Introduction
Myelodysplastic syndromes (MDSs) are a heterogeneous
clonal disease originating in hematopoietic stem cells and
characterized by myeloid dysplasia, impaired differentiation, and
peripheral cytopenia (1), with
estimated incidence rate of 4.3 per 100,000 (2). MDS is recognized as a preleukaemic event
in which neoplastic clones are established. It frequently evolves
to acute leukemia in 20–40% of MDS patients (3). MDS originates from multipotent
hematopoietic stem cells, which can give rise to both lymphoid and
myeloid leukemic cells (4). However,
progression of MDS to acute lymphocytic leukemia (ALL) is extremely
rare, occurring in less than 1% of cases (3,5,6). To date, there are only 29 reported cases
of MDS progressing to ALL. The pathogenesis and prognosis of ALL
transformed from MDS is still unclear.
With the advancement of next-generation sequencing,
it is possible to identify new somatic mutations that have a
potential impact on MDS patients. Mutations in additional sex combs
like-1 (ASXL1), an unfavorable prognostic marker, occurs with a
frequency of 10–20% in MDS, with 70% being frameshift mutations and
30% being heterozygous point mutations (7–9).
Frameshift or nonsense mutations correlated with shorter survival
rates and a higher risk of leukemic transformation (8,10,11). However, the clinical significance of
ASXL1 missense mutations is still questionable. Further research to
the role of ASXL1 missense mutations in MDS is required.
In this report, we describe a Chinese male patient
harboring an ASXL1 missense mutation. He was diagnosed with
pro-B-ALL, transformed from MDS with multiple-lineage dysplasia
(MDS-MLD). Furthermore, in order to gain full insight into the
clinical features and treatment outcome of ALL transformed from
MDS, we analyzed previously reported data from 30 patients for such
progression.
Case report
A 33-year-old Chinese man was evaluated for
thrombocytopenia in July 2013. On examination, he had splenomegaly.
Initial blood counts revealed the following; hemoglobin, 123 g/l;
leukocyte count, 7.23×109/l; and platelet count,
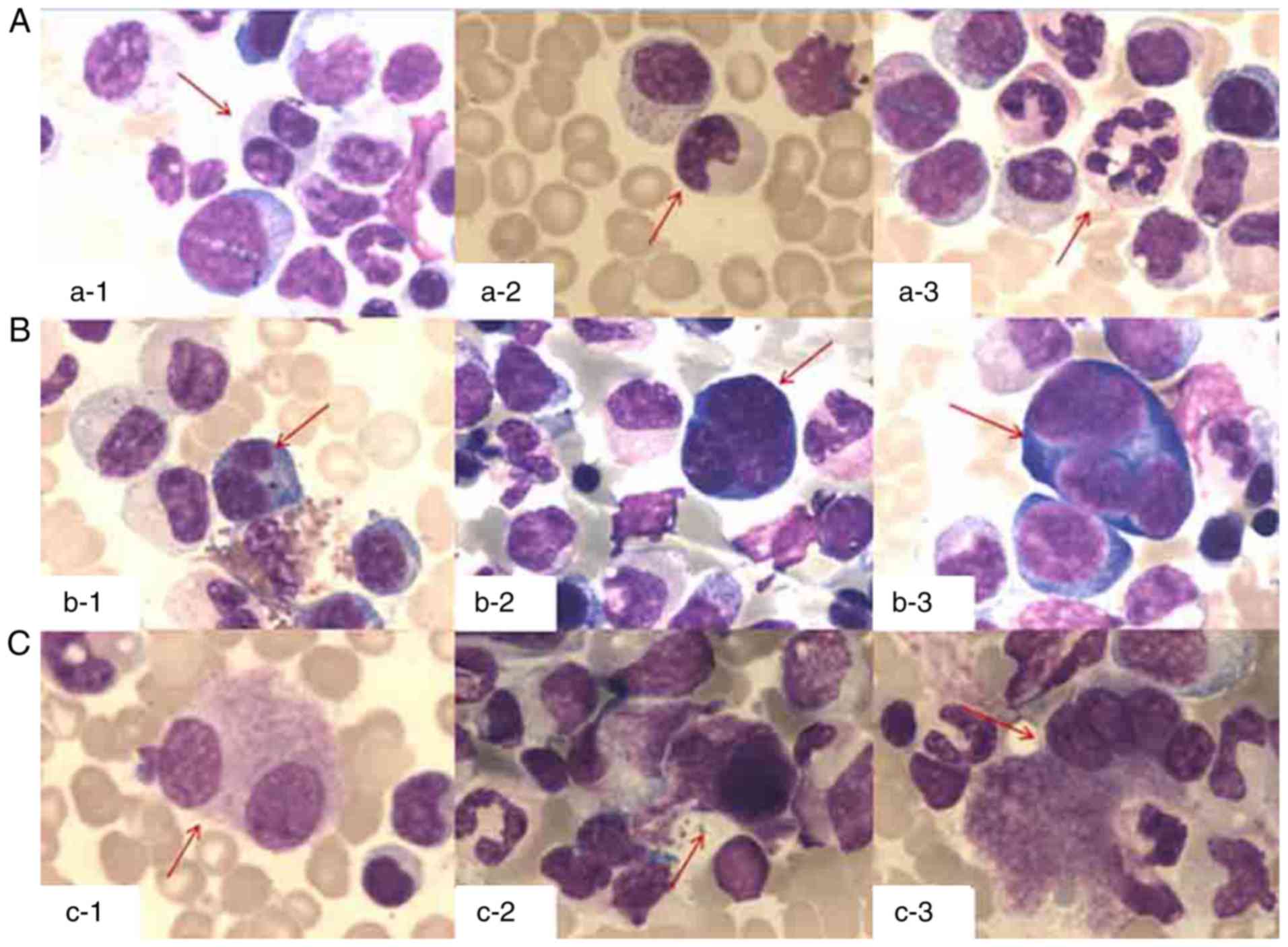
