Introduction
Lymphoma originates from the lymph nodes or
extra-nodal lymphoid tissue. Lymphoma is a malignant tumor that
typically manifests progressive lymph nodes enlargement without
pain and occasionally, the liver, bone and bone marrow may be
involved (1). Lymphoma with bone
marrow involvement is not rare (2).
Some types of lymphoma originate directly from bone marrow.
Lymphoblastic lymphoma (LBL), marginal zone lymphoma and Burkett
lymphoma typically originate from bone marrow (2–4). LBL
originates from immature precursor lymphocytes, it is highly
invasive and divided into B cell LBL and T cell LBL, with the
majority of LBL originating from precursor T cells (2,5,6). Other types of lymphoma rarely originate
from the bone marrow, including diffuse large B cell lymphoma
(DLBCL) and follicular lymphoma. If these types of lymphoma present
with bone marrow involvement as the only clinical manifestation,
they are termed primary bone marrow lymphoma (PBML). Lymphoma
originating from bone marrow typically manifests with diffuse bone
marrow involvement; therefore, it is rare to present with an
isolated mass (2,7,8). In the
present case study, lymphoma originating from the bone marrow was
termed bone marrow lymphoma (BML). The present case study describes
a BML case presenting as an isolated mass and the associated
literature is reviewed.
Case report
A 29-year-old male was admitted into Peking Union
