Introduction
Multidrug resistance (MDR) occurs when cancer cells
are resistant to a number of functionally and structurally
different chemotherapeutic agents, and represents a major obstacle
for successful treatment of cancer (1,2). Efflux
of anticancer drugs by ATP-binding cassette (ABC) transporters
serves a crucial role in the development of the MDR phenotype
(3–5). ABC transporters associated with
chemoresistance include MDR-1, MDR-associated proteins and breast
cancer resistance protein, which are coded for by ABCB1, ABCCs and
ABCG2, respectively (3). ABCB1 is
the most well-known ABC transporter, and is able to extrude natural
toxins, anticancer drugs and drug metabolites across the plasma
membrane to confer an MDR phenotype in cancer cells (6–8).
Similarly, ABCC members have been reported to confer
chemoresistance in cancer cells by translocating a variety of
structurally diverse glutathione conjugates or therapeutic drugs
(4,9). For instance, ABCC1, ABCC2 and ABCC3
cause resistance to hydrophobic anions, including several natural
compounds, whereas ABCC4, ABCC5 and ABCC11 efflux cyclic
nucleotides (10,11). ABCG2, another MDR-associated protein,
is a ubiquitous ABC transporter with an important role in the
distribution, absorption and elimination of its substrate (12,13).
Collectively, these ABC transporters confer the MDR phenotype to
cancer cells by reducing the intracellular concentrations of
anticancer drugs to a nontoxic level. Furthermore, these ABC
transporters are highly expressed in a number of human tumors and
are often associated with poor prognosis (14,15).
Histone deacetylase (HDAC) inhibitors have emerged
as a novel class of anticancer agents due to their significant
anticancer activities, including angiogenesis inhibition, and the
promotion of cell cycle arrest, differentiation and apoptosis
(16,17). HDAC inhibitors may serve as potent
anticancer drugs due to their broad antitumor activity and low
toxicity in normal cells (18).
Several HDAC inhibitors, including trichostatin A (TSA),
suberoylanilide hydroxamic acid (SAHA; also known as vorinostat)
and sodium butyrate, have exhibited potent anticancer activities in
various cancer cells (19). SAHA has
been approved by the US Food and Drug Administration as a treatment
for cutaneous T-cell lymphoma (20).
Furthermore, HDAC inhibitors have been demonstrated to work
synergistically with a variety of antitumor agents, including
gemcitabine, doxorubicin, etoposide, paclitaxel and cisplatin
(21).
Our previous study and other previous research have
demonstrated that treatment of cancer cells with HDAC inhibitors
increases the expression of ABCB1, which results in the development
of an MDR phenotype (22,23). However, the effects of HDAC
inhibitors on other MDR-associated ABC transporters have not
previously been reported. Thus, the aim of the present study was to
investigate the effects of two HDAC inhibitors, namely SAHA and
TSA, on the expression levels of ABCB1, ABCCs and ABCG2 in lung
cancer A549 and colorectal cancer HCT116 cells. The results
indicated that HDAC inhibitors are able to induce differential
expression of these ABC transporters. The present study suggests
that more attention should be paid to drug combinations with HDAC
inhibitors, as ABC transporters have different substrates. DNA
alkylators are substrates of ABCC1. A decrease in ABCC1 protein
level may contribute to the synergism of HDAC inhibitors and DNA
alkylating agents. Conversely, HDAC inhibitors may antagonize the
efficacy of anticancer drugs that are substrates of ABCB1 (14). These differential effects of HDAC
inhibitors on the expression levels of drug transporters support
the necessity for caution in combining these drugs with other
chemotherapeutic agents.
Materials and methods
Chemicals and reagents
Oxaliplatin, 5-fluorouracil, SAHA and TSA were
purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
Primary antibodies against the following were purchased: ABCB1
(cat. no. 13978), purchased from Cell Signaling Technology, Inc.
(MA, Danvers, USA); α-tubulin (cat. no. sc-134237), obtained from
Santa Cruz Biotechnology, Inc. (Dallas, TX, USA); ABCC2 (cat. no.
24893-1-AP), ABCC5 (cat. no. 19503-1-AP) and ABCC6 (cat. no.
27848-1-AP) obtained from ProteinTech Group, Inc. (Chicago, IL,
USA); ABCC1 (cat. no. BS7474) and ABCG2 (cat. no. BS3482) from
Bioworld Technology Co., Ltd. (Nanjing, China); ABCC3 (cat. no.
ab3375), ABCC4 (cat. no. ab77184), ABCC10 (cat. no. ab91451),
ABCC11 (cat. no. ab98979) and ABCC12 (cat. no. ab91453) from Abcam
(Cambridge, MA, USA); HDAC1 (cat. no. ET1605-35), HDAC2 (cat. no.
ET1607-78), HDAC3 (cat. no. ET1610-5) and HDAC4 (cat. no.
ET1612-51), purchased from HuaAn Biotechnology Co., Ltd. (Hangzhou,
China). Horseradish peroxidase (HRP)-conjugated secondary antibody
(cat. nos. SH001X, SH002X and SH003X) were also purchased from
DingGuo Biotechnology Co., Ltd. (Guangzhou, China). The
PrimeScript® RT reagent and SYBR® Premix Ex
Taq™ kits were purchased from Takara Bio, Inc. (Otsu,
Japan).
Cell culture
A549 and HCT116 cells were obtained from the Type
Culture Collection of the Chinese Academy of Sciences (Shanghai,
China). HCT116 cells were maintained in Dulbecco's modified Eagle's
medium/F12 culture medium (Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS).
