Contents
Introduction
Relationships between glycolysis and OXPHOS are
cooperative and competitive
Cancer cells have a diversity of energy production
pathways
Alterations of oncogenes and tumor suppressors drive
cancer cells to aerobic glycolysis
Conclusion
Introduction
Energy consumption from metabolic activities in
normal cells relies primarily on mitochondrial oxidative
phosphorylation (OXPHOS), which is efficient and generates more
adenosine triphosphate (ATP) than glycolysis. However, one of the
metabolic features of cancer cells is to avidly take up glucose for
aerobic glycolysis. This inefficient pathway for energy production
in cancer cells was first described by German scientist Otto
Warburg in the 1920s, and is also known as the Warburg effect
(1).
Warburg originally proposed that the aerobic
glycolysis in cancer cells was due to a permanent impairment of
mitochondrial OXPHOS. However, this view is challenged by recent
investigations which found that defects of mitochondrial OXPHOS are
not common in spontaneous tumors (2) and that the function of mitochondrial
OXPHOS in most cancers is intact (1,3–7).
Aerobic glycolysis in many cancers is a result driven by various
factors, such as activation of oncogenes, loss of tumor
suppressors, the hypoxic microenvironment, mitochondrial DNA
(mtDNA) mutation and the tissue of origin.
Cancers are extremely heterogeneous diseases and
each cancer has its individual metabolic features. Even in a single
cancer, its constituent cells are also heterogeneous and metabolic
phenotypes vary from one cell to another. Although aerobic
glycolysis is often found in malignant tumors, OXPHOS still
contributes to energy production in cancers, and may play a major
role in energy production in some cancers (8). This article reviews the roles of
glycolysis and OXPHOS in the energy metabolism of cancers.
Relationships between glycolysis and OXPHOS
are cooperative and competitive
Before the introduction of free oxygen into the
atmosphere, organisms on earth rely on glycolysis as an energy
source. However, with the increase of atmospheric oxygen, cells
begin to rely primarily on OXPHOS to produce energy since it
generates more ATP per metabolite than the glycolytic pathway.
However, glycolysis, an ancient energy production
pathway preserved in evolution, still affects energy metabolism in
certain human organs and tissues, such as the brain, liver and
muscle (9,10), and is also a major energy production
pathway in anaerobic bacteria. In fact, glycolysis and OXPHOS are
tightly coupled and serve as a molecular interconversion system.
The process of glycolysis is carried out in the cytoplasm and only
produces two ATPs. Pyruvate, the end product of glycolysis, is a
fuel for OXPHOS. Under aerobic conditions pyruvate enters the
mitochondria to be oxidized to acetyl CoA which combines with
oxaloacetate to start the tricarboxylic acid (TCA) cycle and
OXPHOS, which can produce 36 ATPs. Under anaerobic conditions,
pyruvate is reduced to lactate by lactate dehydrogenase A (LDH-A)
in the cytoplasm and then lactate is excreted into the
extracellular space through monocarboxylate transporters
(MCTs).
Since energy production is a response to energy
demand in the cell, the ATP yield varies depending on cellular
conditions and the microenvironment. Mammalian cells rely primarily
on both glycolysis and OXPHOS to produce ATP at present. However,
the contribution ratio of glycolysis versus OXPHOS for the total
ATP yield varies in different cells, growth states and
microenvironments. In normal conditions, the cell metabolism
consumes energy, of which 70% is supplied by OXPHOS. In hypoxia,
however, glycolysis becomes enhanced to compensate for the weakened
function of OXPHOS. Therefore, glycolysis and OXPHOS cooperate to
maintain the cellular energetic balance. Supposing the total ATP is
a constant, if the function of OXPHOS is weakened, the function of
glycolysis must be enhanced in order to maintain a balance of
energy. In contrast, if the function of OXPHOS is normal, it will
regulate glycolytic activity via different pathways to maintain a
balance of energy (11). In
contrast to normal cells, most cancer cells use glycolysis as a
means of energy production whenever normoxia or hypoxia occurs,
which is referred to as aerobic glycolysis (1).
Cancer cells have a diversity of energy
production pathways
Cancer cells are different from most normal tissues
in the energy metabolism and they take up glucose and glutamine at
a high rate for aerobic glycolysis. In addition, cancers are
extremely heterogeneous and each cancer is different in tissue
origin and metabolic phenotype (3).
