Introduction
In women, breast cancer is a major cause of
cancer-associated mortality globally. Each year, ~1.4 million women
are diagnosed with breast cancer, and >0.45 million women
succumb to the disease (1). According
to data from the World Health Organization, since 2008, there has
been an ~20% increase in the number of diagnosed patients with
breast cancer per year. Of the multitude of factors associated with
the tumorigenesis of breast cancer, age is the strongest risk
factor. Unlike numerous cancers that demonstrate an increase in
incidence rate during the fifth decade of life, the incidence rate
for breast cancer increases in the third decade of life, which is
believed to be due to the effects of ovarian hormones on breast
tissue (2–4).
The association between certain hormone levels and
the increased risk of breast cancer indicates the critical function
of hormones in the processes of breast cancer. Estrogens,
particularly 17β-estradiol (E2), have been demonstrated to drive
the tumorigenic processes of breast cancer (5). It has been reported that E2 drives the
tumorigenesis of breast cancer by binding to estrogen receptor α
(ERα) and regulating the expression of the downstream genes
(6–9).
Yager and Davidson (10) described
several potential pathways that may explain how E2 treatment
promotes breast cancer proliferation, migration and invasion.
However, it has also been reported that E2 serves a contradictory
effect on breast cancer cells in a concentration-dependent manner.
Zhao et al (11) demonstrated
that a high concentration of E2 induces apoptosis independent of
the presence of ERα, whereas a low concentration of E2 promotes the
proliferation of breast cancer cells through ERα. A high dose of E2
treatment caused a change in the levels of metastasis-associated
lung adenocarcinoma transcript-1 (non-protein-coding) in MCF7
cells, which consequently caused the inhibition of the
proliferation of breast cancer cells, as well as inhibiting the
migratory, invasive and colony-formation abilities. Further studies
are required to confirm these potential mechanisms.
Stem cells or cells that possess stem-like cell
properties are considered to be fundamental in breast cancer
initiation and progression (12). The
small subpopulation of stem cells that exist within solid tumors,
cancer stem-like cells (CSCs), are heterogeneous and have been
demonstrated to be responsible for the regeneration of breast
tumors (13). In this previous study,
the different mechanisms of CSCs were assessed, including cellular
markers cluster of differentiation
44+/24−/low, aldehyde dehydrogenase 1
expression, and mammosphere formation and self-renewal capacity.
The differential gene expression patterns of breast cancer cells
and the CSCs derived from breast cancer raise the following
question: How does E2 treatment of these two types of cell affect
their physiological processes?
In order to answer this question, in the present
study, the effects of different concentrations of E2 treatment on
breast cancer cells and CSCs were examined. To elucidate the
potential molecular mechanisms underlying the effect of E2 on CSCs,
the levels of the transcription factors associated with
self-renewal capacity were determined. The results of the present
study demonstrated the effects of E2 on CSCs derived from breast
cancer, and the partial underlying molecular mechanism.
Materials and methods
Cell culture
The human breast adenocarcinoma cell line MCF7 was
obtained from the American Type Culture Collection (Manassas, VA,
USA) and frozen in liquid nitrogen (−196°C) in the laboratory.
Cells were kept in 100 cm2 dishes that contained 10 ml
RPMI-1640 medium (Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% fetal bovine serum (Thermo Fisher Scientific,
Inc.) and 1% penicillin/streptomycin (Thermo Fisher Scientific,
Inc.) in a humidified atmosphere containing 5% CO2 at
37°C. The medium was replaced every 3 days.