23×109/l. Bone marrow examination (Fig. 1) revealed augmented cellularity and
trilineage dysplasia with no increase in myeloblasts dysplasia.
Cytogenic testing revealed a normal karyotype 46, XY. Using
specific and comprehensive fluorescent in situ hybridization
(FISH) probe panels showed a lack of demonstrable cytogenetic
abnormalities. The patient was diagnosed with MDS-MLD using the
World Health Organization classification. Using the revised
International Prognostic Scoring System (IPSS-R), he was classified
into the low risk category. The patient exhibited persistent
thrombocytopenia with progressing anemia. He did not receive any
treatment.
After a 47-month history of prolonged bleeding and
fatigue, his condition worsened abruptly. Blood counts demonstrated
hemoglobin levels at 49 g/l; platelets at 5×109/l; and
leukocyte counts at 6.56×109/l. Examination of bone
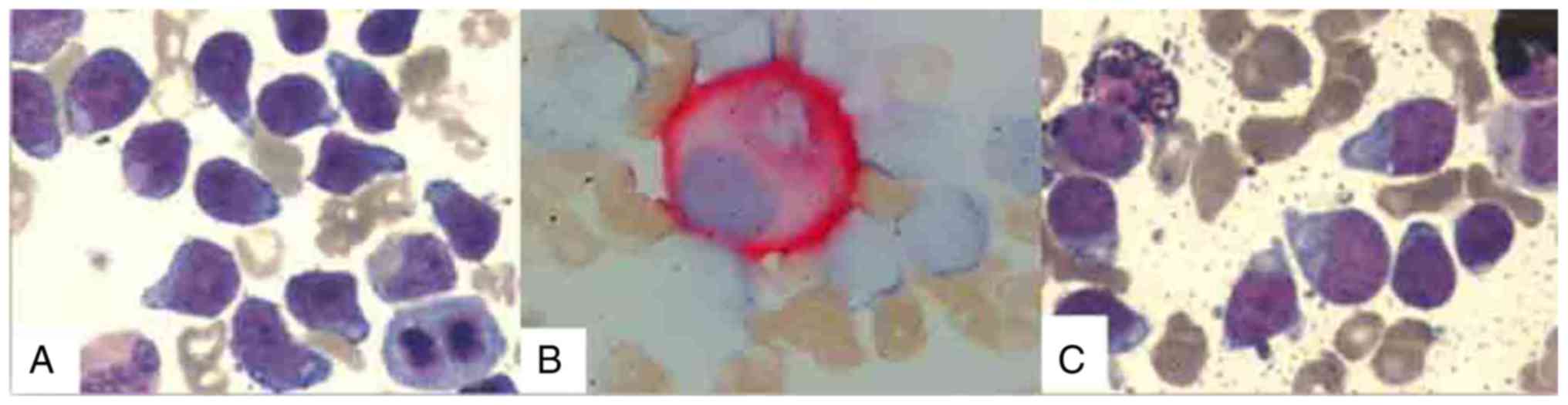
marrow was consistent with a diagnosis of ALL, revealing 63.5%
blasts (Fig. 2A) with numerous
micromegakaryocytes by CD41 monoclonal antibody immune enzyme
staining (Fig. 2B). Cytochemical
staining demonstrated that the blasts were 95% negative for
peroxidase (Fig. 2C). Cell surface
markers analyzed by flow cytometry were positive for CD34, CD19,
cCD79a, CD123, and CD38. The blasts also expressed CD33, a myeloid
marker. Myeloid cell markers for CD117 and CD13 and T cell markers
for cyCD3, CD5, and CD7 were not expressed. The blasts were also
negative for CD10, CD15, CD11b, CD64, CD14, CD20, cTDT, cIgM, CD56
and cyMPO. Based on these markers, the patient was diagnosed with
Pro-B-ALL. Karyotype analysis of bone marrow leukemic cells
revealed normal chromosomes (46, XY). Gene rearrangements were not
detected using reverse transcription-PCR.
In addition, known gene mutations associated with
MDS were screened using next-generation sequencing techniques.
Driver mutant genes include those of RNA splicing (SF3B1,
SRSF2 and U2AF1), DNA methylation (TET2, DNMT3A,
and IDH1/2), chromatin modification (ASXL1 and
EZH2), transcription regulation (RUNX1), DNA repair
(TP53), and signal transduction (NRAS and