A549 cells were cultured in RPMI-1640 culture medium (Gibco; Thermo
Fisher Scientific, Inc.) supplemented with 10% FBS. The two cell
lines were cultured in an incubator at 37°C with an atmosphere of
5% CO2.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) detection
A549 and HCT116 cells were treated with SAHA (0.5
µM) or TSA (100 nM) at 37°C for 24 h. Treatment with DMSO (equal
volume added) was used as the control. Total mRNA was extracted
from A549 and HCT116 cells using TRIzol reagent. RNA sample
concentration was measured using a UV spectrophotometer, and
optical density (OD)260/OD280 was limited to 1.8–2.0. A total of
500 ng RNA was used for cDNA synthesis with the
PrimeScript® RT reagent kit. Subsequently, qPCR was
performed on an ABI 7500 Real-Time PCR system (Thermo Fisher
Scientific, Inc.) using the Takara SYBR® Premix Ex Taq™
kit to quantify the expression of target genes. Primers used in
qPCR experiments are presented in Table
I, with GAPDH serving as an internal reference. The thermal
cycling conditions for qPCR were as follows: Holding stage
conducted at 95°C for 30 sec; cycling stage conducted at 95°C for 5
sec and 60°C for 34 sec for 40 cycles; melt curve stage conducted
at 95°C for 15 sec, 60°C for 60 sec, 95°C for 30 sec and 60°C for
15 sec. Following normalization to the GAPDH gene, the expression
of each target gene was calculated using the comparative cycle
quantification (Cq) method (24). In
correlation analysis, the ΔCq values were calculated according to
the following formula: ΔCq=Cq (gene of interest)-Cq (GAPDH). For
determination of the relative expression, the 2−ΔΔCq
value was calculated according to the following formula: ΔΔCq=ΔCq
(control group)-ΔCq (experimental group).
 | Table I.Primers used in reverse
transcription-quantitative polymerase chain reaction assay. |
Table I.
Primers used in reverse
transcription-quantitative polymerase chain reaction assay.
| Gene | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| ABCB1 |
TGCTCAGACAGGATGTGAGTTG |
AATTACAGCAAGCCTGGAACC |
| ABCC1 |
GCCAAGAAGGAGGAGACC |
AGGAAGATGCTGAGGAAGG |
| ABCC2 |
TGGTGGCAACCTGAGCATAGG |
ACTCGTTTTGGATGGTCGTCTG |
| ABCC3 |
CTTAAGACTTCCCCTCAACATGC |
GGTCAAGTTCCTCTTGGCTC |
| ABCC4 |
GGTTCCCCTTGGAATCATTT |
AATCCTGGTGTGCATCAAACAG |
| ABCC5 |
ACCCGTTGTTGCCATCTTAG |
GCTTTGACCCAGGCATACAT |
| ABCC6 |
GTGGTGTTTGCTGTCCACAC |
ACGACACCAGGGTCAACTTC |
| ABCC10 |
ATTGCCCATAGGCTCAACAC |
AGCAGCCAGCACCTCTGTAT |
| ABCC11 |
GGCTGAGCTACTGGTTGGAG |
TGGTGAAAATCCCTGAGGAG |
| ABCC12 |
GGTGTTCATGCTGGTGTTTGG |
GCTCGTCCATATCCTTGGAA |
| ABCG2 |
TATAGCTCAGATCATTGTCACAGTC |
GTTGGTCGTCAGGAAGAAGAG |
| GAPDH |
GCACCGTCAAGGCTGAGAAC |
TGGTGAAGACGCCAGTGGA |
Western blotting
A549 and HCT116 cells were treated with SAHA (0.5
µM) or TSA (100 nM) at 37°C for 24 h. DMSO treatment was used as
the control. A549 and HCT116 cells were washed three times with
ice-cold PBS and subsequently lysed using western blotting lysis
buffer, containing 50 mM Tris-HCl (pH 7.6), 150 mM NaCl, 1 mM EDTA,
1% NP-40, 0.5% Na-deoxycholate, 5 µg/ml aprotinin, 5 µg/ml
leupeptin and 1 mM phenylmethylsulphonyl fluoride. Cell lysates
were cleared by centrifugation at 12,000 × g at 4°C for 30 min and
denatured by boiling in Laemmli buffer. Bovine serum albumin was
used as the standard for determining the protein concentration
using a Bradford protein assay (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) Equal amounts of protein samples were loaded
onto 8% gels and separated by SDS-PAGE, following which they were
electrophoretically transferred onto polyvinylidene difluoride
(PVDF) membranes. Following blocking with 5% non-fat milk for 2 h
at room temperature, the PVDF membranes were incubated with primary
antibodies (ABCB1, ABCC1, ABCC2, ABCC4, ABCC5, ABCC6, ABCG2,
α-tubulin, HDAC1, HDAC2, HDAC3 and HDAC4 at a dilution of 1:1,000;
ABCC3, ABCC10, ABCC11 and ABCC12 at a dilution of 1:50) at 4°C
overnight. Membranes were then incubated with HRP-conjugated
secondary antibodies (1:5,000 dilution) for 1.5 h at room
temperature. Specific immune complexes were detected using Western
Blotting Plus Chemiluminescence Reagent (Thermo Fisher Scientific,
Inc.). Band intensity was quantified by densitometry analysis using
Image-Pro Plus version 4.5 software (Media Cybernetics, Inc.,
Rockville, MD, USA).
Cell Counting Kit (CCK)-8 assay
Cell viability was measured using a CCK-8 assay.
Briefly, A549 and HCT116 cells (1×104/well) were seeded
into 96-well plates in medium supplemented with 10% FBS (Gibco;
Thermo Fisher Scientific, Inc.). The cells were treated with
different concentrations of SAHA (0, 0.25, 0.5 1, 2 and 4 µM) or
TSA (0, 50, 100, 200, 400 and 800 nM) at 37°C for 48 h. Then, 10 µl
CCK-8 solution (Dojindo Molecular Technologies, Inc., Kumamoto,
Japan) was added to 100 µl medium. The cells were incubated with
CCK-8 in medium at 37°C for 2 h, and the absorbance was then
measured at 450 nm using a microplate reader (Bio-Rad Laboratories,
Inc.).
To investigate the effect of SAHA and TSA on
chemoresistance, HCT116 cells were treated with SAHA (0.5 µM) or
TSA (100 nM) at 37°C for 24 h, then the media were removed and the
cells were treated with different concentrations of oxaliplatin (0,
5, 10, 20, 40 and 80 µg/ml) or 5-fluorouracil (0, 2.5, 5, 10, 20
and 40 µg/ml) at 37°C for 48 h. DMSO treatment was used as a
control. The cells were diluted with 10 µl CCK-8 in medium at 37°C
for 2 h, and the absorbance was then measured at 450 nm using a
microplate reader.