Even in a single cancer, its constituent cells vary in metabolic
phenotype (12). It is worth
mentioning that metabolic phenotypes in cancer are plastic, and
cancer tissues exhibit greater plasticity than normal tissues
(13). Cancer cells may change
their metabolic phenotypes to adapt to microenvironmental changes
and the results of these changes give a selective advantage to
cancer cells under the unfavorable environment (14,15).
Glycolysis versus OXPHOS
Due to their different origin and differentiation,
not all cancers rely primarily on glycolysis, which contributes to
total ATP at a rate of 1–64% in cancer cells (8). For example, Suganuma et al
examined the energy metabolism of four leukemia cell lines using
glycolysis inhibitor 2-deoxy-D-glucose (2-DG) and OXPHOS inhibitor
oligomycin (16). They found that
NB4 cells were more sensitive to 2-DG than the three other cell
lines, hence they regarded NB4 as a ‘glycolytic’ leukemia cell
line. Alternatively, THP-1 cells were resistant to 2-DG and
sensitive to oligomycin, and were regarded as an ‘OXPHOS’ leukemia
cell line (16). These results
suggest that energy metabolic pathways are different in various
cancers. We should first examine energy metabolic pathways in
cancer cells when considering energy metabolism as a target for
cancer therapy in order to obtain good therapeutic results.
Warburg considered that aerobic glycolysis in cancer
cells was irreversibly impaired in its mitochondrial function. This
view is challenged by recent investigations which found that the
function of mitochondrial OXPHOS in many cancers is intact
(1,3–7).
Certain authors consider that the Warburg effect in cancer is due
to enhanced glycolysis suppressing OXPHOS rather than defects in
mitochondrial OXPHOS. If glycolysis is inhibited in cancer cells,
the function of mitochondrial OXPHOS can be restored (4,17–19).
For example, Fantin et al observed that when LDH-A was
suppressed in cancer cells, OXPHOS function could be enhanced to
compensate for reduced ATP by inhibited glycolysis. This
observation suggests that most cancer cells reserve the capacity to
produce ATP by OXPHOS. The glycolytic phenotype in cancer cells is
due to OXPHOS being suppressed by active glycolysis rather than
defects in mitochondrial function. Furthermore, they also found
that proliferation and tumorigenicity of cancer cells were
inhibited when LDH-A activity was suppressed, suggesting that
enhanced OXPHOS is still not sufficient to meet the requirement of
cancer growth and that LDH-A is a target of cancer therapy
(4).
Since the glycolytic contribution to total ATP
production does not generally exceed 50–60% (8), OXPHOS still substantially contributes
to ATP production in tumor cells. Although four human malignant
tumor cell lines (HL60, HeLa, 143B and U937) are cells which rely
on OXPHOS to support the growth of cells (20), this phenotype is altered under
hypoxia. For example, the OXPHOS contribution to total ATP
production is normally 79 and 91% in cervical carcinoma HeLa cells
and breast carcinoma MCF cells, respectively. This contribution,
however, is reduced to 29 and 36% in hypoxia, respectively
(21), suggesting that the
glycolytic phenotype in cancer cells is primarily caused by
hypoxia. In their retrospective review, Moreno-Sánchez et al
point out that although glycolysis plays an important role in
cancer energy metabolism, a considerable amount of cancers use
OXPHOS as a pathway of energy production or a mixture of glycolysis
and OXPHOS (17). In some cases,
the function of OXPHOS in cancer cells is even higher than in
adjacent stromal cells (22).
Researchers from Singapore recently isolated intact mitochondria
from human ovarian and peritoneal cancer tissues, which exhibited
the specific activities of succinate, malate and glutamine
dehydrogenases, and had the capacity of OXPHOS. The cells produced
ATP, but in lower amounts than the human skeletal muscle (6).
Smolkova et al (19) proposed four waves of metabolic
regulation during carcinogenesis. Wave 1: cancer stem cell (CSC)
transformation, primarily due to oncogene-mediated signaling; Wave
2: hypoxia, inducing hypoxia-inducible factor (HIF), AMP-activated
protein kinase (AMPK) and NF-κB signaling. In waves 1 and 2, the
cell metabolism highly favors glycolysis and inhibits OXPHOS due to
the oncogenic and hypoxic controls of gene reprogramming, i.e., the
classic Warburg phenotype. Wave 3: aglycemia, nutrient shortage due
to the high proliferation rate during malignancy. In this wave, the
function of mitochondrial OXPHOS is partially restored due to gene
reprogramming via the LKB1-AMPK-p53 pathway and/or the
PI3K-Akt-mTOR pathway. Myc-mediated glutaminolysis is also involved
in this process. Wave 4: mitochondria revival, retrograde signaling
from revitalized mitochondria may constitute this wave of gene
reprogramming. This working hypothesis indicates that the Warburg
phenotype is not exclusive and that a decrease of mitochondrial
function is not a general feature of cancer cells.