KRAS). The patient harbored the point mutation TCC>TTC in
codon 1231 of ASXL1 gene (Fig. 3),
leading to a serine to phenylalanine substitution (S1231F). Other
genetic aberrations were not detected in this patient.
Subsequently, the patient received chemotherapy beginning the day
after bone marrow examination. Treatment regimen included
prednisone, vincristine, cyclophosphamide and idarubicin.
Unfortunately, the patient passed away due to severe pulmonary
infection during the course of chemotherapy. Written informed
consent for publication of data was obtained from patient's
family.
Discussion
We performed an analysis of 30 patients with MDS
transforming to ALL (summary in Table
I). Clinical characteristics were similar with previous reports
in literature (12,13). Patients were predominantly male
(76.7%, 23/30) with a median age of 56 years (3–90 years). The
median time to transformation was 5.5 months (2–50 months). The
common types of MDS with ALL transformation include MDS-excess
blasts (MDS-EB) (40%, 12/30), MDS with single-lineage dysplasia
(MDS-SLD) (30%, 9/30) and MDS with ring sideroblasts (MDS-RS)
(16.7%, 5/30). Majority of the patients transformed to B-cell
(66.7%, 16/24) followed by T-cell (33.3%, 8/24) ALL. Using
immunophenotyping analysis of our 33 year old male patient, we
confirmed the diagnosis of pro-B-ALL due to the bright uniform
expression of CD19 and the absence of CD10 surface antigen. The
coexpression of CD34 and CD33 myeloid antigens was consistent with
the immunophenotypic subtype of pro-B-ALL. Our patient had absence
of MLL1/AF4 and BCR/ABL fusion genes, which correlates with a
significantly better clinical outcome (14). Because of this, our patient was
expected to have a good prognosis. However, after a period of
prolonged fatigue and bleeding, his condition rapidly deteriorated
and he eventually died of severe pulmonary infection due to excess
myelosuppression during ALL-directed chemotherapy. His death may be
attributed to the nature of MDS transformation.
 | Table I.Previously reported cases of lymphoid
transformation of myelodysplastic syndrome. |
Table I.