Statistical analysis
Data are presented as the mean ± standard deviation
of three independent experiments. One-way analysis of variance was
used to assess the differences among multiple groups. Data were
analyzed using two-tailed unpaired Student's t-tests for
differences between any two groups. These analyses were performed
using GraphPad Prism software, version 5.0 (GraphPad Software,
Inc., La Jolla, CA, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
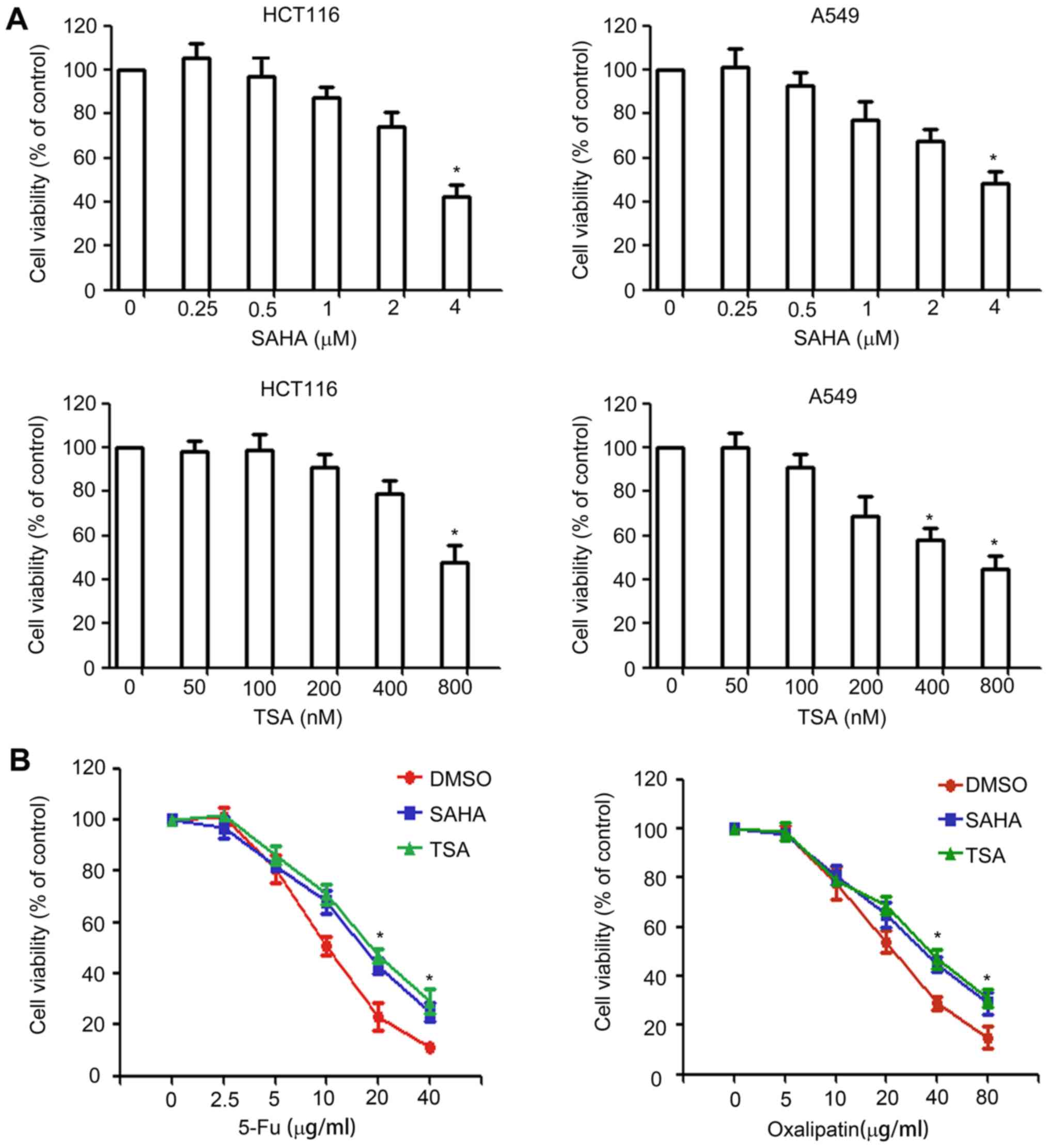
HDAC inhibitors induce drug resistance
in HCT116 cells
HDAC inhibitors are known to have a potent
anticancer activity. The present study attempted to investigate the
effect of two HDAC inhibitors, SAHA and TSA, on the expression of
ABC transporters and drug resistance in cancer cells. The
concentration of SAHA and TSA used to treat cancer cells should
have no significant effect on cell survival. In order to determine
the appropriate concentration, SAHA and TSA at concentrations of
0–4 µM and 0–800 nM, respectively, were used in the cell viability
assay, which are below their IC50 values. HCT116 and
A549 cells were treated with different concentrations of SAHA or
TSA for 48 h, and the cell viability was assessed using a CCK-8
assay. As shown in Fig. 1A, slight
inhibition was observed for concentrations >0.5 µM for SAHA and
>100 nM for TSA. Therefore, 0.5 µM SAHA and 100 nM TSA were
selected for use in subsequent experiments.
Next, the study investigated whether HDAC inhibitors
affect drug resistance by performing a CCK-8 assay. HCT116 cells
were pretreated with 0.5 µM SAHA or 100 nM TSA for 24 h, following
which cells were exposed to various concentrations of oxaliplatin
or 5-fluorouracil for 48 h, and cell viability was assessed using a
CCK-8 assay. The results revealed that, compared with the
DMSO-treated group, SAHA and TSA pretreatment significantly
decreased the sensitivity of HCT116 cells to anticancer drugs
(Fig. 1B).
Effect of HDAC inhibitors on ABCB1,
ABCC5, ABCC6, ABCC10, ABCC11 and ABCC12 expression
ABC transporter-induced drug efflux is closely
associated with acquisition of chemo-resistance in cancer cells
(3). To investigate the effect of
HDAC inhibitors on the expression of ABC transporters, HCT116 and
A549 cells were treated with 0.5 µM SAHA and 100 nM TSA for 24 h.
