Introduction
Epidermal growth factor receptor (EGFR) is a 170 kDa
transmembrane tyrosine kinase that belongs to the erbB family of
receptor tyrosine kinases and contains three domains: an
extracellular domain, a cross-membrane domain and an intracellular
domain. Further, the intracellular domain can be divided into three
sub-domains: the approximate membrane, tyrosine kinase and
C-terminal sub-domain (1). EGFR has
been strongly implicated in the biology of human epithelial
malignancies, with therapeutic applications in cancers of the
colon, head and neck, lung and pancreas (2). Its mechanism of activation relies on
receptor dimerization and auto-phosphorylation (3). The activation of EGFR triggers several
signal transduction pathways, including the MAPK-mediated signaling
pathway (4). Activated EGFR
promotes the binding between adapter protein Grb2 and Sos1
(5). The activated Sos1 can lead to
the activation of small G protein Ras. Ras proteins act as
molecular switches that alternate between inactive GDP-bound and
active GTP-bound states. The small GTPase Ras has a prominent role
in the signaling pathways leading from activated growth factor
receptors to ERKs (6–8).
cGMP-dependent protein kinases (PKGs) are
serine/threonine kinases. Two main classes of PKG have been
indentified: type I PKG (PKG I) and type II PKG (PKG II) (9,10). PKG
II is a membrane-bound enzyme primarily found in the epithelial
cells of the intestine (11). PKG
phosphorylates target effectors, including vasodilator-stimulated
phosphoprotein (VASP), inositol-1,4,5-trisphosphate (IP3)
receptor-associated cGMP kinase substrate (IRAG) and thromboxane A2
(TXA2) receptor (12–15). Recent research has revealed that
PKGs were associated with the proliferation of tumor cells
(16). Recently, our experiments
indicated that increased exogenous PKG II activity inhibited the
proliferation of gastric cancer cells (17). Our further study showed that the
inhibitory effects of exogenous PKG II on EGF-triggered
proliferation were associated with its effects on the MAPK/ERK
signal transduction pathway (18).
In light of this, we carried out further experiments to elucidate
whether the endogenous PKG activity is able to reverse the
EGF-induced MAPK/ERK signaling pathway, and to investigate its
possible mechanism.
Materials and methods
Cell line
The human small cell lung carcinoma (SCLC) cell line
was provided by the Institute of Cell Biology (Shanghai, China).
The study was approved by the ethics committee of Jiangsu
University, Jiangsu, China.
Reagents
Antibodies against p-MEK (Ser217/221) and p-EGFR
(Tyr1068) were purchased from Cell Signaling Technology, Inc.
(Danvers, MA, USA). Antibodies against pan-Ras, Sos1, Grb2, VASP
and p-VASP (Ser239) were from Santa Cruz Biotechnology, Inc. (Santa
Cruz, CA, USA). Antibodies against pERK1/2 (Thr202/Tyr204) and
GAPDH were from Bioworld Technology Co., Ltd. (St. Louis Park, MN,
USA). Horseradish peroxidase (HRP)-conjugated secondary antibodies
were from Jackson ImmunoResearch Laboratories, Inc. (West Grove,
PA, USA). The cellular permeable cGMP analog 8-pCPT-cGMP was from
Calbiochem (San Diego, CA, USA). Electrochemiluminescence (ECL)
reagent was from Millipore (Billerica, MA, USA). Dulbecco’s
modified Eagle’s medium (DMEM) and new-born calf serum (NBCS) were
from Gibco (Invitrogen Life Technologies, Grand Island, NY,
USA).