Culture of CSCs from MCF7 cells
The suspended MCF7 cells were diluted to a density
of 106 cells/ml in sphere-forming medium (SFM; Gibco;
Thermo Fisher Scientific, Inc.) which was supplemented with 10
ng/ml basic fibroblast growth factor (bFGF; PeproTech, Inc., Rocky
Hill, NJ, USA), 20 ng/ml epidermal growth factor (EGF; PeproTech,
Inc.) and 2% B27 (Thermo Fisher Scientific, Inc.). The medium was
half-replaced every 3 days and the cells were passaged every 10–15
days.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
In order to detect the expression levels of ERα,
octamer-binding transcription factor 4 (Oct4), sex-determining
region Y-box 2 (Sox2), Krüppel-like factor 4 (Klf4) and MYC
proto-oncogene (c-Myc), total RNA was isolated using TRIzol reagent
(Thermo Fisher Scientific, Inc.) following the manufacturer's
protocol. Total RNA (0.5 µg) was added to the RT reaction mixture
in a final volume of 25 µl using the RevertAid RT Reverse
Transcription kit (Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. cDNA was used for qPCR using SYBRGreen
SuperMix (Thermo Fisher Scientific, Inc.) on a ABI7500 device
(Applied Biosystems; Thermo Fisher Scientific, Inc.). For each
cycle: 10 sec at 95°C for denaturation, 45 sec at 60°C for
annealing and extension, repeat 35 cycles. The primer pairs used
for amplification were as follows: ERα forward,
5′-CCCACTCAACAGCGTGTCTC-3′ and reverse,
5′-CGTCGATTATCTGAATTTGGCCT-3′; Oct4 forward,
5′-CTGGGTTGATCCTCGGACCT-3′ and reverse, 5′-CCATCGGAGTTGCTCTCCA-3′;
Sox2 forward, 5′-GCCGAGTGGAAACTTTTGTCG-3′ and reverse,
5′-GGCAGCGTGTACTTATCCTTCT-3′; Klf4 forward,
5′-CCCACATGAAGCGACTTCCC-3′ and reverse,
5′-CAGGTCCAGGAGATCGTTGAA-3′; c-Myc forward,
5′-GGCTCCTGGCAAAAGGTCA-3′ and reverse, 5′-CTGCGTAGTTGTGCTGATGT-3′.
For data analysis, the DDCq method was used (14). All experiments were performed three
times.
Cell counting Kit-8 (CCK-8) assay
The proliferation of CSCs was measured using CCK-8
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) according to the
manufacturer's protocol. CSC spheres were signalized using
TrypLE™ Express (Life Technologies, Grand Island, NY,
USA) and a total amount of 5×103 CSCs were seeded and
incubated in 96-well plates for 24 h. Subsequently, the 0, 1, 10 or
50 nM of E2 (Sigma-Aldrich; Merck KGaA) was added and co-incubated
with the CSCs for 1–5 days at 37°C in 5% CO2 incubator.
Each day, 10 µl CCK-8 reagent was added to each well and incubated
for 4 h at 37°C. Absorbance was then measured at 450 nm. All
experiments were performed in triplicate and repeated at least
twice.
Caspase-3/7 activity assay
Target cells were seeded in 96-well plates at a
concentration of 5×103 cells/ml. Following exposure to
0, 1, 10 or 50 nM E2 for 24 h at 37°C, caspase-3/7 activity was
analyzed using Caspase-Glu™3/7 assay kit (Promega
Corporation, Madison, WI, USA) by following the manufacturer's
protocol. Briefly, Caspase-Glu™ reagents were added and
incubated with cells for 1 h at 37°C and the absorbance at a
wavelength at 520–530 nm was determined using a microplate reader
(Synergy 2 Multi-Mode Microplate Reader; BioTek Instruments, Inc.,
Winooski, VT, USA).
Immunofluorescence analysis
Cells were fixed in 4% paraformaldehyde for 15 min
at room temperature, and incubated with PBS supplemented with 0.1%
Triton X-100 for 10 min. Permeabilized cells were blocked with 5%
bovine serum albumin (BSA; Sigma-Aldrich; Merck KGaA) and incubated
with antibody against ERα (cat. no. ab32063; Abcam, Cambridge, UK)
at a dilution of 1:2,000 overnight at 4°C. Cells were washed and
further stained with Alex Fluor® 594-conjugated goat
anti-mouse secondary antibodies (cat. no. R37121; Life
Technologies, Grand Island, NY, USA) at a dilution of 1:1,000 for 2
h in darkness. Following washing with PBS, cells were imaged under
a X71 (U-RFL-T) fluorescence microscope (Olympus Corporation,
Tokyo, Japan) at a magnification of ×400.
Western blot
Cells were pelleted and washed three times with PBS
and resuspended with lysis buffer (50 mM Tris, 150 mM NaCl, 1%
Nonidet P40 (NP-40), and 0.25% sodium deoxycholate). Lysate was
centrifuged for 5 min at 12,000 × g, 4°C to remove cell debris. The
supernatant was removed into a fresh tube before sample buffer was
added (Guangzhou RiboBio Co., Ltd., Guangzhou, China). Following
incubation at 100°C for 10 min, samples were separated SDS-PAGE and
transferred to polyvinylidene difluoride membranes (Bio-Rad,
Hercules, CA, USA), which were pre-treated with PBS containing 5%
BSA and 0.3% Tween 20. Membranes were probed with antibodies
against human ERα (Cat. No.: ab32063), activated caspase-3 (Cat.