Previously reported cases of lymphoid
transformation of myelodysplastic syndrome.
| No. | Age (y) | Sex | Time to
transformation (months) | Initial diagnosis of
MDS | Phenotype of blast at
transformation | Chromosomal
abnormalities | Outcome | Survival | (Refs.) |
|---|
| 1 | 68 | M | 31 | RS | B-cell | +8 | Early death | – | (6) |
| 2 | 68 | F | 5 | EB | B-cell | 45XX, −3,
del(5)(q13q31)[4]/46,XX[16] | Death during
induction | – | (17) |
| 3 | 43 | M | 11 | Eosinophilic MDS | B-cell | 45,XY,
del(5q)(q22q23), −7, add(12)(p13)[5] | Death during
induction | – | (18) |
| 4 | 58 | M | 20 | RA | L | not available | Death during
induction | – | (19) |
| 5 | 33 | M | 47 | MLD | B-ALL | Normal | Death during
induction | – | Current case |
| 6 | 67 | M | 18 | EB | T-cell | 45XY, −7 | Fail | – | (15) |
| 7 | 78 | M | 5 | RS | T-cell | – | Fail | – | (15) |
| 8 | 54 | M | 30 | SLD | B-cell |
46,XY,t(9;22)(q34;q11), 20q- | Refractory | – | (20) |
| 9 | 28 | F | 11 | SLD | B-cell | Normal | Refractory | – | (21) |
| 10 | 65 | M | 18 | SLD | B-cell | +13, partially
tetraploid | PR |
| (22) |
| 11 | 53 | F | 2 | MDS with
myelofibrosis | B cell | Normal | CR | Alive | (12) |
| 12 | 3 | M |
| EB | Pre-B-cell |
| CR | – | (23) |
| 13 | 4 | M | NA | EB | ALL | 5q- | CR | 19 months | (24) |
| 14 | 5 | M | 4 | EB | B cell | not available | CR | Alive | (13) |
| 15 | 46 | M | 2 | EB | Pre B cell | – | CR | Alive | (25) |
| 16 | 50 | M | 4 | SLD | B-cell | Complex | CR | – | (26) |
| 17 | 67 | M | 24 | RS | – | Aneuploidy | CR | 12 months | (27) |
| 18 | 53 | M | 50 | RS | Null cell | not available | CR | 11 months | (28) |
| 19 | 20 | M | 5 | EB | T cell | not available | CR | 24 months | (29) |
| 20 | 50 | F | 3 | EB | cALL | Normal | CR | Alive | (30) |
| 21 | 65 | M | 4 | EB | T-cell | 46XY[7]/47XY,
+8[7]/48XY, +8,+13[1]/48XY, +8,+20[1] | CR | 51 months | (15) |
| 22 | 72 | F | 5 | RS | T-cell | Normal | CR | 4.5 months | (15) |
| 23 | 69 | M | 6 | SLD | T-cell | 46XY[9]/47XY,
+8[1] | CR | 15 months | (15) |
| 24 | 72 | M | 5 | EB | T-cell | Normal | CR | 12 months | (15) |
| 25 | 62 | F | 2 | SLD | B-cell | – | CR | 25 months | (31) |
| 26 | 9 | M | 21 | SLD | B-cell | not available | not available | – | (32) |
| 27 | 90 | M | 5 | EB | cALL | not done | not available | – | (33) |
| 28 | 75 | M | 18 | EB | T-cell | Normal | not available | – | (34) |
| 29 | 70 | M | 22 | SLD | B-cell | – | not available | – | (35) |
| 30 | 8 | F | 3 | RCC | B cell | Normal | not available | – | (36) |
Although AML evolved from MDS has a poor prognosis,
ALL following MDS has not been associated with a poor prognosis as
reported in the literature (15).