Medical College Hospital (Beijing, China) on October the 12th 2013,
presenting with a 6-month history of pain in the right elbow and a
4-month history of fever. Previous examinations of the patient
revealed pancytopenia, no tumor cell in bone marrow aspiration
(BMA) and no abnormality in a computed tomography (CT) scan of the
right upper limb. The patient had received antibiotics, but no
improvement was observed. The patient also had a weight loss of ~4
kg. In addition, the patient reported a seafood allergy. Physical
examination revealed a fever (37.8°C), a rapid heartbeat (112
beats/min) and a low blood pressure (89/56 mmHg). No superficial
lymphadenopathy was determined, and the spleen could be touched
under the ribs. A complete blood count (CBC) demonstrated decreased
levels of white blood cells (WBC) (2.0×109 cells/l;
normal range, 4.0–10.0×109 cells/l), neutrophils
(1.2×109 cells/l; normal range, 2.0–7.5×109
cells/l), hemoglobin (8.6 g/dl; normal range, 12.0–16.0 g/dl) and
platelets (36×109 cells/l; normal range,
100–300×109 cells/l). The activity of natural killer
(NK) cells, determined as the activity to kill fluorescent plasmid
transfected cells, was identified to be low (15.0%; normal range,
31.5–41.6%). Soluble cluster of differentiation (CD)25 was
>44,000 pg/ml (normal, <6,400 pg/ml). An abdominopelvic CT
revealed splenomegaly (9.0×23.0 cm; normal, 4.0×12.0 cm). An