Cloning and transfection
As described by Geese et al, mutagenesis of
the VASP phosphorylation site Ser239 (S239A) was performed using
the QuickChange site-directed mutagenesis kit (Stratagene, La
Jolla, CA, USA) according to the manufacturer’s instructions
(18). The primers used for
pMSCV+EGFP-VASP SAT S239A were CTCAGGAAAGTCGCCAAGCAGGAGGAG-GCC
(forward) and GGCCTCCTCCTGCTTGGCGACTTT CCT-GAG (reverse). A
BamHI restriction site was introduced into the 5-end of the
S239A-S primer, and an EcoRI restriction site was introduced
into the 5-end of the S239A-A primer. The S239A fragment was then
subcloned into lentiviral transfer vector FUGW (F, HIV-1 flap
sequence; U, human polyubiquitin promoter; G, green fluorescent
protein; W, woodchuck hepatitis virus post-transcriptional
regulatory element) by BamHI and EcoRI double
digestion. Recombinant lentiviral particles were produced in 293T
cells by transient cotransfection involving a three-plasmid
expression system (20), and cells
were transfected as described previously (21). The expression levels of p-VASP
Ser239 were verified by western blotting analysis.
Ras activity assay
The activity of Ras was detected with the pull-down
method as described previously (22). In brief, cells growing on 100-mm
culture plates were washed three times with cold phosphate-buffered
saline (PBS) and lysed by adding 400 μl of the lysis buffer [25 mM
HEPES (pH 7.5), 150 mM NaCl, 1% NP40, 10% glycerol, 25 mM NaF, 10
mM MgCl2, 0.25% sodium deoxycholate, 1 mM EDTA, 1 mM
Na3VO4, 10 μg/ml aprotinin and 10 μg/ml
leupeptin]. The sample was collected and centrifuged (14,000 × g,
4°C, 10 min) to get rid of the debris. The supernatant was
incubated with glutathione sepharose beads and glutathione
S-transferase-Ras-RBD (GST-RBD) at 4°C for 1 h. The beads were
washed 3 times with lysis buffer and heated in boiled water to
release proteins. The protein samples (containing active Ras) were
analyzed by western blotting with an antibody against pan-Ras.
Co-immunoprecipitation
The cells growing on the 100-mm culture plate were
washed twice with cold PBS and lysed by adding 1 ml RIPA buffer [50
mM Tris-HCl (pH 7.4), 1% Triton X-100, 1 mM EDTA, 1 mM leupeptin, 1
mM phenylmethylsulfonyl fluoride, 10 mM NaF, 1 mM
Na3VO4) per plate. Antibodies against Grb2
and Sos1 and isotype matched IgG were used for
immunoprecipitation.
Western blot assay
Sample proteins were separated on SDS-PAGE gels and
blotted onto polyvinyl difluoride (PVDF) membrane. The PVDF
membrane was blocked with 3% (w/v) bovine serum albumin (BSA) in
TBS-T for 1 h at room temperature. The incubation with the primary
antibody was at 4°C overnight, and with the secondary antibody was
1 h at room temperature, with three washes after each incubation.
Electrochemiluminescence reagents were used to show the positive
bands on the membrane. The bands were detected by Typhoon 9400 (GE
Healthcare, Waukesha, WI, USA).