However, due to the limited number of cases (9 cases), it is
difficult to make any conclusions regarding prognosis. From the 25
cases of MDS transforming to ALL reported in the literature
(Table I), 15 patients achieved
complete remission (CR) and 1 achieved partial remission (PR). Two
cases were refractory and 2 patients failed to achieve a CR after
induction treatment. Four patients died during induction therapy
and 1 died before therapy. Our results confirm that the complete
remission (CR) rate (75%, 15/20) with ALL-directed chemotherapy and
the median remission duration of 15 months (range 4.5 to 51 months)
are similar with previous reports (15). However, ALL following MDS is
characterized by a higher rate of early mortality (20%, 5/25).
Genetic aberrations appear to play an important role
in the pathogenesis of MDS. ASXL1 gene mutations are one of the
characteristic mutations observed for MDS. It encodes a highly
conserved protein that belongs to the enhancer of trithorax and
polycomb (ETP) genes, a gene family with dual functions in both
epigenetic activation and repression of gene transcription
(16). It contains several nuclear
receptor binding motifs and a carboxy-terminal plant homeodomain
(PHD) that is predicted to be truncated by frameshift mutations or
nonsense mutations (10,11). However, the clinical significance of
missense mutations in ASXL1 is unknown. An ASXL1 missense mutation
was detected in our patient. This ASXL1 missense mutation did not
correspond with other gene mutations identified as adverse
prognostic factors. We hypothesize that it may play a substantial
role in the progression of our case. The function of ASXL1 is
expected to be impaired by this missense mutation.
These novel genetic aberrations in MDS need to be
further evaluated regarding their prognostic impact. In our
patient, the time to transformation after primary diagnosis was
much longer than the median observed. It seems that this missense
mutation, unlike frameshift mutations of ASXL1, was not an
unfavorable prognostic marker for patients with MDS, which is
consistent with previous reports (9).
Additional studies needs to be conducted to elucidate the influence
of ASXL1 missense mutations on the prognosis of patients with
MDS.
In summary, we report an ASXL1 missense mutation
potentially leading to MDS-MLD transforming to pro-B ALL. This
mutation might provide insight into the pathogenesis and prognosis
of MDS. ALL following MDS responds well to ALL-directed therapy,
but higher rates of early mortality are observed. The prognosis of
ALL following MDS needs to be further validated using more
uniformly treated patients within a given protocol. Identification
of specific gene mutations will hopefully provide for better
treatment strategies for this group of patients.
Acknowledgements
The authors would like to thanks the faculty members
who analyzed the cytogenetic data.
Funding
Nor applicable.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
LX and ZG analyzed and interpreted the patient data
regarding the hematological disease. ZG was a major contributor in
designing the work and writing the manuscript. YT and XC performed
the PCR and next-generation sequencing. ZX analyzed the flow
cytometric data. JL performed the bone marrow examination. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
The study was granted an exemption from requiring
ethics approval from the ethics committee of the Second Hospital of
Shanxi Medical University.
Consent for publication
Informed consent for publication was obtained from
the family of the patient reported in current study.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Nimer SD: Myelodysplastic syndromes.
Blood. 111:4841–4851. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Visconte V, Selleri C, Maciejewski JP and
Tiu RV: Molecular pathogenesis of myelodysplastic syndromes. Transl
Med UniSa. 8:19–30. 2014.PubMed/NCBI
|
|
3
|
Miguel San JF, Hernández JM,
González-Sarmiento R, González M, Sánchez I, Orfao A, Cañizo MC and
Borrasca López A: Acute leukemia after a primary myelodysplastic
syndrome: Immunophenotypic, genotypic, and clinical
characteristics. Blood. 78:768–774. 1991.PubMed/NCBI
|
|
4
|
Janssen JW, Buschle M, Layton M, Drexler
HG, Lyons J, van den Berghe H, Heimpel H, Kubanek B, Kleihauer E,
Mufti GJ, et al: Clonal analysis of myelodysplastic syndromes:
Evidence of multipotent stem cell origin. Blood. 73:248–254.