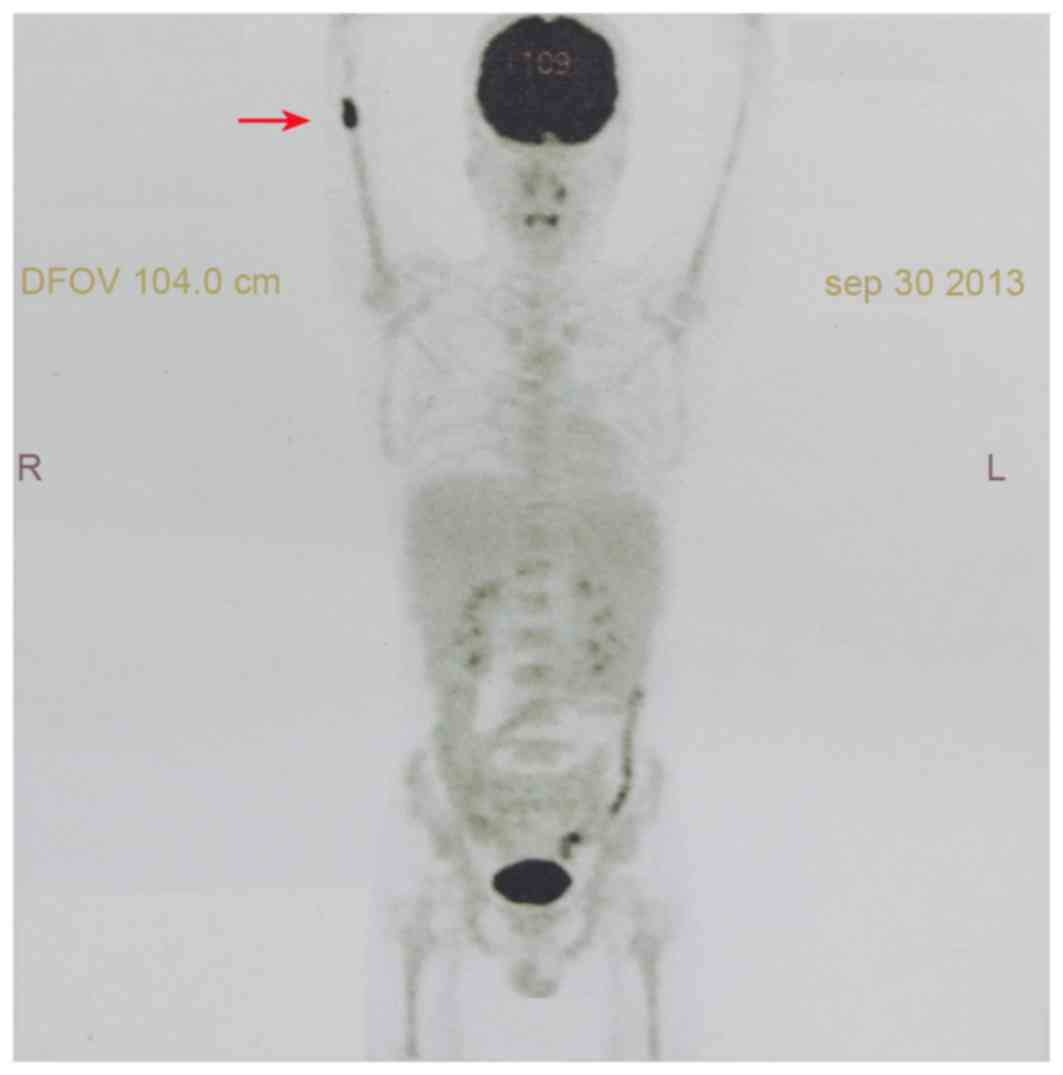
18F-fluorodeoxyglucose (FDG) positron emission
tomography (PET) scan demonstrated an increased uptake of FDG in
medullary space of the right distal humerus (maximum standardized
uptake value, 9.3) without evidence of dissemination at other sites
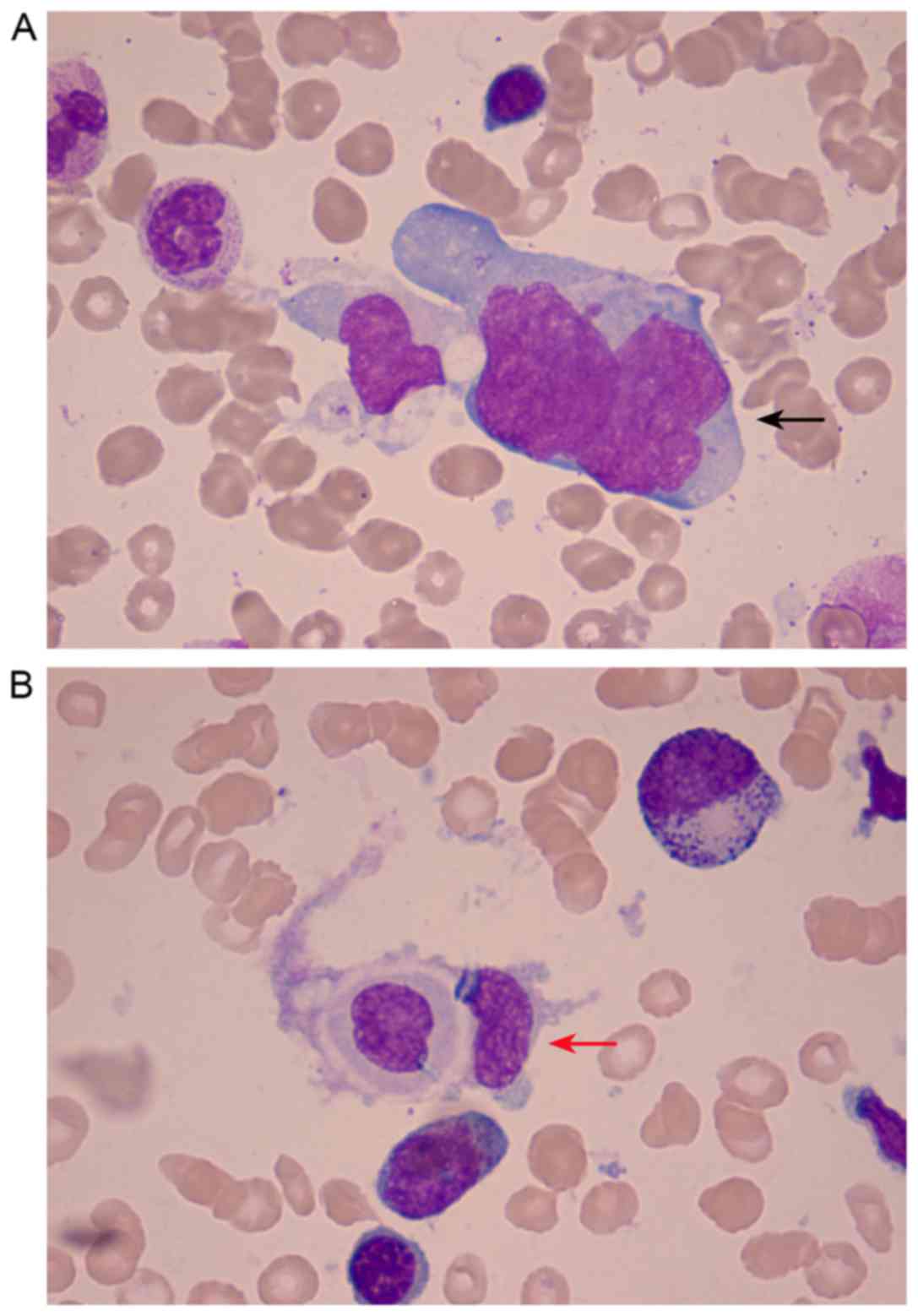
(Fig. 1). BMA, using Wright-Giemsa
staining, revealed 4% lymphoma cells and phagocytes engulfing
hemocytes (optical microscope; magnification, ×1,000) (Fig. 2). Bone marrow biopsy (BMB) was
embedded with paraffin and stained with ready-to-use hematoxylin
and eosin for between 30 and 40 min, and incubated between 24 and
28°C (optical microscope; magnification, ×100) (Fig. 3). The proportion of hematopoietic
tissue in bone marrow increased and the BMB revealed scattered and
focal CD20-positive cells. According to the results of BMB, B cell
lymphoma involving bone marrow was not excluded. Gene rearrangement
detection, using multiplex polymerase chain reaction (IGH PCR
assay; Invivoscribe, San Diego, CA, USA), identified a
rearrangement in immunoglobulin κ
(Vk−Kde+INTR−Kde+). The
bone marrow karyotype was 46, XY [20]. Flow cytometric (FCM)
analysis revealed that CD45+CD19+ cells,
which were potentially abnormal B lymphocytes, expressed human
leukocyte antigen-antigen d related, CD5, CD11c, CD20, CD22, CD38,
limited FCM-7 antigen, no κ- or λ-polyclone, and accounted for 0.5%
of nuclear cells.