The mRNA and protein expression levels of various ABC transporters
were detected using RT-qPCR and western blotting, respectively. The
RT-qPCR results indicated that SAHA markedly upregulated mRNA
expression of ABCB1, ABCC5, ABCC10, ABCC11 and ABCC12 in A549 cells
and ABCB1, ABCC10 and ABCC12 in HCT116 cells. TSA markedly
upregulated mRNA levels of ABCB1, ABCC5, ABCC6, ABCC10, ABCC11 and
ABCC12 in A549 cells and HCT116 cells (Fig. 2A). Similarly, the results of western
blotting revealed that SAHA and TSA upregulated ABCB1, ABCC10,
ABCC11 and ABCC12 protein expression. However, SAHA and TSA had no
clear effect on the expression levels of ABCC5 and ABCC6 (Fig. 2B).
 | Figure 2.Histone deacetylase inhibitors
increased the ABCB1, ABCC5, ABCC6, ABCC10, ABCC11 and ABCC12
expression levels. A549 and HCT116 cells were treated with DMSO,
SAHA (0.5 µM) or TSA (100 nM) for 24 h. (A) mRNA expression levels
of ABCB1, ABCC5, ABCC6, ABCC10, ABCC11 and ABCC12 were measured
using reverse transcription-quantitative polymerase chain reaction.
(B) Protein expression levels of ABCB1, ABCC5, ABCC6, ABCC10,
ABCC11 and ABCC12 were detected using western blotting. *P<0.05
and **P<0.01, vs. DMSO group. ABC, ATP-binding cassette; SAHA,
suberoylanilide hydroxamic acid; TSA, trichostatin A. |
Effect of HDAC inhibitors on ABCC1,
ABCC2, ABCC3, ABCC4 and ABCG2 expression
Treatment with HDAC inhibitors affected the
expression of ABCC1, ABCC2, ABCC2 and ABCC4 differently, as
compared with ABCB1, ABCC5, ABCC6, ABCC10, ABCC11 and ABCC12. It
was observed that SAHA and TSA treatment downregulated the
expression levels of ABCC1, ABCC3 and ABCC4 mRNA (Fig. 3A). Similarly, the western blotting
results indicated that the ABCC1 and ABCC3 protein expression
levels were also decreased (Fig.
3B). Notably, while SAHA and TSA decreased ABCC2 mRNA
expression, its protein expression in A549 and HCT116 cells was
found to be increased (Fig. 3A and
B). SAHA and TSA exerted no obvious effects on the protein
expression levels of ABCC4 (Fig.
3B).
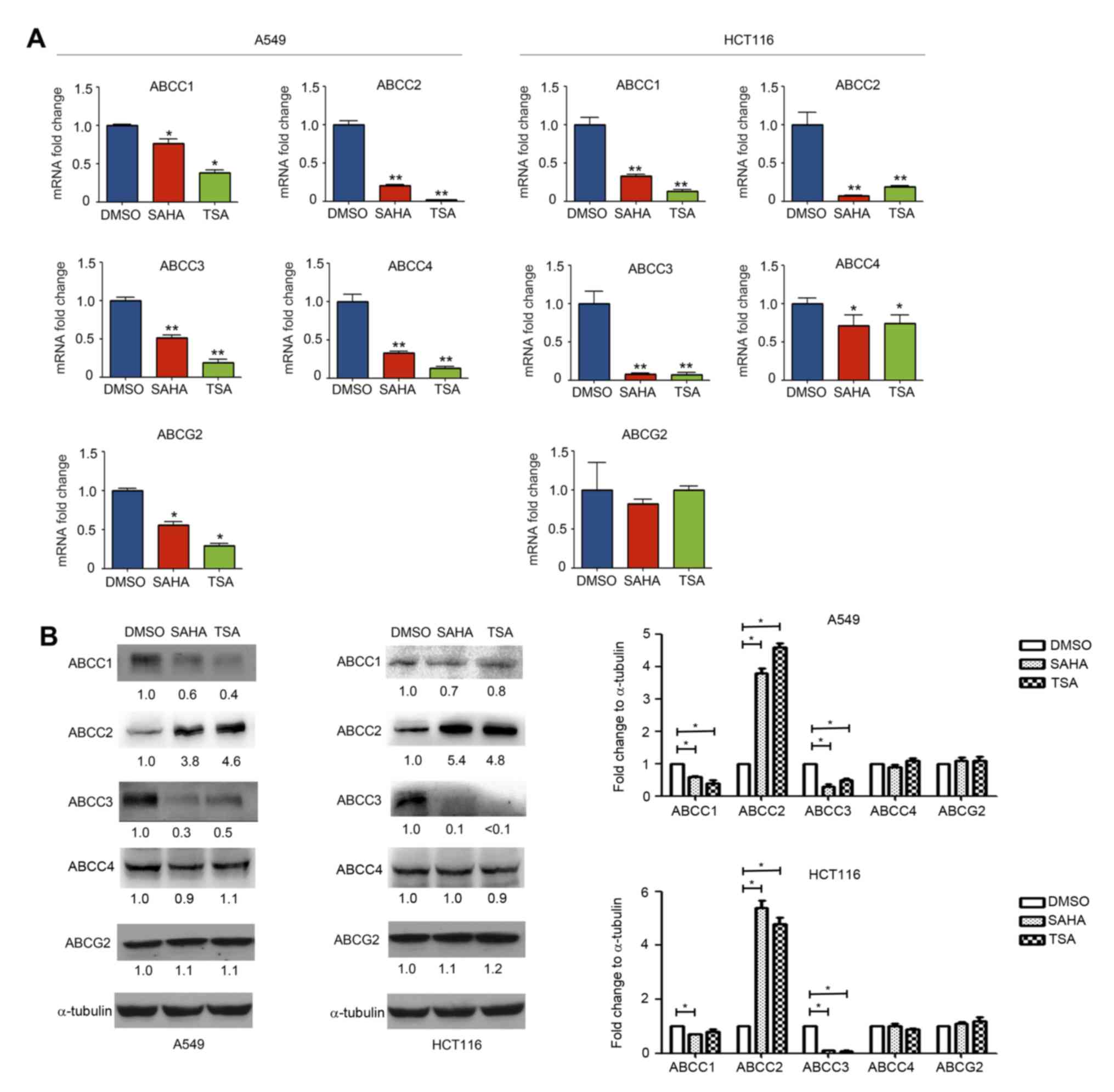 | Figure 3.Effect of histone deacetylase
inhibitors on ABCC1, ABCC2, ABCC3, ABCC4 and ABCG2 expression
levels. A549 and HCT116 cells were treated with DMSO, SAHA (0.5 µM)
or TSA (100 nM) for 24 h. (A) mRNA and (B) protein expression
levels of ABCC1, ABCC2, ABCC3, ABCC4 and ABCG2 were measured using
reverse transcription-quantitative polymerase chain reaction and
western blotting, respectively. *P<0.05 and **P<0.01, vs.