An emerging hypothesis of the so-called reverse
Warburg effect also supports the function of OXPHOS in cancer cells
(22). In the reverse Warburg
effect, epithelial cancer cells induce aerobic glycolysis in
carcinoma-associated fibroblasts (CAFs) which produce lactate,
ketones and pyruvate that enter the TCA cycle in cancer cells for
OXPHOS (22,23). Previously, it was consdiered that
the behavior of cancer was determined by cancer cells, but more
recently stromal cells have also been demonstrated to be involved
in the growth of cancer cells. This is possible due to the
co-evolution between cancer cells and stromal cells in
tumorigenesis. For example, under the ‘education’ of cancer cells
and inflammatory cytokines, stromal fibroblasts become CAFs,
macrophages become tumor-associated macrophages (TAMs) and
neutrophils become tumor-associated neutrophils (TANs), and so on.
In fact, these tumor-associated stromal cells are already different
from their original cells and have genetic and epigenetic changes
which lead to alterations in expression and metabolic profiles.
Therefore, tumor cells and stromal cells not only influence each
other in cytokines and growth factors, but also in energy
metabolites. The relationship between cancer cells and
tumor-associated stromal cells is to promote each other, unlike the
original inhibitory relationship between normal epithelial and
stromal cells (24). These studies
suggest that caution should be applied when using lactate in cancer
patients, and also provide a theoretical basis for using stromal
cells as a target of cancer therapy.
An increasing number of recent studies show that
lactate released by glycolysis in hypoxic tumor cells and/or
stromal cells is not discharged as a waste product, but can be
taken up by oxygenated tumor cells as energy fuel. Lactate is
converted to pyruvate by LDH-B and then enters the mitochondria for
OXPHOS to generate ATP (3,14,22,25–28).
Metabolic symbiosis is not cancer-specific. In fact, the
cooperation between different cells in energy production is also
observed in normal tissues. For example, neurons rely on OXPHOS to
meet their energy demands, whereas astrocytes rely on glycolysis to
meet their energy needs. Lactate released by astrocytes can be
taken up by neurons, used as energy fuel and form the so-called
astrocyte-neuron lactate shuttle (ANLS) (29).
Cancer cells prefer to use aerobic glycolysis for
ATP production while still retaining the function of OXPHOS for the
following reasons: i) Glycolysis is more suitable for cancer
growth. Since proliferation of cancer tissues is faster than normal
tissues, it not only needs energy, but also needs metabolic
intermediates for the biosynthesis of macromolecules. Many
intermediates from glycolysis and the truncated TCA cycle can be
used to synthesize macromolecules, such as nucleic acids, lipids
and proteins, which are required for cancer growth and
proliferation (2,30). ii) Too efficient products of ATP may
not a good thing for cancer cells. If cancer cells use
high-efficiency glucose, ADP is converted to ATP. The high
concentration of ATP will inhibit phosphofructokinase 1 (PFK1), the
rate-limiting enzyme in glycolysis and pyruvate kinase 1 (PK1), and
glycolysis will be inhibited. Inhibited glycolysis is unfavorable
for cancer cell growth. Although glycolysis yields less ATP than
OXPHOS, the speed of ATP generation in the former is quicker than
in the latter, which is suited to the energy demands of rapid
proliferation tissues such as cancer and embryonic tissues
(11). Generally speaking, rapid
proliferation tissues rely more on glycolysis for ATP production
whereas differentiation tissues rely primarily on OXPHOS for energy
production (13,31). If using glycolysis inhibitor
3-bromopyruvate (3-BP) treats tumors, it is more efficient for
rapid growth of tumors than slow growth of tumors. iii) Hypoxia is
often observed in cancer tissues, and glycolysis offers growth
advantage of cancers under this hypoxic environment (4). Glycolysis produces lactate which is
released into the extracellular space. An acidic microenvironment
provides a growth advantage to cancer tissues over normal tissues
and enhances the invasion and metastasis of cancer cells (32,33).