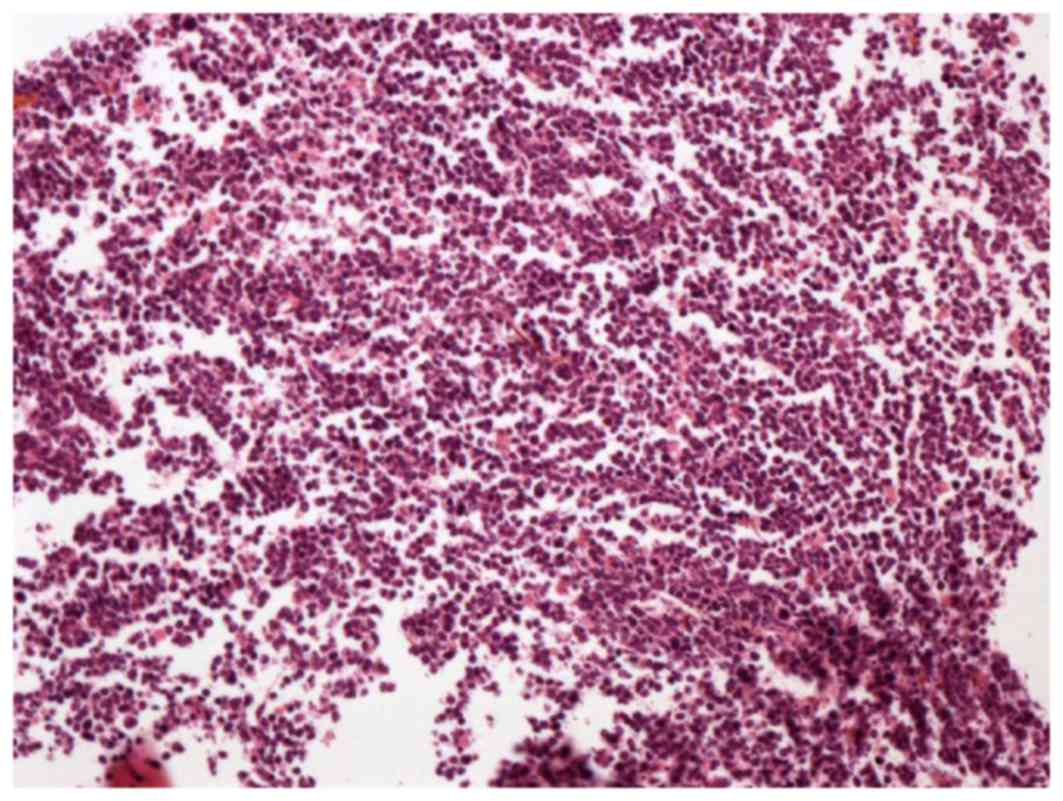 | Figure 3.Biopsy of the mass in the bone marrow
cavity of the right distal humerus revealed abnormal lymphoid cells
gathering in clusters and hemosiderin pigment (hematoxylin and
eosin staining; optical microscope; magnification, ×100). Antigen
Ki-67 index was 60%. The cells were positive for CD5, CD3, CD20,
mutated melanoma-associated antigen 1 and CD99, and negative for
anion exchanger 1/3, B cell lymphoma 6, CD10, myeloperoxidase,
terminal deoxynucleotidyl transferase and cyclin D1. CD, cluster of
differentiation. |
Subsequently, the patient received fenestration
surgery, a biopsy and bone reconstruction on the right distal
humerus. In surgery, it was identified that the bone cortex of the
right distal humerus became thin without periosteal reaction, and a
tender, dark red, isolated mass was identified in the medullary
space. Without the capsule, the size of the mass was ~4.0×2.0×2.0
cm, and it was not able to distinguish the mass from the
surrounding tissue. A curettage biopsy was performed and subsequent
immunohistochemistry (IHC) was performed. The IHC indicated the
following: Gathering of abnormal lymphoid cells in cluster,
hemosiderin pigment, anion exchanger 1/3−,
CD5+, CD3+ (scattered), CD20+,
CD23−, mutated melanoma-associated antigen
1+, B cell lymphoma 6−, CD10−,
myeloperoxidase−, antigen Ki-67 (index, 60%), terminal
deoxynucleotidyl transferase−, CD99+ and
cyclin D1− (Fig. 3). The
patient was diagnosed with B cell LBL and secondary hemophagocytic
lymphohistiocytosis (HLH). Subsequently, the patient was
administered the following chemotherapy: 3 courses of
cyclophosphamide (CTX), epirubicin hydrochloride, vindesine
sulfate, prednisone (CHOP), a course of high-dose methotrexate,
vindesine and pegaspargase (HD-MTX/VL) and a course of etoposide,
ara-c, pegaspargase and CTX (CLEA) (Table
I).
 | Table I.Details of chemotherapy regimen. |
Table I.
