and Jaeschke H: The mechanism underlying
acetaminophen-induced hepatotoxicity in humans and mice involves
mitochondrial damage and nuclear DNA fragmentation. J Clin Invest.
122:1574–1583. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hu J, Ramshesh VK, McGill MR, Jaeschke H
and Lemasters JJ: Low dose acetaminophen induces reversible
mitochondrial dysfunction associated with transient c-Jun
N-terminal kinase activation in mouse liver. Toxicol Sci.
150:204–15. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen X, Yang S and Ma S: Drug induced
hepatotoxicity in targeted therapy for lung cancer. Zhongguo Fei Ai
Za Zhi. 17:685–688. 2014.(In Chinese). PubMed/NCBI
|
|
24
|
Xue T, Luo P, Zhu H, Zhao Y, Wu H, Gai R,
Wu Y, Yang B, Yang X and He Q: Oxidative stress is involved in
Dasatinib-induced apoptosis in rat primary hepatocytes. Toxicol
Appl Pharmacol. 261:280–291. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang X, Wang J, Dai J, Shao J, Ma J, Chen
C, Ma S, He Q, Luo P and Yang B: Autophagy protects against
dasatinib-induced hepatotoxicity via p38 signaling. Oncotarget.
6:6203–6217. 2015. View Article : Google Scholar : PubMed/NCBI
|