DMSO group. ABC, ATP-binding cassette; SAHA, suberoylanilide
hydroxamic acid; TSA, trichostatin A. |
The effect of HDAC inhibitors on ABCG2 expression
was further investigated in A549 and HCT116 cells. As shown in
Fig. 3A, SAHA and TSA downregulated
ABCG2 mRNA expression in A549 cells, while no marked effect was
observed in HCT116 cells. Furthermore, western blotting revealed
that SAHA and TSA had no evident effect on the expression of ABCG2
protein in A549 and HCT116 cells (Fig.
3B).
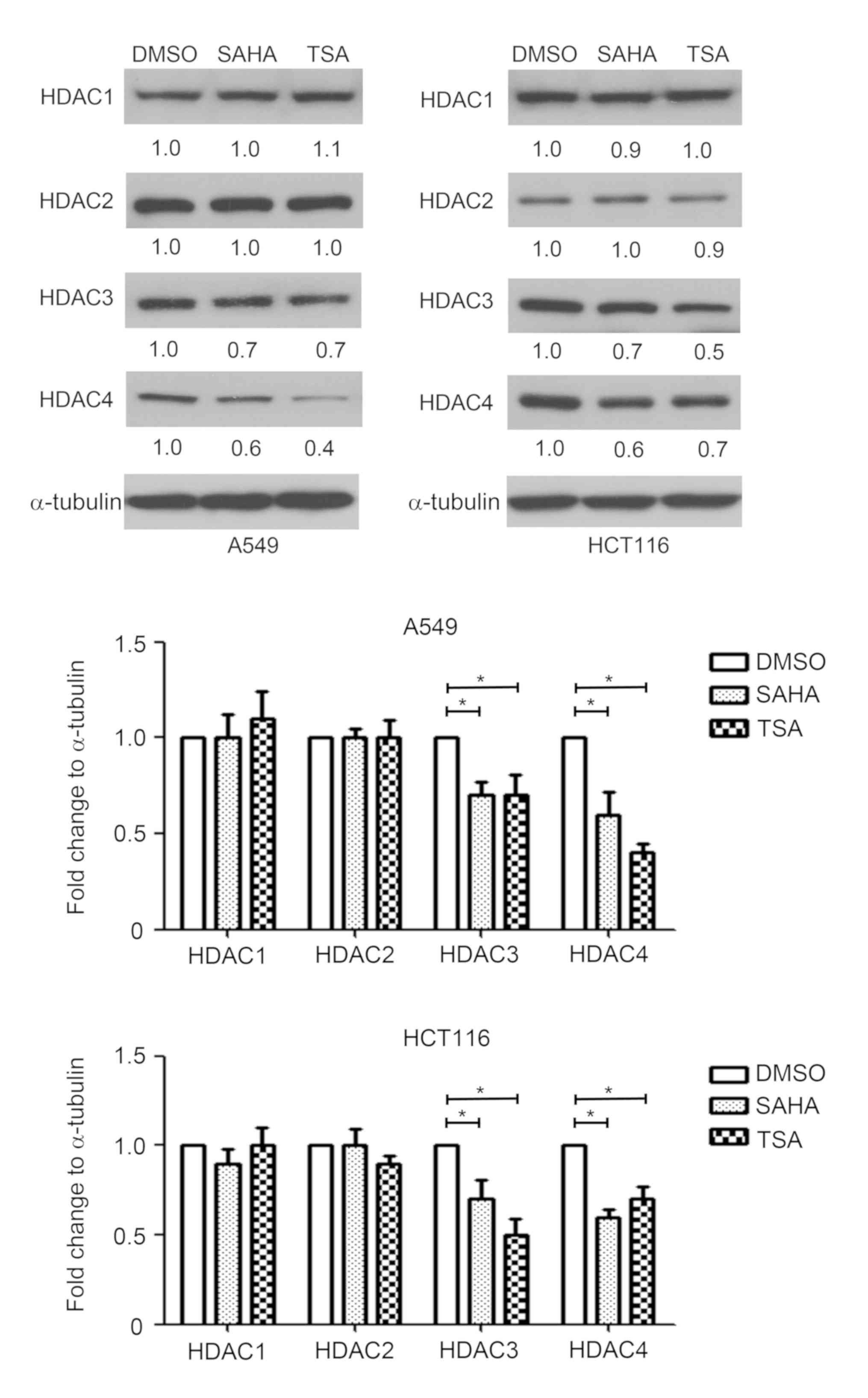
Effect of HDAC inhibitors on the
expression of HDAC1, HDAC2, HDAC3 and HDAC4
Since HDACs serve an important role in the
regulation of gene transcription, the effect of HDAC inhibitors on
HDAC1, HDAC2, HDAC3 and HDAC4 expression was measured in A549 and
HCT116 cells. HDAC1-4 exert their function depending on the
protein; therefore, protein expression was detected by western
blotting. As shown in Fig. 4, SAHA
and TSA decreased the protein expression levels of HDAC3 and HDAC4,
compared with the DMSO-treated group. By contrast, SAHA and TSA had
no marked effects on HDAC1 and HDAC2 expression levels in A549 and
HCT116 cells (Fig. 4).
Discussion
Chemotherapy is a common strategy used for the
treatment of malignant tumors. However, the efficacy of
chemotherapy is limited by the development of an MDR phenotype due
to overexpression of ABC transporters in cancer cells.
Chemoresistance-associated ABC transporters mainly include ABCB1,
ABCC1-6, ABCC10-12 and ABCG2 (3).