Details of chemotherapy regimen.
| Date | Acronym of
regimen | Regimen | Notes |
|---|
| 25/10/13 | CHO | CTX, 1.3 g, day 1;
epirubicin hydrochloride, 130 mg, day 1; vindesine sulfate, 4 mg,
day 1 | Rituximab (100 mg
infusion) was ceased on day 1 due to an allergy, and the regimen
did not include prednisone due to a wound in the right upper
limb |
| 06/11/13 | CHOP | CTX, 1.3 g, day 1;
epirubicin hydrochloride, 130 mg, day 1; vindesine sulfate, 4 mg,
day 1; prednisone, 100 mg, days 1–5 | Lumbar puncture and
intrathecal injection (50 mg cytarabine and 5 mg dexamethasone)
were performed on November 6, 2013. |
| 28/11/13 | CHOP | CTX, 1.3 g, day 1;
epirubicin hydrochloride, 130 mg, day 1; vindesine sulfate, 4 mg,
day 1; prednisone, 100 mg, days 1–5 | N/A |
| 19/12/13 | HD-MTX/VL | MTX, 8.9 g, day 1 and
15; VDS, 4 mg, day 1 and 15; pegaspargase, 3,750 U, day 5 and
19 | Lumbar puncture and
intrathecal injection (50 mg cytarabine and 5 mg dexamethasone)
were performed on December 19, 2013 and January 2, 2014. |
| 23/01/14 | CLEA | VP16, 100 mg, days
1–5; ara-c, 130 mg, days 1–5; pegaspargase, 3,750 U, day 1 and 15;
CTX, 1.8 g, day 1 | Lumbar puncture and
intrathecal injection (50 mg cytarabine and 5 mg dexamethasone)
were performed on January 24, 2014. |
The patient achieved complete remission following 3
courses of CHOP and a course of HD-MTX/VL (Table I), and during treatment, the patient
did not experience discomfort and cerebrospinal fluid was normal.
However, 4 months following the initial diagnosis, the patient
experienced disease relapse, which manifested as acute leukemia.
The patient did not respond to repeated CHOP regimen and succumbed.
The present case report obtained informed consent from the
patient's next of kin.
Discussion
Lymphoma is a type of malignant tumor and originates
from the lymph nodes or extra-nodal lymphoid tissue (5). A study revealed there was 36.4%
extra-nodal involvement in non-Hodgkin lymphoma (NHL) (9). Lymphoma arising directly from bone
marrow is not rare (2). Although
arising from bone marrow, LBL, marginal zone lymphoma and Burkett
lymphoma are not classified as PBML (2–4). Other
types of lymphoma rarely originate from the bone marrow, including
DLBCL and follicular lymphoma (10).
If DLBCL or follicular lymphoma initially arises from bone marrow,
it is termed PBML (10). In
accordance with the World Health Organization's classification of
lymphoid neoplasms and criteria (11), PBML is rare. The most common
histological subtype of PBML is DLBCL, which accounts for 1.16% of
lymphoma and 2.65% of all DLBCL (7).
Patients with BML typically present with diffuse bone marrow
involvement and leukemia syndrome, increased WBC and immature
cells, at the initial diagnosis. To the best of our knowledge,
there was no case report of a patient with BML presenting with an
isolated mass in the bone marrow cavity.
The patient in the present case report presented
with pain in the right distal humerus, a repeated fever of unknown
origin, splenomegaly and pancytopenia. Following the completion of
examinations, the patient was diagnosed with HLH. Secondary HLH was
initially considered due to the age of the patient. Clinicians
attempted to identify the cause of secondary HLH and lymphoma was
included in consideration. Following overall examination and
biopsy, the patient was diagnosed with LBL. LBL which arises from
extra-nodal and extramedullary tissue is not rare; for instance, B
cell LBL typically involves the skin, liver and bone (12,13).
Initially, a number of patients exhibit normal CBC and BMA results
at an early stage. However, patients typically develop diffuse bone
marrow infiltration and acute lymphocytic leukemia (ALL)
transformation in a short time (14).