1989.PubMed/NCBI
|
|
5
|
Orfao A, Ortuño F, de Santiago M, Lopez A
and Miguel San J: Immunophenotyping of acute leukemias and
myelodysplastic syndromes. Cytometry A. 58:62–71. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Disperati P, Ichim CV, Tkachuk D, Chun K,
Schuh AC and Wells RA: Progression of myelodysplasia to acute
lymphoblastic leukaemia: Implications for disease biology. Leuk
Res. 30:233–239. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gelsi-Boyer V, Trouplin V, Adélaïde J,
Bonansea J, Cervera N, Carbuccia N, Lagarde A, Prebet T, Nezri M,
Sainty D, et al: Mutations of polycomb-associated gene ASXL1 in
myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br
J Haematol. 145:788–800. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Argote Alvarez J and Dasanu CA: ASXL1
mutations in myeloid neoplasms: Pathogenetic considerations, impact
on clinical outcomes and survival. Curr Med Res Opin. 1–7.
2017.(Epub ahead of print).
|
|
9
|
Thol F, Friesen I, Damm F, Yun H,
Weissinger EM, Krauter J, Wagner K, Chaturvedi A, Sharma A,
Wichmann M, et al: Prognostic significance of ASXL1 mutations in
patients with myelodysplastic syndromes. J Clin Oncol.
29:2499–2506. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fisher CL, Pineault N, Brookes C, Helgason
CD, Ohta H, Bodner C, Hess JL, Humphries RK and Brock HW:
Loss-of-function Additional sex combs like 1 mutations disrupt
hematopoiesis but do not cause severe myelodysplasia or leukemia.
Blood. 115:38–46. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vainchenker W, Delhommeau F,
Constantinescu SN and Bernard OA: New mutations and pathogenesis of
myeloproliferative neoplasms. Blood. 118:1723–1735. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Algarni AA, Akhtari M and Fu K:
Myelodysplastic syndrome with myelofibrosis transformed to a
precursor B-cell acute lymphoblastic leukemia: A case report with
review of the literature. Case Rep Hematol.
2012:2075372012.PubMed/NCBI
|
|
13
|
Gupta V and Bhatia B: Transformation of
myelodysplastic syndrome to acute lymphoblastic leukemia in a
child. Indian J Hematol Blood Transfus. 26:111–113. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cimino G, Elia L, Mancini M, Annino L,
Anaclerico B, Fazi P, Vitale A, Specchia G, Di Raimondo F, Recchia
A, et al: Clinico-biologic features and treatment outcome of adult
pro-B-ALL patients enrolled in the GIMEMA 0496 study: Absence of
the ALL1/AF4 and of the BCR/ABL fusion genes correlates with a
significantly better clinical outcome. Blood. 102:2014–2020. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Escudier SM, Albitar M, Robertson LE,
Andreeff M, Pierce S and Kantarjian HM: Acute lymphoblastic
leukemia following preleukemic syndromes in adults. Leukemia.
10:473–477. 1996.PubMed/NCBI
|
|
16
|
Fisher CL, Randazzo F, Humphries RK and
Brock HW: Characterization of Asxl1, a murine homolog of Additional
sex combs, and analysis of the Asx-like gene family. Gene.
369:109–118. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sato N, Nakazato T, Kizaki M, Ikeda Y and
Okamoto S: Transformation of myelodysplastic syndrome to acute
lymphoblastic leukemia: A case report and review of the literature.
Int J Hematol. 79:147–151. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Follows GA, Owen RG, Ashcroft AJ and
Parapia LA: Eosinophilic myelodysplasia transforming to acute
lymphoblastic leukaemia. J Clin Pathol. 52:388–389. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Inoshita T: Acute lymphoblastic leukemia
following myelodysplastic syndrome. Am J Clin Pathol. 84:233–237.
1985. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kohno T, Amenomori T, Atogami S, Sasagawa
I, Nakamura H, Kuriyama K and Tomonaga M: Progression from
myelodysplastic syndrome to acute lymphoblastic leukaemia with
Philadelphia chromosome and p190 BCR-ABL transcript. Br J Haematol.