These transporters function as efflux pumps that energetically
translocate chemotherapeutic drugs from inside to outside of cancer
cells, reducing the intracellular drug concentration and resulting
in an MDR phenotype (3). In order to
overcome the MDR phenotype induced by HDAC inhibitors, it is
crucial to understand the effect of HDAC inhibitors on the
expression of chemoresistance-associated ABC transporters. In the
present study, it was demonstrated that treatment with HDAC
inhibitors increased the expression levels of ABCB1, ABCC2, ABCC5,
ABCC6, ABCC10, ABCC11 and ABCC12 in cancer cells, whereas it
decreased the expression levels of ABCC1, ABCC3 and ABCC4. To the
best of our knowledge, this is the first systematic investigation
on the effect of HDAC inhibitors on the expression of
chemoresistance-associated ABC transporters.
ABCB1, the most well-known ABC transporter, extrudes
natural toxins, anticancer drugs and drug metabolites across the
plasma membrane, conferring an MDR phenotype in various cancer
cells (25,26). A number of studies have reported that
HDAC inhibitors may promote ABCB1 expression in several types of
cancer cells (27,28). For instance, the HDAC inhibitors
SAHA, TSA and phenylbutyrate were reported to increase ABCB1
expression in acute myeloid leukemia (29). Our previous study revealed that SAHA,
TSA and sodium butyrate induced ABCB1 expression by transcriptional
activation of STAT3, and stabilized ABCB1 mRNA in lung and
colorectal cancer (22,23). Consistently, the present study also
demonstrated that SAHA and TSA significantly increase ABCB1 mRNA
and protein expression in A549 and HCT116 cells. Recent research
has suggested that continuous stimulation with the HDAC inhibitor
FK228 may active ABCG2, another ABC transporter, at the mRNA and
protein levels in renal and colon cancer cells to give an MDR
phenotype (30). Furthermore, SAHA,
TSA and phenylbutyrate have been reported to increase ABCG2
expression in acute myeloid leukemia (19,29).
However, in the present study it was observed that SAHA decreased
ABCG2 mRNA expression in A549 cells, while neither SAHA nor TSA had
any evident influence on ABCG2 protein expression in A549 and
HCT116 cells.
ABCC members have been demonstrated to confer
chemoresistance in cancer cells by translocating a variety of
structurally diverse glutathione conjugates or therapeutic drugs
(4). A previous study suggested that
SAHA, TSA and valproate were able to increase ABCC11 expression by
promoting its transcription in acute myeloid leukemia (29). In the present study, the data
revealed that SAHA and TSA increased ABCC5, ABCC6, ABCC10, ABCC11
and ABCC12 mRNA expression levels, as well as ABCC10, ABCC12
protein expression levels. By contrast, SAHA and TSA significantly
decreased ABCC1, ABCC3 and ABCC4 mRNA expression levels, as well as
ABCC1 and ABCC3 protein levels. A recent study reported that the
HDAC inhibitors SAHA and belinostat downregulated ABCC1 and
upregulated ABCB1 expression in T-cell lymphoma and T-cell
prolymphocytic leukemia (31).
Of the nine ABCC proteins investigated in the
present study, ABCC4, ABCC5, ABCC11 and ABCC12 have a typical ABC
structure, with four domains (MSD1, MSD2, NBD1 and NBD2). The other
ABCCs, namely ABCC1, ABCC2, ABCC3, ABCC6 and ABCC10 have an
additional fifth domain, MSD0 (4).
It can thus be speculated that HDAC inhibitors may induce different
effects on ABCC expression in cancer cells due to their different
structures. Notably, the current study results revealed that SAHA
and TSA significantly decreased ABCC2 mRNA expression, but
evidently increased its protein expression in A549 and HCT116
cells. Post-transcriptional regulation, such as mRNA stability
alternations and protein stability modifications, serves an
important role in protein expression (20,32,33).
Therefore, HDAC inhibitors may influence the post-transcriptional
regulation processes that regulate ABCC2 mRNA and protein
expression.
According to the findings of the current study, it
can be speculated that the underlying mechanisms responsible for
the differential effects of HDAC inhibitors on ABC transporters may
be associated with differences in site-specific
acetylation/methylation of histones. For instance, acetylation and
mono-methylation of histone 3 at Lys-9 activates gene
transcription, whereas di- and tri-methylation of the same residue
results in gene suppression (34–36).
Several HDAC family members are aberrantly expressed in various
cancer types and may have potential as target molecules for
anticancer treatments (37). Valdez
et al (31) also reported
that the HDAC inhibitors SAHA, TSA and phenylbutyrate downregulated
the expression levels of HDAC3, HDAC4 and HDAC6 in T-cell lymphoma
and T-cell prolymphocytic leukemia. In the present study, it was
observed that SAHA and TSA treatment decreased HDAC3 and HDAC4
expression levels, but had no significant effect on HDAC1 or HDAC2
expression. Furthermore, an earlier study indicated that the HDAC3
and HDAC4 complex stimulated the transcriptional activity of
mineralocorticoid receptor (MR), and HDAC4 served an important role
as a scaffold between MR and HDAC3 (38). Acetylation occurs, in part, due to
decreased HDAC3 and HDAC4 protein expression, while methylation is
most likely to occur due to the functional interaction between
histone methyltransferases and deacetylases (39). Nevertheless, it remains unclear which
histone modifications contribute to the differential effects of
HDAC inhibitors on the expression of ABC transporters.
ABC transporters have different substrates; for
instance, ABCC1 is known to pump GSH-conjugated DNA alkylators out
of cells (4,40), while decreased ABCC1 activity may
cause cellular accumulation of DNA alkylating agents and thus
enhanced cytotoxicity. Cancer cells with high levels of ABCC1 are
more resistant to DNA alkylating agents, such as busulfan and
chlorambucil. It has been reported that busulfan exerts synergistic
cytotoxicity when used in combination with HDAC inhibitors
(41,42). However, the present study
demonstrated that SAHA and TSA increased ABCB1 expression, which is
known to pump its substrates, such as doxorubicin, vincristine and
prednisone, out of cells (25,30). The
increased ABCB1 expression induced by HDAC inhibitors may lead to
reduced anticancer activity of ABCB1 substrates. Therefore, the
results of the present study indicate that it is important to
select appropriate drugs in combination with HDAC inhibitors.
In conclusion, the present study demonstrated that
HDAC inhibitors have different effects on the expression of ABC
transporters in A549 and HCT116 cells. SAHA and TSA increased the
expression levels of ABCB1, ABCC2, ABCC5, ABCC6, ABCC10, ABCC11 and
ABCC12, whereas they downregulated ABCC1, ABCC3 and ABCC4.