Other patients with onset of bone marrow LBL, directly present with
diffuse bone marrow involvement and ALL transformation (14). The present case is unique due to the
onset of an isolated mass in the bone marrow cavity. Local pain in
right distal humerus was the only complaint at the early stage and
2 months after the initial presentation, the patient experienced
HLH manifestation. Subsequently, 6 months after the initial
presentation, the patient exhibited diffuse bone marrow involvement
without ALL transformation (6). The
bone marrow karyotype of the patient was normal. The patient
exhibited BML, which did not originate from extramedullary tissue
or diffuse bone marrow, but originated from an isolated mass in the
bone marrow cavity of the right distal humerus. In addition,
pathology revealed B cell LBL with negative Philadelphia
chromosome. According to a search by the authors, BML as an
isolated mass in bone marrow cavity was not reported in PubMed.
The present case was difficult to diagnose due to
the onset of an isolated mass in the bone marrow cavity. At an
early stage, CT did not reveal abnormal findings in the right
distal humerus, and BMA and BMB results were identified as normal.
PET examination served a critical role in identifying the abnormal
region early and enabled treatment to begin, despite the CT
revealing no abnormality. Although BMA and BMB results revealed the
presence of lymphoma cells at a later stage, the number of lymphoma
cells was limited for validation of the pathological type. Surgical
removal and pathology of the primary abnormality were important for
diagnosis and determining the pathological type. Therefore, PET
examination may be used to identify an abnormal region and provide
a target for surgery and pathology, which may help validate a
pathological diagnosis.
Despite the onset of an isolated mass in the bone
marrow cavity, the patient developed bone marrow infiltration at a
later stage. Additionally, bone marrow involvement is an important
characteristic in patients with BML (15). BMA and BMB are important methods to
diagnose this type of disease. The morphology of marrow lymphoma
cells, observed in the patient, was typical of the disease, with
incisura and folding in the round, oval, irregular or rare double
nuclei. The coarse granular chromatin was deep purple and the
nuclear membrane was thick, determined using Wright-Giemsa
staining. There were nucleoli in a limited number of cells, and the
lymphoma cells were rich in cytoplasm. Furthermore, there was
cytoplasmic extension. IHC of BMA and BMB are required for
diagnosis of lymphoma (16). In the
present case, IHC of BMB enabled a definitive diagnosis to be made.
IHC was able to mark the signal molecules on the lymphoma cells,
and the signal molecules may provide information about the
characteristics of tumor cells. For example, as a ligand of
immunoglobulin on the cell membrane, CD5 mediates the adhesion
between cells (17). Therefore,
CD5+ may indicate increased adhesion and invasion of
lymphoma cells. For a number of cases with a limited number of
lymphoma cells involving bone marrow, FCM may assist with diagnosis
(18,19). However, there may be different results
between BMA/BMB and FCM (20,21).
Patients with NHL, in particular those with BML, may
present with HLH. Previous studies have identified that ~43%
(21/49) patients with PBML exhibited HLH (22,23).
Patients with HLH exhibit NK cells with decreased activity,
rendering NK cells unable to eliminate excessive activated T cells,
which release a number of cytokines, including interferon-γ and
interleukin-10. Excessive cytokines may activate an increased
number of macrophages, which serve an important function in
cytophagy, leading to HLH (24,25).
Therefore, patients with HLH manifest with decreased activity of NK
cells and an increased level of CD25, indicating the excessive
activation of T cells. To the best of our knowledge, the underlying
molecular mechanism of HLH remains unknown and requires additional
investigation (26,27). Although the present patient did not
exhibit PBML, the patient presented with symptom of HLH including
fever, pancytopenia, splenomegaly, a decreased activity of NK
cells, an increased level of soluble CD25 and phagocytes engulfing
hemocytes in BMA. The manifestation of splenomegaly at the early
stage was due to HLH, not lymphoma infiltration, as the PET
examination revealed no increased uptake in the spleen. Previous
studies have demonstrated that patients with B cell lymphoma and
HLH exhibit a poor prognosis, with a median survival time between 8
and 11 months (28,29). In the present report, the patient had
a poor prognosis. Although treated with a standard ALL strategy,
the patient rapidly relapsed and succumbed following a transient
complete remission.