In addition, lactic acidosis inhibits glycolysis and favors aerobic
respiration as a means of energy generation (14). iv) Due to the decrease of
mitochondrial OXPHOS, less reactive oxygen species (ROS) are
generated, which are cytotoxic to cancer cells (34,35).
Although cancer cells may retain OXPHOS function, it
does not mean that cancer cells have no defects in mitochondrial
respiration. Enhanced glycolysis in certain cancers is due to an
impairment of mitochondrial function (36,37),
including decreased expression of mitochondrial oxidative enzymes
and transporters, truncated TCA cycle, a lowering in the amount of
mitochondria per cell and defective respiratory chain, an increased
amount in the natural inhibitors of the mitochondrial ATP synthase
and a higher sensitivity of mtDNA to oxidative stress (17,38).
Certain cancer cells may use glutamine as
energy fuel
Although it is widely accepted that glucose is the
dominant energy fuel for most cancers, it is not the only one.
Glutaminolysis may be an alternative pathway for energy production
in certain cancers since it is known that elevation in glutamine
consumption is frequently observed in cancer (39,40).
In 1979, Reitzer et al reported that glutamine, not sugar,
was the major energy source for cultured HeLa cells (41). Since then, several reports have
shown that glutamine may be used as the energy fuel for cancer
cells (42–45). In contrast to glucose, glutamine as
an energy fuel is only observed in a few cancer cell lines
(27) and plays an important role
in compensating for the shortage of glucose in some cases.
After entering cells via membrane transport ASCT2,
glutamine is hydrolyzed to glutamate and ammonia by glutaminase
(GLS). Glutamate combines with cysteine and glycine to form reduced
glutathione (GSH) which is found in all human cells. GSH is
involved in regulating the redox state and is a major antioxidant
in cells.
Glutamate may also be converted into α-ketoglutarate
(α-KG) and enters the TCA cycle to supply intermediates and energy
for cell growth. It is particularly useful in the truncated TCA
cycle which cannot efficiently use glucose to generate energy
(2). This process provides fuel for
the passive TCA cycle due to a lack of isocitrate (2,7,39,46–48).
Therefore, it also demonstrates that cancer cells are able to use
OXPHOS for ATP production.
Glutamine not only supplies energy in certain cancer
cells, but also provides precursors (such as citrate) for the
synthesis of lipids. For example, Mullen et al recently
showed that tumor cells with mutations in complex I or III of the
electron transport chain (ETC) used glutamine-dependent reductive
carboxylation as the major pathway of citrate formation, suggesting
that glutamine supports tumor growth in various pathways in
defective mitochondria (49).
It is worth mentioning that elevation of glutamine
consumption in cancer cells is closely related to Myc activation
(see below). When mouse embryonic fibroblasts are introduced by the
Myc gene, the transfected cells will increase
glutaminolysis. If RNA interference (RNAi) downregulates Myc
expression, the tumor cells will reduce their dependency on
glutamine (39). Other research
also supports the effects of Myc on glutaminolysis. For example,
human fibroblasts in medium without glucose will die. This process
is not related to Myc activity and occurs by a mechanism other than
apoptosis. However, these cells in medium without glutamine induced
Myc-dependent apoptosis (43),
suggesting that interfering with glutaminolysis may obtain better
therapeutic effects than interfering with glucose in certain
cancers. In fact, the situation is more complex in vivo.
Cells also obtain energy through other pathways, such as fatty
acid, lactate and ketone oxidation, as well as unidentified sources
(22,23,42,50).