No.: ab2302), β-actin (Cat. No.: ab8226), GAPDH (Cat. No.: ab8245)
which were bought from Abcam (Cambridge, UK) at dilution of
1:1,000. The signals were visualized using a enhanced
chemiluminescence substrate (Supersignal West Femto
Luminal/Enhancer Solution; Thermo Fisher Scientific, Inc.) and
blotted on X-ray films in a dark room. To quantify the western
blots, ImageJ software (Version. 1.48a; National Institutes of
Health, Bethesda, MD, USA) was used to quantitatively measure the
bands and normalized using β-actin.
Transwell migration assay
The migration of CSCs was quantified using a
Transwell assay (EMD Millipore, Billerica, MA, USA). Cells
(1×104) were suspended with RPMI-1640 medium containing
0, 1 or 10 nM E2 and seeded onto the surface of the upper chamber.
RPMI-1640 medium supplemented with 10% fetal bovine serum (Life
Technologies, Grand Island, NY, USA) was added to the lower well.
The plates were incubated for 24 h at 37°C and, subsequently,
migrated cells were stained with 0.5% crystal violet at room
temperature for 30 min followed by three washes with PBS and imaged
under a X71 (U-RFL-T) fluorescence microscope (Olympus Corporation)
at a magnification of ×200.
Self-renewal capacity assay
A total of 2×103 signalized CSCs were
plated into 24-well plates. The cells were cultured in the SFM in
the presence of 0, 1 or 10 nM E2 for 7–15 days. The spheres >40
µm in diameter were counted under an X71 (U-RFL-T) fluorescence
microscope (Olympus Corporation) at a magnification of ×40.