BML with the onset of an isolated mass in bone
marrow cavity is extremely rare (14). At the early stage, the patient
presented only with HLH and no local bone damage or diffuse bone
marrow involvement, which made the present case difficult to
diagnose. PET examination enabled a region of focus to be
identified early and guided biopsy, which was necessary to validate
the diagnosis. In spite of treatment with ALL combined
chemotherapy, the present patient relapsed, developed ALL and
succumbed. Therefore, the identification of an appropriate
treatment strategy is required.
References
|
1
|
Lambertenghi-Deliliers G, Annaloro C,
Soligo D, Oriani A, Pozzoli E, Quirici N, Luksch R and Polli EE:
Incidence and histological features of bone marrow involvement in
malignant lymphomas. Ann Hematol. 65:61–65. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cortelazzo S, Ferreri A, Hoelzer D and
Ponzoni M: Lymphoblastic lymphoma. Crit Rev Oncol Hematol.
113:304–317. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Piris MA, Onaindia A and Mollejo M:
Splenic marginal zone lymphoma. Best Pract Res Clin Haematol.
30:56–64. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Linch DC: Burkitt lymphoma in adults. Br J
Haematol. 156:693–703. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Vardiman JW: The World Health Organization
(WHO) classification of tumors of the hematopoietic and lymphoid
tissues: An overview with emphasis on the myeloid neoplasms. Chem
Biol Interact. 184:16–20. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bassan R, Maino E and Cortelazzo S:
Lymphoblastic lymphoma: An updated review on biology, diagnosis,
and treatment. Eur J Haematol. 96:447–460. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chang H, Hung YS, Lin TL, Wang PN, Kuo MC,
Tang TC, Wu JH, Dunn P and Shih LY: Primary bone marrow diffuse
large B cell lymphoma: A case series and review. Ann Hematol.
90:791–796. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kung TA, Smith LB and Chung KC: Atypical
presentation of isolated peripheral T-cell lymphoma in the hand:
Case report. J Hand Surg Am. 39:732–736. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shahid R, Gulzar R, Avesi L, Hassan S,
Danish F and Mirza T: Immunohistochemical Profile of Hodgkin and
Non-Hodgkin Lymphoma. J Coll Physicians Surg Pak. 26:103–107.
2016.PubMed/NCBI
|
|
10
|
Martinez A, Ponzoni M, Agostinelli C,
Hebeda KM, Matutes E, Peccatori J, Campidelli C, Espinet B, Perea
G, Acevedo A, et al: Primary bone marrow lymphoma: An uncommon
extranodal presentation of aggressive non-hodgkin lymphomas. Am J
Surg Pathol. 36:296–304. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Campo E, Swerdlow SH, Harris NL, Pileri S,
Stein H and Jaffe ES: The 2008 WHO classification of lymphoid
neoplasms and beyond: Evolving concepts and practical applications.
Blood. 117:5019–5032. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen CC, Weng HH, Hwang CE, Lu CH, Chen PT
and Gau JP: Acute leukemia presenting with extramedullary diseases
and completely normal hemogram: An extremely unusual manifestation
unique to pre-B ALL. Am J Hematol. 85:729–731. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lee WJ, Moon HR, Won CH, Chang SE, Choi
JH, Moon KC and Lee MW: Precursor B- or T-lymphoblastic lymphoma
presenting with cutaneous involvement: A series of 13 cases
including 7 cases of cutaneous T-lymphoblastic lymphoma. J Am Acad
Dermatol. 70:318–325. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Park JH, Pahk K, Kim S, Lim SM, Cheon GJ,
Park YH, Lee SS and Choe JG: Fluorine-18 fluorodeoxyglucose
positron emission tomography imaging of T-lymphoblastic lymphoma
patients. Oncol Lett. 12:1620–1622. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Alvares CL, Matutes E, Scully MA,
Swansbury J, Min T, Gruszka-Westwood AM, Atkinson S, Hilditch B,
Morilla R, Wotherspoon AC and Catovsky D: Isolated bone marrow
involvement in diffuse large B cell lymphoma: A report of three
cases with review of morphological, immunophenotypic and
cytogenetic findings. Leuk Lymphoma. 45:769–775. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Strauchen JA: Primary bone marrow B-cell
lymphoma: Report of four cases. Mt Sinai J Med. 70:133–138.