Furthermore, SAHA and TSA induced drug resistance in HCT116 cells,
and decreased HDAC3 and HCAC4 expression levels. In future studies,
drug resistance mediated by SAHA and TSA in A549 cells will be
investigated. ABC transporters have different substrates (14). Differential effects of HDAC
inhibitors on the expression of drug transporters support the
necessity for caution in combining these drugs with other
chemotherapeutic agents. The present study seems timely in light of
an ongoing clinical trial (no. NCT01280526) testing the value of
combining romidepsin, a HDAC inhibitor, with cyclophosphamide,
doxorubicin, vincristine and prednisone (31). Particularly, efflux of doxorubicin,
vincristine and prednisone may increase with HDAC
inhibitor-mediated upregulation of ABCB1 (43). The present study highlighted the
importance of understanding the mechanism of drug combination to
achieve more efficient cytotoxicity to cancer cells. However, the
molecular mechanisms of the different effects of ABC transporter
expression induced by HDAC inhibitors remain unclear. It is
necessary to investigate the underlying mechanisms in future
studies.
Acknowledgements
Not applicable.
Funding
The present study was funded by the National Natural
Science Foundation of China (grant no. 81502599), the Natural
Science Foundation of Anhui Province (grant no. 1608085QH217), the
Natural Science Foundation of Shandong Province (grant no.
ZR2013HM43), the China Postdoctoral Science Foundation funded
project (grant no. 2016M592040) and the Anhui Province Postdoctoral
Science Foundation funded project (grant no. 2016B142).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
HW contributed to the study design, data acquisition
and analysis, and drafted the manuscript. BJW participated in the
study design, data acquisition and revision of the manuscript. CHC,
YZ, BS and RJ assisted in the performance of the statistical
analysis. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mohammad IS, He W and Yin L: Understanding
of human ATP binding cassette superfamily and novel multidrug
resistance modulators to overcome MDR. Biomed Pharmacother.
100:335–348. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang L, Tan RZ, Zhang ZX, Yin R, Zhang YL,
Cui WJ and He T: Association between Twist and multidrug resistance
gene-associated proteins in Taxol®-resistant MCF-7 cells
and a 293 cell model of Twist overexpression. Oncol Lett.
15:1058–1066. 2018.PubMed/NCBI
|
|
3
|
Fukuda Y and Schuetz JD: ABC transporters
and their role in nucleoside and nucleotide drug resistance.
Biochem Pharmacol. 83:1073–1083. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen ZS and Tiwari AK: Multidrug
resistance proteins (MRPs/ABCCs) in cancer chemotherapy and genetic
diseases. FEBS J. 278:3226–3245. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu YR, Liang L, Zhao JM, Zhang Y, Zhang
M, Zhong WL, Zhang Q, Wei JJ, Li M, Yuan J, et al: Twist1 confers
multidrug resistance in colon cancer through upregulation of
ATP-binding cassette transporters. Oncotarget. 8:52901–52912.
2017.PubMed/NCBI
|
|
6
|
Norouzi-Barough L, Sarookhani M, Salehi R,
Sharifi M and Moghbelinejad S: CRISPR/Cas9, a new approach to
successful knockdown of ABCB1/P-glycoprotein and reversal of
chemosensitivity in human epithelial ovarian cancer cell line. Iran
J Basic Med Sci. 21:181–187. 2018.PubMed/NCBI
|
|
7
|
Ceballos MP, Rigalli JP, Cere LI, Semeniuk
M, Catania VA and Ruiz ML: ABC transporters: Regulation and
association with multidrug resistance in hepatocellular carcinoma
and colorectal carcinoma. Curr Med Chem. 2018.
|
|
8
|
Maia RC, Vasconcelos FC, Souza PS and
Rumjanek VM: Towards comprehension of the ABCB1/P-glycoprotein role
in chronic myeloid leukemia. Molecules. 23:2018. View Article : Google Scholar
|
|
9
|
Assaraf YG: Molecular basis of antifolate
resistance. Cancer Metastasis Rev. 26:153–181. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Borst P, Evers R, Kool M and Wijnholds J:
A family of drug transporters: The multidrug resistance-associated
proteins. J Natl Cancer Inst. 92:1295–1302. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dean M, Hamon Y and Chimini G: The human
ATP-binding cassette (ABC) transporter superfamily. J Lipid Res.
42:1007–1017. 2001.PubMed/NCBI
|
|
12
|
Wang J, Yunyun Z, Wang L, Chen X and Zhu
Z: ABCG2 confers promotion in gastric cancer through modulating
downstream CRKL in vitro combining with biostatistics mining.
Oncotarget. 8:5256–5267. 2017.PubMed/NCBI
|
|
13
|
Kaehler M, Ruemenapp J, Gonnermann D,
Nagel I, Bruhn O, Haenisch S, Ammerpohl O, Wesch D, Cascorbi I and
Bruckmueller H: MicroRNA-212/ABCG2-axis contributes to development
of imatinib-resistance in leukemic cells. Oncotarget.
8:92018–92031. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gottesman MM, Fojo T and Bates SE:
Multidrug resistance in cancer: Role of ATP-dependent transporters.
Nat Rev Cancer. 2:48–58. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ling V: Multidrug resistance: Molecular
mechanisms and clinical relevance. Cancer Chemother Pharmacol (40
Suppl). S3–S8. 1997. View Article : Google Scholar
|
|
16
|
Xu W, Liu H, Liu ZG, Wang HS, Zhang F,
Wang H, Zhang J, Chen JJ, Huang HJ, Tan Y, et al: Histone
deacetylase inhibitors upregulate Snail via Smad2/3 phosphorylation
and stabilization of Snail to promote metastasis of hepatoma cells.
Cancer Lett. 420:1–13. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jiang GM, Wang HS, Zhang F, Zhang KS, Liu
ZC, Fang R, Wang H, Cai SH and Du J: Histone deacetylase inhibitor
induction of epithelial-mesenchymal transitions via up-regulation
of Snail facilitates cancer progression. Biochim Biophys Acta.