Results
Increased endogenous PKG activity
reverses EGF-induced Tyr1068 phosphorylation of EGFR in SCLC
cells
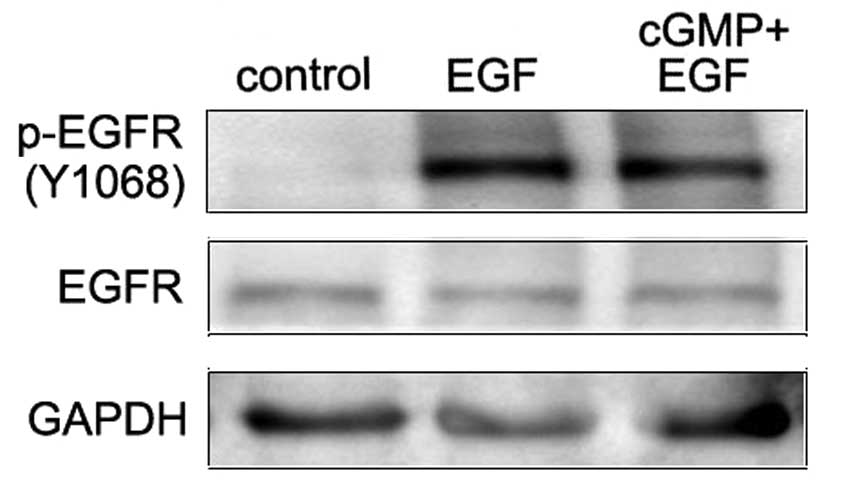
Stimulation of EGFR by its natural ligand EGF causes
the dimerization and the auto-phosphorylation of tyrosine of the
receptors. Tyrosine 1068 (Y1068) is one of the auto-phosphorylation
sites of EGFR. Phosphorylation of this site is associated with
MAPK-mediated signaling. In this study, western blot assay was
applied to investigate the effect of endogenous PKG activity on the
phosphorylation of EGFR (Y1068) in EGF-treated SCLC cells. Results
showed that EGF treatment notably increased the EGFR
phosphorylation at Y1068. In cells with the treatment of cGMP
followed by EGF, this phosphorylation was significantly decreased
compared with the EGF treatment alone (Fig. 1). This demonstrates that increased
endogenous PKG activity inhibited the Y1068 phosphorylation of EGFR
induced by EGF.
Increased endogenous PKG activity
prevents binding between adaptor protein Grb2 and Sos1 in SCLC
cells
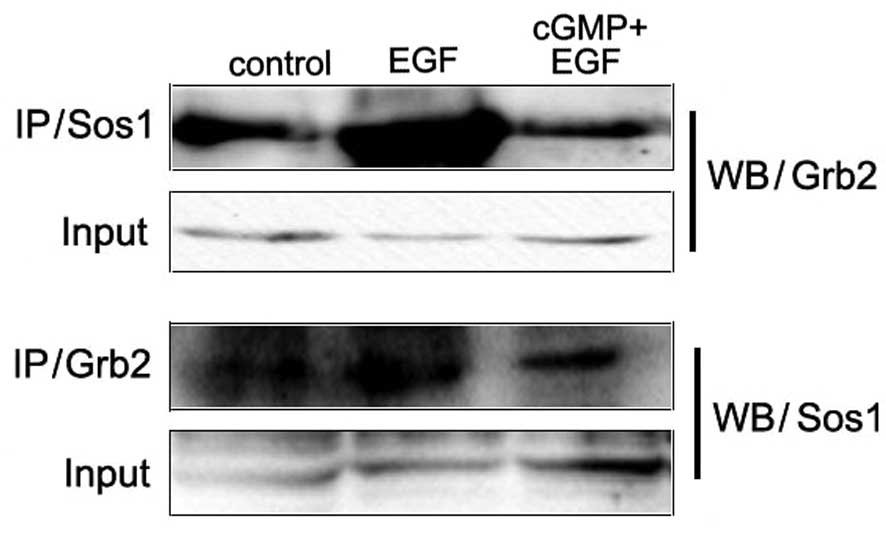
The phosphorylated tyrosines on EGFR provide binding
sites for adaptor proteins. Among them, phosphorylated Tyr1068 is
associated with the activation of the MAPK-mediated signaling
pathway. Grb2 is a growth factor receptor-bound protein and Sos1 is
a guanine nucleotide exchange factor (GEF). They play a role as
adaptor proteins in the MAPK-mediated signaling pathway. The bind
between them activates the downstream signal component Ras.
Co-immunoprecipitation assay was applied to detect the binding
between Grb2 and Sos1. Results showed that with the treatment of
EGF, this binding between Grb2 and Sos1 increased. In the cells
stimulated with cGMP followed by EGF, this binding decreased more
than in cells with treatment of EGF only (Fig. 2). These results revealed that
increased endogenous PKG activity inhibited the EGF-induced binding
between Grb2 and Sos1.