2003.PubMed/NCBI
|
|
17
|
Pospisil R, Fitts MG and Mage RG: CD5 is a
potential selecting ligand for B cell surface immunoglobulin
framework region sequences. J Exp Med. 184:1279–1284. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xu Y, McKenna RW and Kroft SH: Comparison
of multiparameter flow cytometry with cluster analysis and
immunohistochemistry for the detection of CD10 in diffuse large
B-Cell lymphomas. Mod Pathol. 15:413–419. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Manabe N, Yamaoka G, Ohnishi H, Arai T,
Nakaishi H, Kajikawa T, Kubota Y, Tanaka T, Kitanaka A, Waki M, et
al: Sub-classification of diffuse large B-cell lymphoma by
semi-quantification of the CD5 expression with flow cytometric
analysis. Rinsho Byori. 50:906–911. 2002.(In Japanese). PubMed/NCBI
|
|
20
|
Sah SP, Matutes E, Wotherspoon AC, Morilla
R and Catovsky D: A comparison of flow cytometry, bone marrow
biopsy, and bone marrow aspirates in the detection of lymphoid
infiltration in B cell disorders. J Clin Pathol. 56:129–132. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yokote T, Akioka T, Oka S, Hara S,
Kobayashi K, Nakajima H, Yamano T, Ikemoto T, Shimizu A, Tsuji M
and Hanafusa T: Flow cytometric immunophenotyping of adult T-cell
leukemia/lymphoma using CD3 gating. Am J Clin Pathol. 124:199–204.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wong KF, Chan JK, Ng CS, Chu YC, Li LP and
Chan CH: Large cell lymphoma with initial presentation in the bone
marrow. Hematol Oncol. 10:261–271. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kajiura D, Yamashita Y and Mori N: Diffuse
large B-cell lymphoma initially manifesting in the bone marrow. Am
J Clin Pathol. 127:762–769. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shimazaki C, Inaba T and Nakagawa M:
B-cell lymphoma-associated hemophagocytic syndrome. Leuk Lymphoma.
38:121–130. 2000.PubMed/NCBI
|
|
25
|
Ohno T, Miyake N, Hada S, Hirose Y, Imura
A, Hori T, Uchiyama T, Saiga T, Mizumoto T and Furukawa H:
Hemophagocytic syndrome in five patients with Epstein-Barr virus
negative B-cell lymphoma. Cancer. 82:1963–1972. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Murase T, Nakamura S, Tashiro K, Suchi T,
Hiraga J, Hayasaki N, Kimura M, Murakami M, Mizoguchi Y, Suzuki T
and Saito H: Malignant histiocytosis-like B-cell lymphoma, a
distinct pathologic variant of intravascular lymphomatosis: A
report of five cases and review of the literature. Br J Haematol.
99:656–664. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Murase T, Nakamura S, Kawauchi K,
Matsuzaki H, Sakai C, Inaba T, Nasu K, Tashiro K, Suchi T and Saito
H: An Asian variant of intravascular large B-cell lymphoma:
Clinical, pathological and cytogenetic approaches to diffuse large
B-cell lymphoma associated with haemophagocytic syndrome. Br J
Haematol. 111:826–834. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Takahashi N, Chubachi A, Miura I, Nakamura
S and Miura AB: Lymphoma-associated hemophagocytic syndrome in
Japan. Rinsho Ketsueki. 40:542–549. 1999.(In Japanese). PubMed/NCBI
|
|
29
|
Shimazaki C, Inaba T, Okano A, Hatsuse M,
Takahashi R, Hirai H, Sudo Y, Ashihara E, Adachi Y, Murakami S, et
al: Clinical characteristics of B-cell lymphoma-associated
hemophagocytic syndrome (B-LAHS): Comparison of CD5+
with CD5-B-LAHS. Intern Med. 40:878–882. 2001. View Article : Google Scholar : PubMed/NCBI
|