Statistical analysis
Data are presented as the mean ± standard error of
the mean. The data were evaluated statistically using one-way
analysis of variance followed by the Tukey test for paired
observations. The two-tailed Student's t-test was used to compare
two groups. P<0.05 was considered to indicate a statistically
significant difference. All experiments were performed at least
three times independently.
Results
CSCs derived from MCF7 express a lower
ERα level compared with MCF7 cells
To isolate the CSC subpopulation from MCF7 cells,
1×106 MCF7 cells were incubated in Dulbecco's modified
Eagle's medium/Ham's F12 supplemented with B27, bFGF and EGF for 25
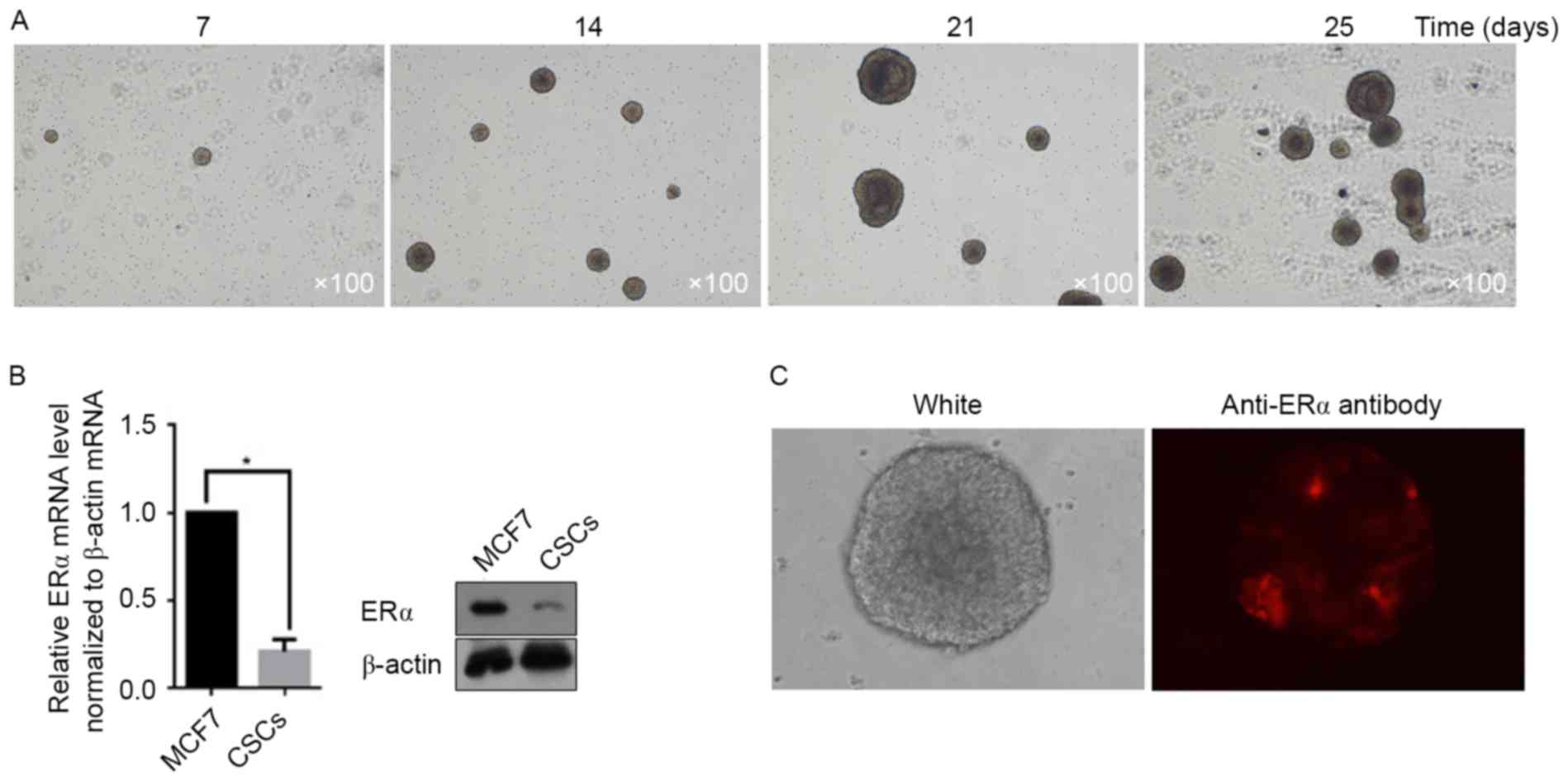
days. Images were taken at days 7, 14, 21 and 25. As presented in
Fig. 1A, the CSC spheres rapidly
increased in size. Owing to the presence of ERα on the surface of
MCF7 cells, the potential for CSCs derived from MCF7 to express ERα
was examined. According to the RT-qPCR and semi-quantitative
western blot assays, the total amount of mRNA and protein from ERα
in CSCs decreased markedly when compared with that in MCF7 cells
(Fig. 1B). In order to determine
whether the decrease in ERα mRNA and protein levels occurred in
each CSC, immunofluorescent staining was utilized to demonstrate
the ERα-positive cells in spheres. As presented in Fig. 1C, a small section of ERα-positive CSCs
in the sphere was detectable, whereas further cells exhibited an
ERα-negative status.