Increased endogenous PKG activity
inhibits EGF-activated Ras and the phosphorylation of MEK and ERK
in SCLC cells
Ras is the key component in the MAPK-mediated
signaling pathway. Once Ras is in GTP-bound form, it binds and
activates Raf-1 and starts the consequent activations of the
serine/threonine kinase in the signaling pathway. Raf-1 is a
regulator upstream of ERK in MAPK-mediated signal transduction
pathway. Activated Raf-1 activates MEK by inducing its
phosphorylation of two serine residues at positions 217 and 221.
MEK1 and MEK2 are members of the dual specificity protein kinase
family, which acts as a MAPK or ERK kinase. Pull-down assay and
western blotting were applied to analyze the Ras activity induced
by EGF or the combination of cGMP with EGF. Results showed that EGF
activates Ras, while the later treatment reverses its effect on Ras
activity (Fig. 3A). Furthermore,
western blot analysis was applied to detect the phosphorylation of
MEK and ERK induced by EGF or the combination of cGMP with EGF.
Results indicated that with treatment of EGF alone, the
phosphorylation level of both MEK and ERK increased, while with the
treatments of cGMP followed by EGF, the phosphorylation level of
MEK and ERK notably decreased compared with EGF treatment, and
there was a significant increase in the phosphorylation level of
VASP at Ser239 (Fig. 3B). These
results demonstrate that increased endogenous PKG activity prevents
EGF-activated Ras and the phosphorylation of MEK and ERK in SCLC
cells.
Downregulating the level of p-VASP Ser239
blocks the effects of endogenous PKG on EGF-induced MAPK/ERK signal
transduction
VASP is known to be a substrate for several protein
kinases in a variety of cells, including PKG (23). VASP harbors three phosphorylation
sites: Ser157, located N-terminally to the central proline-rich
region (PRR); and Ser239 and Tyr278, located in the Ena/VASP
homology domain 2 (EVH2 domain) (24–26).
Since p-VASP Ser239 increased while the phosphorylation level of
MEK and ERK decreased in cells treated with cGMP followed by EGF,
it was important to investigate whether endogenous PKG exerted its
effects on the EGF-induced MAPK/ERK signaling pathway through
phosphorylating VASP at Ser239. For the present study, we designed
lentivirus-mediated EGFP-VASP Ser239 site mutation (239A) to block
its phosphorylation in SCLC cells. The expression of EGFP (green)
was used to present the positive infected cells, the infected
efficiency was more than 95% (Fig.
4A), and cells stably expressing 239A were named SCLC-239A.
Furthermore, western blotting was applied to detect the level of
p-VASP Ser239 following treatment with cGMP in SCLC-239A cells.
Results showed that increased endogenous PKG activity by cGMP
treatment did not trigger the phosphorylation of VASP at Ser239 in
SCLC-239A cells (Fig. 4B). Finally,
we analyzed the role of p-VASP Ser239 in endogenous PKG activity in
the EGF-induced MAPK/ERK signaling pathway. The SCLC-239A cells
were applied and the effects of cGMP on EGF-induced MAPK/ERK
signaling were measured. Western blotting was applied to detect the
effect of endogenous PKG activity on the phosphorylation of EGFR
(Y1068) in EGF-treated SCLC-239A cells. The results showed that
treatment with cGMP did not inhibit the EGF-induced phosphorylation
of EGFR (Y1068; Fig. 4C). In order
to analyze the binding between Grb2 and Sos1 in SCLC-239A cells,
co-immunoprecipitation assay was applied. Results showed that,
compared with EGF treatment alone, there was no detectable
alteration on this binding with the treatment of cGMP (Fig. 4D). Pull-down assay and western
blotting were applied to detect the Ras activity in SCLC-239A
cells. Results showed that cGMP treatment did not inhibit Ras
activation induced by EGF (Fig.