1833:663–671. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Botrugno OA, Santoro F and Minucci S:
Histone deacetylase inhibitors as a new weapon in the arsenal of
differentiation therapies of cancer. Cancer Lett. 280:134–144.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ni X, Li L and Pan G: HDAC
inhibitor-induced drug resistance involving ATP-binding cassette
transporters (Review). Oncol Lett. 9:515–521. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Di Costanzo A, Del Gaudio N, Conte L,
Dell'Aversana C, Vermeulen M, de Thé H, Migliaccio A, Nebbioso A
and Altucci L: The HDAC inhibitor SAHA regulates CBX2 stability via
a SUMO-triggered ubiquitin-mediated pathway in leukemia. Oncogene.
37:2559–2572. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Stimson L and La Thangue NB: Biomarkers
for predicting clinical responses to HDAC inhibitors. Cancer Lett.
280:177–183. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang H, Huang C, Zhao L, Zhang H, Yang JM,
Luo P, Zhan BX, Pan Q, Li J and Wang BL: Histone deacetylase
inhibitors regulate P-gp expression in colorectal cancer via
transcriptional activation and mRNA stabilization. Oncotarget.
7:49848–49858. 2016.PubMed/NCBI
|
|
23
|
Zhao L, Bin S, He HL, Yang JM, Pu YC, Gao
CH, Wang H and Wang BL: Sodium butyrate increases P-gp expression
in lung cancer by upregulation of STAT3 and mRNA stabilization of
ABCB1. Anticancer Drugs. 29:227–233. 2018.PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sharom FJ: The P-glycoprotein multidrug
transporter. Essays Biochem. 50:161–178. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Han Z and Shi L: Long non-coding RNA
LUCAT1 modulates methotrexate resistance in osteosarcoma via
miR-200c/ABCB1 axis. Biochem Biophys Res Commun. 495:947–953. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Robey RW, Zhan Z, Piekarz RL, Kayastha GL,
Fojo T and Bates SE: Increased MDR1 expression in normal and
malignant peripheral blood mononuclear cells obtained from patients
receiving depsipeptide (FR901228, FK228, NSC630176). Clin Cancer
Res. 12:1547–1555. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tabe Y, Konopleva M, Contractor R, Munsell
M, Schober WD, Jin L, Tsutsumi-Ishii Y, Nagaoka I, Igari J and
Andreeff M: Up-regulation of MDR1 and induction of doxorubicin
resistance by histone deacetylase inhibitor depsipeptide (FK228)
and ATRA in acute promyelocytic leukemia cells. Blood.
107:1546–1554. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hauswald S, Duque-Afonso J, Wagner MM,
Schertl FM, Lübbert M, Peschel C, Keller U and Licht T: Histone
deacetylase inhibitors induce a very broad, pleiotropic anticancer
drug resistance phenotype in acute myeloid leukemia cells by
modulation of multiple ABC transporter genes. Clin Cancer Res.
15:3705–3715. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
To KK, Polgar O, Huff LM, Morisaki K and
Bates SE: Histone modifications at the ABCG2 promoter following
treatment with histone deacetylase inhibitor mirror those in
multidrug-resistant cells. Mol Cancer Res. 6:151–164. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Valdez BC, Li Y, Murray D, Brammer JE, Liu
Y, Hosing C, Nieto Y, Champlin RE and Andersson BS: Differential
effects of histone deacetylase inhibitors on cellular drug
transporters and their implications for using epigenetic modifiers
in combination chemotherapy. Oncotarget. 7:63829–63838. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ménez C, Mselli-Lakhal L, Foucaud-Vignault
M, Balaguer P, Alvinerie M and Lespine A: Ivermectin induces
P-glycoprotein expression and function through mRNA stabilization
in murine hepatocyte cell line. Biochem Pharmacol. 83:269–278.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim SH, Kang HJ, Na H and Lee MO:
Trichostatin A enhances acetylation as well as protein stability of
ERalpha through induction of p300 protein. Breast Cancer Res.
12:R222010. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Barski A, Cuddapah S, Cui K, Roh TY,
Schones DE, Wang Z, Wei G, Chepelev I and Zhao K: High-resolution
profiling of histone methylations in the human genome. Cell.
129:823–837. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Koch CM, Andrews RM, Flicek P, Dillon SC,
Karaöz U, Clelland GK, Wilcox S, Beare DM, Fowler JC, Couttet P, et
al: The landscape of histone modifications across 1% of the human
genome in five human cell lines. Genome Res. 17:691–707. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rosenfeld JA, Wang Z, Schones DE, Zhao K,
DeSalle R and Zhang MQ: Determination of enriched histone
modifications in non-genic portions of the human genome. BMC
Genomics. 10:1432009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Witt O, Deubzer HE, Milde T and Oehme I:
HDAC family: What are the cancer relevant targets? Cancer Lett.
277:8–21. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lee HA, Song MJ, Seok YM, Kang SH, Kim SY
and Kim I: Histone deacetylase 3 and 4 complex stimulates the
transcriptional activity of the mineralocorticoid receptor. PLoS
One. 10:e01368012015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Vaute O, Nicolas E, Vandel L and Trouche
D: Functional and physical interaction between the histone methyl
transferase Suv39H1 and histone deacetylases. Nucleic Acids Res.
30:475–481. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Johnson ZL and Chen J: ATP binding enables
substrate release from multidrug resistance protein 1. Cell.
172:81–89.e10. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Valdez BC, Nieto Y, Murray D, Li Y, Wang
G, Champlin RE and Andersson BS: Epigenetic modifiers enhance the
synergistic cytotoxicity of combined nucleoside analog-DNA
alkylating agents in lymphoma cell lines. Exp Hematol. 40:800–810.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Nieto Y, Valdez BC, Thall PF, Ahmed S,
Jones RB, Hosing C, Popat U, Shpall EJ, Qazilbash M, Gulbis A, et
al: Vorinostat combined with high-dose gemcitabine, busulfan, and
melphalan with autologous stem cell transplantation in patients
with refractory lymphomas. Biol Blood Marrow Transplant.
21:1914–1920. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Köck K, Grube M, Jedlitschky G, Oevermann
L, Siegmund W, Ritter CA and Kroemer HK: Expression of adenosine
triphosphate-binding cassette (ABC) drug transporters in peripheral
blood cells: Relevance for physiology and pharmacotherapy. Clin
Pharmacokinet. 46:449–470. 2007. View Article : Google Scholar : PubMed/NCBI
|