E2 treatment regulates the
proliferation and apoptosis of CSCs in a dose-dependent manner
It has been reported that the effect of E2 treatment
varies depending on the concentration. Low doses or high doses of
E2 treatment have opposing effects on cell proliferation. This
indicates that, considering the decrease in ERα in CSCs, E2
treatment results in differential effects on CSCs according to the
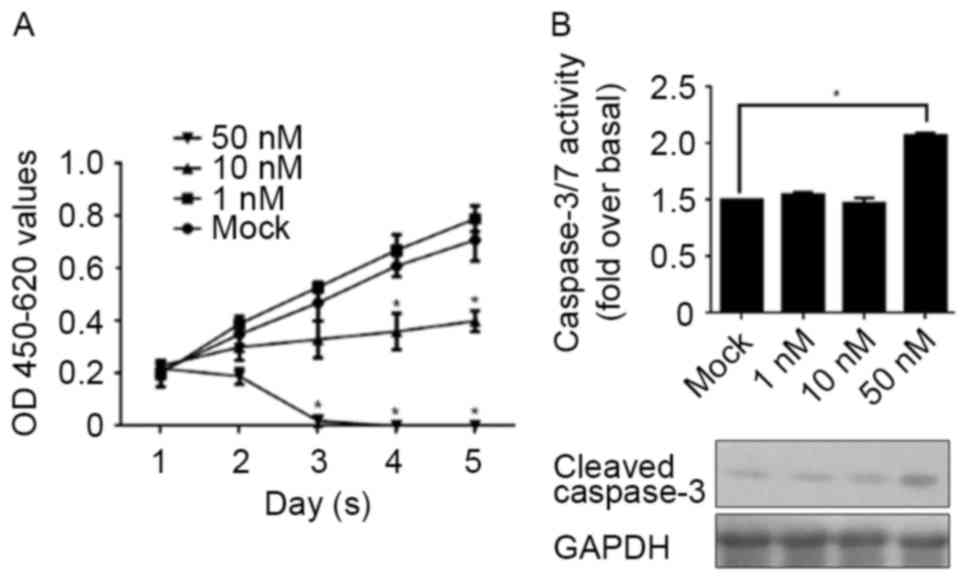
concentration. CSCs were treated with 1, 10 and 50 nM E2 for 1–5
days and assessed using a CCK-8 assay for cell proliferation. In
Fig. 2A, 1 nM E2 treatment was
demonstrated to present no detectable effect on cell proliferation,
whereas 10 nM E2 treatment markedly decreased cell proliferation.
Notably, 50 nM E2 treatment directly eliminated all cells, meaning
that this concentration of E2 treatment is fatal to CSCs. In order
to identify whether the elimination of CSCs following 50 nM E2
treatment was due to the induction of apoptosis, the activity of
caspase-3/7 and the cleaved form of caspase-3 were detected
separately. As expected, the results demonstrated that 50 nM E2
treatment increased the activity of caspase-3/7, accompanied by the
increase in the levels of the cleaved form of caspase-3 (Fig. 2B).
E2 treatment affects migration,
self-renewal capacity and colony formation, potentially due to the
regulation of Sox2
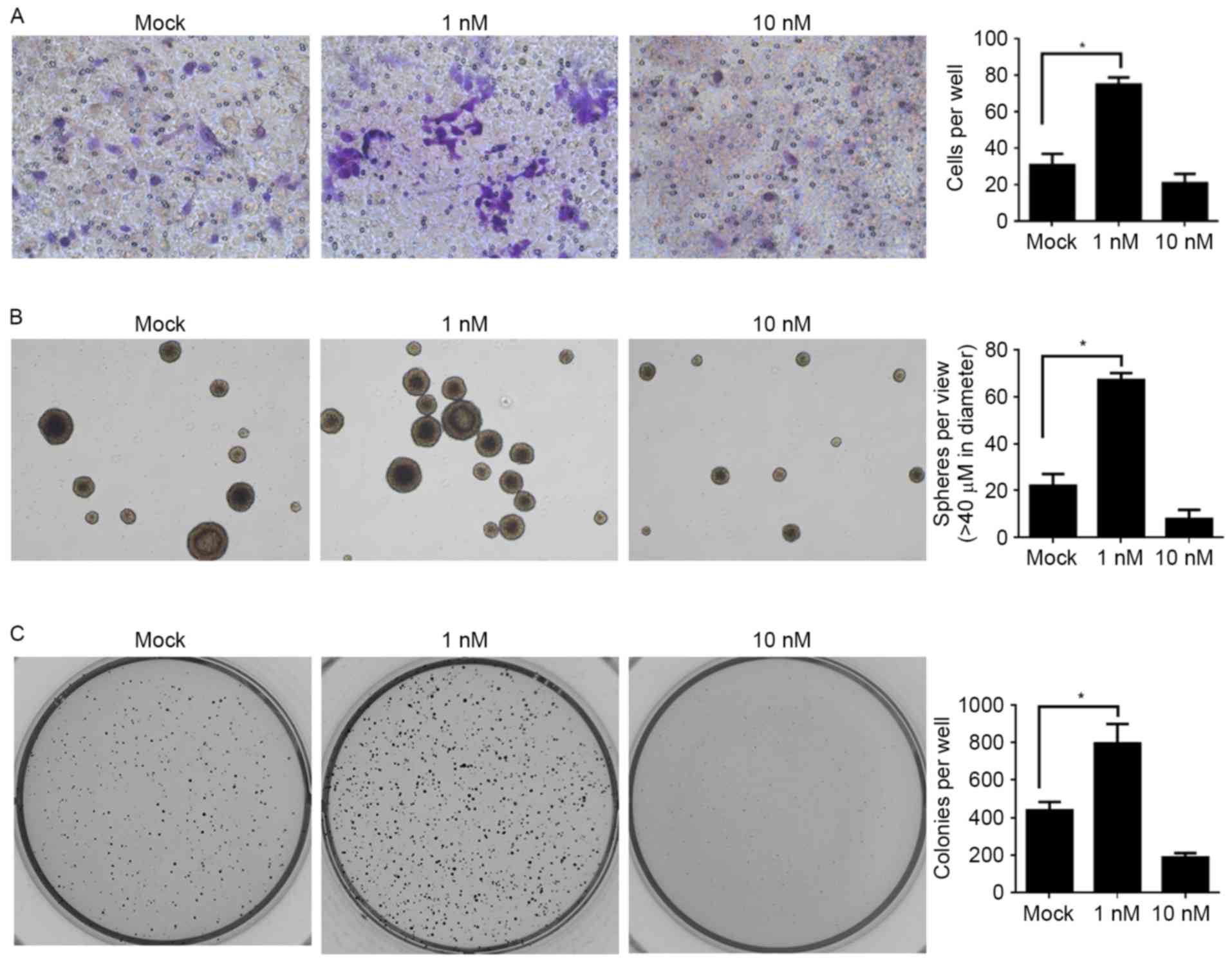
In order to further investigate the effects of E2
treatment on the physiological processes of CSCs, the effects on
migration, self-renewal capacity and colony formation were
assessed. Taking into consideration the fatal effect of the 50 nM
E2 treatment, Mock, 1 and 10 nM E2 treatments were employed for the
following assays: For the migration assay, a Transwell assay
without Matrigel coating was used. Compared with the Mock group,
the 1 nM E2-treated group promoted the migration of CSCs and, in
contrast, the 10 nM E2 treatment inhibited the migration of CSCs,