93:389–391. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bonati A, Delia D and Starcich R:
Progression of a myelodysplastic syndrome to pre-B acute
lymphoblastic leukaemia with unusual phenotype. Br J Haematol.
64:487–491. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ascensao JL, Kay NE, Wright JJ, Arthur D,
Finkel B, Rydell R and Kaplan ME: Lymphoblastic transformation of
myelodysplastic syndrome. Am J Hematol. 22:431–434. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rossbach HC, Sutcliffe MJ, Chamizo W, Haag
MM, Grana NH, Washington KR and Barbosa JL: Pre-B acute
lymphoblastic leukemia in a 3-year-old boy with pre-acute
myelogenous leukemia myelodysplastic syndrome: Cytogenetic evidence
of common early progenitor cell ontogeny. J Pediatr Hematol Oncol.
20:347–352. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bader-Meunier B, Miélot F, Tchernia G,
Buisine J, Delsol G, Duchayne E, Lemerle S, Leverger G, de Lumley L
and Manel AM: Myelodysplastic syndromes in childhood: Report of 49
patients from a French multicentre study. French society of
paediatric haematology and immunology. Br J Haematol. 92:344–350.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Abruzzese E, Buss D, Rainer R, Pettenati
MJ and Rao PN: Progression of a myelodysplastic syndrome to pre-B
acute lymphoblastic leukemia: A case report and cell lineage study.
Ann Hematol. 73:35–38. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ikeda T, Sato K, Yamashita T, Kanai Y,
Kuwada N, Matsumura T, Nakamura Y, Kimura F and Motoyoshi K:
Burkitt's acute lymphoblastic leukaemia transformation after
myelodysplastic syndrome. Br J Haematol. 115:69–71. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hussein KK, Salem Z, Bottomley SS and
Livingston RB: Acute leukemia in idiopathic sideroblastic anemia:
Response to combination chemotherapy. Blood. 59:652–656.
1982.PubMed/NCBI
|
|
28
|
Barton JC, Conrad ME and Parmley RT: Acute
lymphoblastic leukemia in idiopathic refractory sideroblastic
anemia: Evidence for a common lymphoid and myeloid progenitor cell.
Am J Hematol. 9:109–115. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Naithani R, Kumar R, Saxena R and
Mahapatra M: Transformation of myelodysplastic syndrome to T-cell
acute lymphoblastic leukemia in a young adult. Pediatr Hematol
Oncol. 26:100–102. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Berneman ZN, Van Bockstaele D, De Meyer P,
Van der Planken M, Vertessen F, De Bock R and Peetermans ME: A
myelodysplastic syndrome preceding acute lymphoblastic leukaemia.
Br J Haematol. 60:353–354. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lima CS, de Souza CA, Cardinalli IA and
Lorand-Metze I: Lymphoblastic transformation of myelodysplastic
syndrome. Sao Paulo Med J. 115:1508–1512. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Goel R, Kumar R and Bakhshi S:
Transformation of childhood MDS-refractory anemia to acute
lymphoblastic leukemia. J Pediatr Hematol Oncol. 29:725–727. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nagler A, Brenner B and Tatarsky I:
Secondary refractory anemia with excess of blasts in transformation
terminating as acute lymphoblastic leukemia. Acta Haematol.
76:164–165. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pereira AM, de Castro Tavares J, Santos
EG, Perloiro MC and Catovsky D: T lymphoblastic transformation of
refractory anaemia with excess of blasts. Clin Lab Haematol.
7:89–95. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pajor L, Matolcsy A, Vass JA, Méhes G,
Marton E, Szabó F and Iványi JL: Phenotypic and genotypic analyses
of blastic cell population suggest that pure B-lymphoblastic
leukemia may arise from myelodysplastic syndrome. Leuk Res.
22:13–17. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Koh YR, Cho EH, Park SS, Park MY, Lee SM,
Kim IS and Lee EY: A rare case of transformation of childhood
myelodysplastic syndrome to acute lymphoblastic leukemia. Ann Lab
Med. 33:130–135. 2013. View Article : Google Scholar : PubMed/NCBI
|