4E). Furthermore, western blot analysis was applied to
investigate the effects of cGMP on EGF-induced phosphorylation of
MEK and ERK in SCLC-239A cells. Results showed that with the
treatment of cGMP, there was no detectable decrease in MEK and ERK
phosphorylation (Fig. 4F).
Together, these data demonstrate that p-VASP Ser239 is critical for
endogenous PKG activity in the EGF-induced MAPK/ERK signal
transduction pathway.
Discussion
EGFR, a transmembrane tyrosine kinase, can be
selectively activated through the binding with ligands belonging to
the EGF family of peptide growth factors. Its mechanism of
activation depends on the receptor dimerization and
auto-phosphorylation. The adaptor proteins bind EGFR at
phospho-Tyr1068 and phospho-Tyr1086 and activate the MAP kinase and
PI3K/Akt pathways (27,28). The majority of human epithelial
cancers are marked by the activation of EGFR, and it was the first
growth factor receptor to be proposed as a target for cancer
therapy. Anti-EGFR therapies have been recently developed for the
treatment of multiple cancer types.
PKG II is membrane-anchored, with high levels in
brain and intestinal mucosa and low levels in certain human cancer
cell lines (29). Previous research
indicated that PKG II was associated with proliferation and
apoptosis in certain cells (30,31).
Our previous study revealed that exogenous PKG II suppressed
EGF-induced MAPK/ERK-mediated signal transduction in gastric cancer
cells (18). However, the
mechanisms involved remained unknown. In order to investigate its
functions further, we focused on the endogenous PKG as well as
exogenous PKG. In this study, we aimed to investigate the role of
endogenous PKG on EGF-triggered MAPK/ERK signaling, and further
focused on its possible mechanism in this signaling pathway.
In the present study, PKG-selective cGMP analog
8-pCPT-cGMP was applied to increase the endogenous PKG activity.
Through confirming the effect of EGF on the MAPK/ERK signaling
pathway, we analyzed the role of endogenous PKG on the
EGF-triggered MAPK/ERK signal transduction pathway. Compared with
the EGF treatment alone, 8-pCPT-cGMP treatment prior to EGF
treatment notably blocked EGF-induced alteration, including the
phosphorylation of EGFR, MEK and ERK, the binding between Sos1 and
Grb2, and the Ras activity. Notably, with the treatment of cGMP
followed by EGF, the phosphorylation of EGFR, MEK and ERK
decreased, while the phosphorylation of VASP at Ser239 increased.
VASP is a major substrate of PKG. In order to test the role of
p-VASP Ser239 during this process, we designed lentivirus-mediated
EGFP-VASP Ser239 site mutation (239A) to block its phosphorylation
in SCLC cells. The results showed that the effects of endogenous
PKG activity on EGF-induced MAPK/ERK signaling were notably
attenuated. In SCLC-239A cells, with treatment of cGMP to increase
the endogenous PKG activity, there was no increase in p-VASP
Ser239, and compared with EGF treatment alone, increased endogenous
PKG activity did not notably block the effect of EGF on MAPK/ERK
signaling.
Our interpretation of the data presented in this
study is that increased endogenous PKG activity inhibits the
EGF-triggered MAPK/ERK signal transduction pathway, and p-VASP
Ser239 plays a critical role. However, the exact role of p-VASP
Ser239 in PKG/MAPK/ERK signal transduction requires further
investigation.
Acknowledgements
This study was supported by the
National Natural Science Foundation of China (Grant No. 31100974
and 81001100) and the Specialized Research Fund for Senior
Personnel Program of Jiangsu University (Grant No. 11JDG032).