but the difference was not significant (Fig. 3A). The assays for self-renewal
capacity and colony formation revealed similar tendencies: When
compared with the Mock group, the lower dose of E2 treatment (1 nM)
significantly promoted these processes and the higher dose of E2
treatment (10 nM) inhibited these processes (Fig. 3B and C).
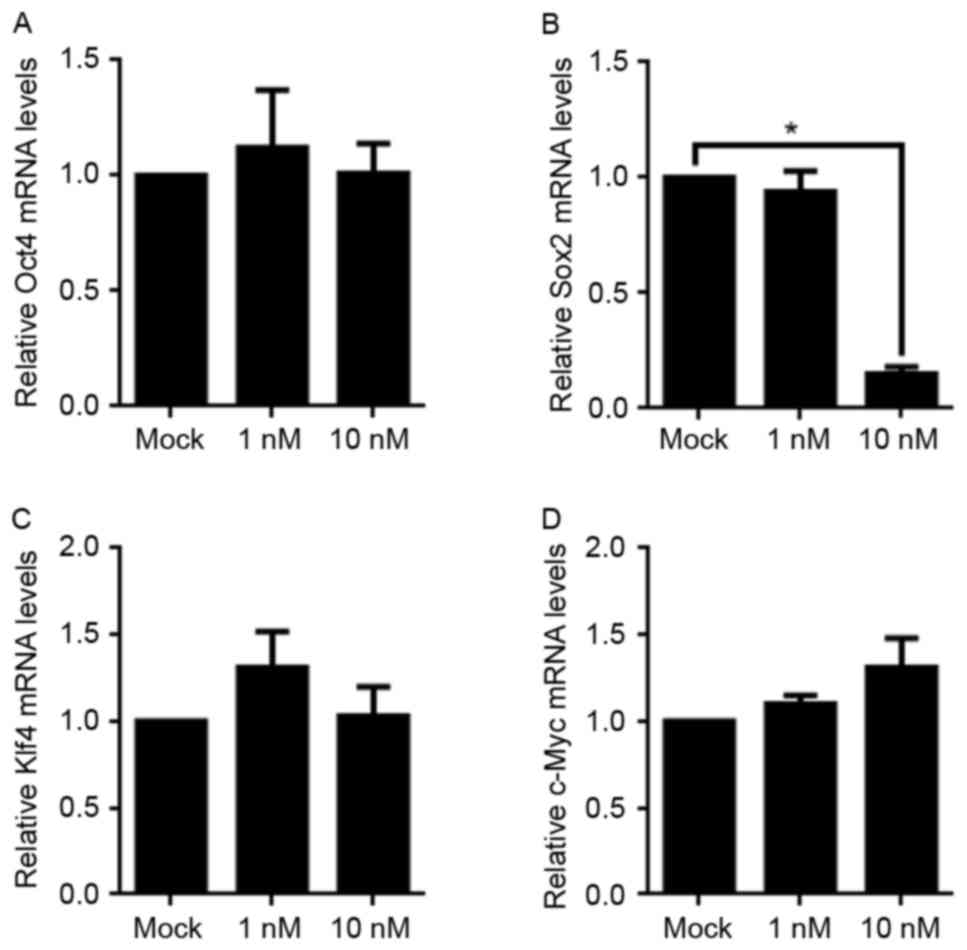
Owing to the fact that Oct4, Sox2, Klf4 and c-Myc
serve critical functions in maintaining cell stemness, the
aforementioned result which revealed the decreased stemness
following 10 nM E2 treatment prompted an interest in detecting the
changes of the mRNA levels of these four factors. Using RT-qPCR,
although Oct4, Klf4, and c-Myc levels were not altered, the mRNA
level of Sox2 was significantly decreased. This indicated that a
decrease in Sox2 mRNA expression may be the potential underlying
molecular mechanism for the loss of stemness following E2 treatment
(Fig. 4).
Discussion
E2 is believed to regulate the physiological
processes of normal breast cells or breast cancer cells, depending
on the presence of ER (15,16). This is supported by the fact that ERα
is frequently highly expressed in ER-positive breast cancer cells,
and thus regulates the cell cycle in these cells, indicating that
E2-ERα signaling serves a critical function in cell proliferation
(17,18). Notably, Zhao et al (11) reported that E2 also performs a
regulatory function on breast cancer cells independent of ERα.
Consistently, in their results, a low level of E2 (1 nM) was
demonstrated to affect the proliferation of ER-positive MCF7 breast
cancer cells, but not that of ER-negative MB231 breast cancer
cells. However, a high dose of E2 (50–100 nM) markedly blocked
proliferation and induced apoptosis in these two types of breast
cancer cell.
In the present study, the difference in ERα
expression between the CSCs derived from MCF7 cells and original
MCF7 cells, and the effects of E2 treatment at a range of