References
|
1.
|
A WellsEGF receptorInt J Biochem Cell
Biol31637643199910.1016/S1357-2725(99)00015-1
|
|
2.
|
DL WheelerEF DunnPM HarariUnderstanding
resistance to EGFR inhibitors-impact on future treatment
strategiesNat Rev Clin
Oncol7493507201010.1038/nrclinonc.2010.9720551942
|
|
3.
|
PO HackelE ZwickN PrenzelA
UllrichEpidermal growth factor receptors: critical mediators of
multiple receptor pathwaysCurr Opin Cell
Biol11184189199910.1016/S0955-0674(99)80024-610209149
|
|
4.
|
H JiangMO GrenleyMJ BravoRZ BlumhagenBA
EdgarEGFR/Ras/MAPK signaling mediates adult midgut epithelial
homeostasis and regeneration in DrosophilaCell Stem
Cell88495201110.1016/j.stem.2010.11.02621167805
|
|
5.
|
T YamazakiK ZaalD HaileyJ PresleyJ
Lippincott-SchwartzLE SamelsonRole of Grb2 in EGF-stimulated EGFR
internalizationJ Cell Sci11517911802200211956311
|
|
6.
|
J DownwardCell cycle: routine role for
RasCurr Biol7258260199710.1016/S0960-9822(06)00116-39162506
|
|
7.
|
W KolchMeaningful relationships: the
regulation of the Ras/Raf/MEK/ERK pathway by protein
interactionsBiochem
J2289305200010.1042/0264-6021:351028911023813
|
|
8.
|
JD StockandJG MeszarosAldosterone
stimulates proliferation of cardiac fibroblasts by activating
Ki-RasA and MAPK1/2 signalingAm J Physiol Heart Circ
Physiol284H176H184200310.1152/ajpheart.00421.200212388314
|
|
9.
|
JL MartindaleNJ HolbrookCellular response
to oxidative stress: signaling for suicide and survivalJ Cell
Physiol192115200210.1002/jcp.1011912115731
|
|
10.
|
S OrstavikV NatarajanK TaskénT JahnsenM
SandbergCharacterization of the human gene encoding the type Ialpha
and type Ibeta cGMP-dependent protein kinase
(PRKG1)Genomics42311318199710.1006/geno.1997.47439192852
|
|
11.
|
T MarkertAB VaandragerS GambaryanD PöhlerC
HäuslerU WalterHR De JongeT JarchauSM LohmannEndogenous expression
of type II cGMP-dependent protein kinase mRNA and protein in rat
intestine. Implications for cystic fibrosis transmembrane
conductance regulatorJ Clin Invest96822830199510.1172/JCI118128
|
|
12.
|
RJ HaslamNT DickinsonEK JangCyclic
nucleotides and phosphodiesterases in plateletsThromb
Haemost82412423199910605732
|
|
13.
|
J SchlossmannA AmmendolaK AshmanX ZongA
HuberG NeubauerGX WangHD AllescherM KorthM WilmF HofmannP
RuthRegulation of intracellular calcium by a signalling complex of
IRAG, IP3 receptor and cGMP kinase
IbetaNature404197201200010.1038/3500460610724174
|
|
14.
|
A AmmendolaA GeiselhöringerF HofmannJ
SchlossmannMolecular determinants of the interaction between the
inositol 1,4,5-trisphosphate receptor-associated cGMP kinase
substrate (IRAG) and cGMP kinase IbetaJ Biol
Chem2762415324159200110.1074/jbc.M101530200
|
|
15.
|
I RussoG DoronzoA De SalveL MattielloM
TrovatiG AnfossiThe activity of constitutive nitric oxide synthase
is increased by the pathway cAMP/cAMP-activated protein kinase in
human platelets. New insights into the antiaggregating effects of
cAMP-elevating agentsThromb
Res114265273200410.1016/j.thromres.2004.06.036
|
|
16.
|
FJ SwartlingM FerlettaM KastemarWA WeissB
WestermarkCyclic GMP-dependent protein kinase II inhibits cell
proliferation, Sox9 expression and Akt phosphorylation in human
glioma cell
linesOncogene2831213131200910.1038/onc.2009.16819543319
|
|
17.
|
YC ChenF RenJR SangY TaoWX XuType II
cGMP-dependent protein kinase inhibits proliferation of the gastric
cancer cell line BGC-823Mol Med Report3361366201021472248
|
|
18.