concentrations, were investigated. Initially, CSCs were obtained
using a serum-free maintenance system and a confirmatory assay for
their self-renewal capacity was performed (Fig. 1A). RT-qPCR and semi-quantitative
western blot assays demonstrated that the mRNA and protein levels
of ERα in MCF7 cells were significantly increased compared with
that in CSCs derived from MCF7 cells (Fig. 1B). Notably, the fluorescent staining
of ERα in the CSC sphere demonstrated that a small proportion of
the CSCs presented an ERα-positive signal, indicating the existence
of two subpopulations of CSCs: An ERα-positive subpopulation and an
ERα-negative subpopulation (Fig. 1C).
These two subpopulations of CSCs may be derived from two separate
subpopulations of MCF7, or may be derived from the same population
and subsequently differentiated into two subpopulations.
In the present study, the effects of different
concentrations of E2 on a mixture of ERα-positive and ERα-negative
CSCs were tested due to the failure to separate these two
subpopulations (data not shown). Owing to the unknown ratio of
ERα-positive and ERα-negative CSCs, no detectable promotion of
proliferation was observed following 1 nM E2 treatment and, as
expected, 10 nM E2 treatment resulted in inhibition of
proliferation, and 50 nM E2 treatment directly eliminated cell
viability in 48 h by inducing apoptosis (Fig. 2A and B). Despite the lack of clarity
regarding whether low doses of E2 treatment promote the
proliferation of CSC in these two subpopulations, it was confirmed
that a high dose of E2 treatment is fatal for them, independent of
the existence of ERα.
Because of the fatal effects of 50 nM E2 treatment,
10 nM E2 treatment was employed for further investigation of
migration, self-renewal capacity and colony formation. It was
observed that 10 nM E2 treatment universally inhibited these
processes, whereas 1 nM E2 treatment had an opposing effect on them
(Fig. 3).
The decreased maintenance of stemness of CSCs
prompted an interest in detecting the expressional changes of Oct4,
Sox2, Klf4 and c-Myc, which are critical for stemness maintenance
(19), and it was revealed that the
Sox2 mRNA level was significantly decreased by 50 nM E2 treatment
(Fig. 4).
In summary, the results of the present study
confirmed the regulatory effects of different concentration of E2
treatment on CSCs derived from MCF7. It was identified that there
were two subpopulation of CSCs derived from MCF7 and this may have
resulted in the differential effects following E2 treatment. In
addition, a high dose of E2 treatment may inhibit the malignancy of
CSCs by decreasing their stemness through the downregulation of the
Sox2 expression level.
Acknowledgements
The authors would like to thank Miss Changjin Chen
for the English editing.
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Landis MD, Lehmann BD, Pietenpol JA and
Chang JC: Patient-derived breast tumor xenografts facilitating
personalized cancer therapy. Breast Cancer Res. 15:2012013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hulka BS and Moorman PG: Breast cancer:
Hormones and other risk factors. Maturitas. 38:103–116. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Moore NM and Nagahara LA: Physical biology
in cancer. 1. Cellular physics of cancer metastasis. Am J Physiol
Cell Physiol. 306:C78–C79. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Osborne CK and Schiff R: Estrogen-receptor
biology: Continuing progress and therapeutic implications. J Clin
Oncol. 23:1616–1622. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Doisneau-Sixou SF, Sergio CM, Carroll JS,
Hui R, Musgrove EA and Sutherland RL: Estrogen and antiestrogen
regulation of cell cycle progression in breast cancer cells. Endocr
Relat Cancer. 10:179–186. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cunliffe HE, Ringnér M, Bilke S, Walker
RL, Cheung JM, Chen Y and Meltzer PS: The gene expression response
of breast cancer to growth regulators: Patterns and correlation
with tumor expression profiles. Cancer Res. 63:7158–7166.
2003.PubMed/NCBI
|
|
8
|
Basu A and Rowan BG: Genes related to
estrogen action in reproduction and breast cancer. Front Biosci.
10:2346–2372. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Deroo BJ and Korach KS: Estrogen receptors
and human disease. J Clin Invest. 116:561–570. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yager JD and Davidson NE: Estrogen
carcinogenesis in breast cancer. N Engl J Med. 354:270–282. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhao Z, Chen C, Liu Y and Wu C:
17β-Estradiol treatment inhibits breast cell proliferation,
migration and invasion by decreasing MALAT-1 RNA level. Biochem
Biophys Res Commun. 445:388–393. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lamb R, Lehn S, Rogerson L, Clarke RB and
Landberg G: Cell cycle regulators cyclin D1 and CDK4/6 have
estrogen receptor-dependent divergent functions in breast cancer
migration and stem cell-like activity. Cell Cycle. 12:2384–2394.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Al-Hajj M, Wicha MS, Benito-Hernandez A,
Morrison SJ and Clarke MF: Prospective identification of
tumorigenic breast cancer cells. Proc Natl Acad Sci USA.
100:3983–3988. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Miyoshi Y, Murase K, Saito M, Imamura M
and Oh K: Mechanisms of estrogen receptor-α upregulation in breast
cancers. Med Mol Morphol. 43:193–196. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tyson JJ, Baumann WT, Chen C, Verdugo A,
Tavassoly I, Wang Y, Weiner LM and Clarke R: Dynamic modelling of
oestrogen signalling and cell fate in breast cancer cells. Nat Rev
Cancer. 11:523–532. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Berger CE, Qian Y, Liu G, Chen H and Chen
X: p53, a target of estrogen receptor (ER) α, modulates DNA
damage-induced growth suppression in ER-positive breast cancer
cells. J Biol Chem. 287:30117–30127. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Platet N, Cathiard AM, Gleizes M and
Garcia M: Estrogens and their receptors in breast cancer
progression: A dual role in cancer proliferation and invasion. Crit
Rev Oncol Hematol. 51:55–67. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Takahashi K and Yamanaka S: Induction of
pluripotent stem cells from mouse embryonic and adult fibroblast
cultures by defined factors. Cell. 126:663–676. 2006. View Article : Google Scholar : PubMed/NCBI
|