|
Y WuYC ChenR QuT LanJR SangType II
cGMP-dependent protein kinase inhibits EGF-triggered signal
transduction of the MAPK/ERK-mediated pathway in gastric cancer
cellsOncol Rep27553558201222012247
|
|
19.
|
M GeeseJJ LoureiroJE BearJ WehlandFB
GertlerAS SechiContribution of Ena/VASP proteins to intracellular
motility of listeria requires phosphorylation and proline-rich core
but not F-actin binding or multimerizationMol Biol
Cell1323832396200210.1091/mbc.E02-01-005812134077
|
|
20.
|
C LoisEJ HongS PeaseEJ BrownD
BaltimoreGermline transmission and tissue-specific expression of
transgenes delivered by
lentiviralvectorsScience295868872200210.1126/science.106708111786607
|
|
21.
|
XY ZhangVF La RussaJ ReiserTransduction of
bone-marrow-derived mesenchymal stem cells by using lentivirus
vectors pseudotyped with modified RD114 envelope glycoproteinsJ
Virol7812191229200410.1128/JVI.78.3.1219-1229.200414722277
|
|
22.
|
J de RooijJL BosMinimal Ras-binding domain
of Raf1 can be used as an activation-specific probe for
RasOncogene1462362519979053862
|
|
23.
|
R DraijerAB VaandragerC NolteHR de JongeU
WalterVW van HinsberghExpression of cGMP-dependent protein kinase I
and phosphorylation of its substrate, vasodilator-stimulated
phosphoprotein, in human endothelial cells of different originCirc
Res77897905199510.1161/01.RES.77.5.897
|
|
24.
|
E ButtK AbelM KriegerD PalmV HoppeJ HoppeU
WaltercAMP- and cGMP-dependent protein kinase phosphorylation sites
of the focal adhesion vasodilator-stimulated phosphoprotein (VASP)
in vitro and in intact human plateletsJ Biol
Chem26914509145171994
|
|
25.
|
FB GertlerK NiebuhrM ReinhardJ WehlandP
SorianoMena, a relative of VASP and Drosophila Enabled, is
implicated in the control of microfilament
dynamicsCell87227239199610.1016/S0092-8674(00)81341-08861907
|
|
26.
|
A LambrechtsAV KwiatkowskiLM LanierJE
BearJ VandekerckhoveC AmpeFB GertlercAMP-dependent protein kinase
phosphorylation of EVL, a Mena/VASP relative, regulates its
interaction with actin and SH3 domainsJ Biol
Chem2753614336151200010.1074/jbc.M00627420010945997
|
|
27.
|
S LiAD CouvillonBB BrasherRA Van
EttenTyrosine phosphorylation of Grb2 by Bcr/Abl and epidermal
growth factor receptor: a novel regulatory mechanism for tyrosine
kinase signalingEMBO
J2067936804200110.1093/emboj/20.23.679311726515
|
|
28.
|
GA RodriguesM FalascaZ ZhangSH OngJ
SchlessingerA novel positive feedback loop mediated by the docking
protein Gab1 and phosphatidylinositol 3-kinase in epidermal growth
factor receptor signalingMol Cell
Biol2014481459200010.1128/MCB.20.4.1448-1459.200010648629
|
|
29.
|
J SchlossmannR FeilF HofmannInsights into
cGMP signalling derived from cGMP kinase knockout miceFront
Biosci1012791289200510.2741/161815769624
|
|
30.
|
AL CookJM HaynesProtein kinase G
II-mediated proliferative effects in human cultured prostatic
stromal cellsCell
Signal16253261200410.1016/S0898-6568(03)00134-714636895
|
|
31.
|
AL CookJM HaynesPhosphorylation of the PKG
substrate, vasodilator-stimulated phosphoprotein (VASP), in human
cultured prostatic stromal cellsNitric
Oxide161017200710.1016/j.niox.2006.09.00317049286
|