Alterations of oncogenes and tumor
suppressors drive cancer cells to aerobic glycolysis
Cancer cells still use aerobic glycolysis, despite
it being an inefficient way to generate ATP. Increasing evidence
shows that the alterations of oncogenes and tumor suppressors in
tumorigenesis play a key role in aerobic glycolysis of cancer
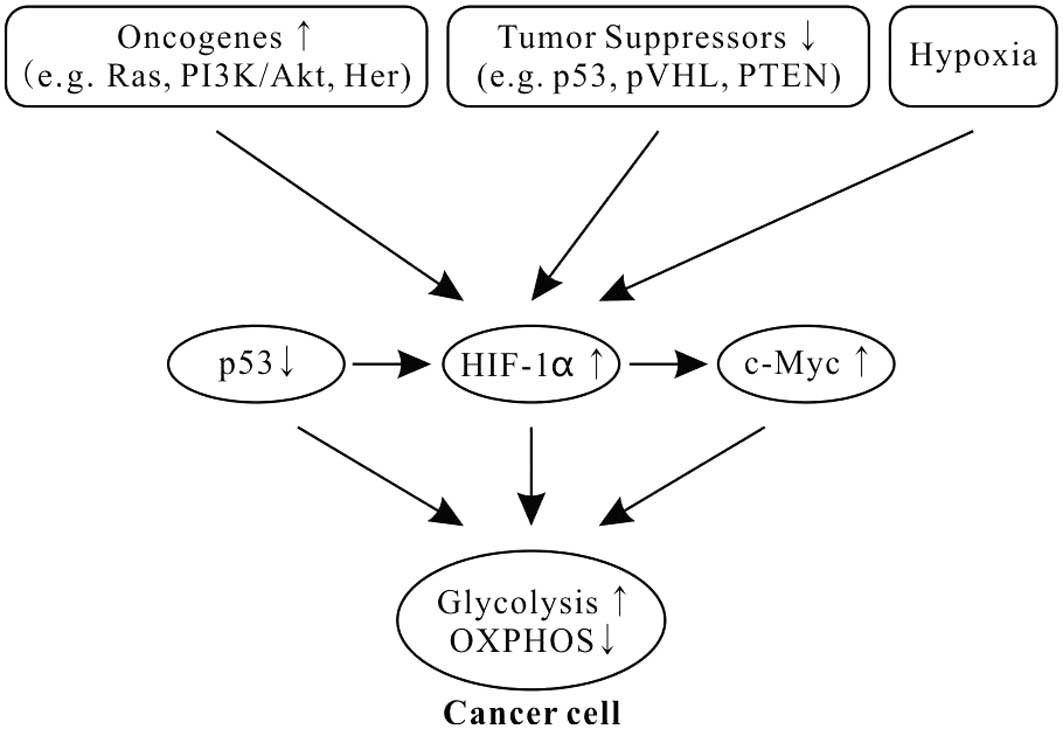
(Fig. 1) (51–53).
Oncogene Ras mutations are often found in
many types of human cancers and drive the metabolic phenotype of
cancer cells toward aerobic glycolysis (54). Ras activates the mammalian target of
rapamycin (mTOR) via the PI3K-Akt-mTOR signaling pathway and mTOR
promotes glycolysis through inducing HIFs (34,55–58).
HIFs, induced transcription factors to facilitate cellular
adaptation to hypoxic environments, play critical roles in shifting
from the OXPHOS to the glycolytic phenotype in cancer (Fig. 1) (34,55,59,60).
HIFs are heterodimers consisting of an oxygen-labile α subunit and
a stable β subunit. In mammals, HIFs have three isoforms: HIF1,
HIF2 and HIF3. HIF1 is ubiquitously expressed in all cells, whereas
HIF2 and HIF3 are selectively expressed in certain tissues
(61). As a transcription factor,
HIF1 regulates over one hundred different genes, including vascular
endothelial growth factor (VEGF), hepatocyte growth factor
receptor (c-Met), erythropoietin (epo), transforming
growth factor-α (TGF-α), platelet-derived growth factor-β
(PDGF-β) and glucose transporter GLUT1, and
influences many cellular activities, including angiogenesis,
glycolysis and cell survival.
In energy metabolism, HIF1 induces GLUT1 and
GLUT3 expression and upregulates 9 of the 10 enzymes that
function in glycolysis (52). It
also inhibits the conversion of pyruvate to acetyl-CoA via the
activation of pyruvate dehydrogenase kinase 1 (PDK1), leading to a
decrease of mitochondrial OXPHOS.
Recent studies show that mTOR upregulation of
pyruvate kinase M2 (PKM2) is critical for aerobic glycolysis and
tumor growth (62). M type pyruvate
kinase (PK) has two isoforms: PKM1 and PKM2. PKM1 is expressed in
most adult tissue, whereas PKM2 is only expressed in embryonic and
proliferating tissues. When cells are transformed, PKM1 expression
is inhibited, and PKM2 expression is restored. The metabolic switch
from OXPHOS to aerobic glycolysis in tumor tissues is considered to
be a shift from PKM1 to PKM2 expression (63). mTOR upregulates PKM2 via HIF1 and
Myc (62), being consistent with
Myc upregulation of glycolysis. Myc can also upregulate the PKM2
expression via polypyrimidine tract-binding protein (PTB),
heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1) and hnRNPA2
(64). Notably, new data also show
that PKM2 may be a strong partner for HIF1 transcription activity
in hypoxia, and since PKM2 gene transcription is also activated by
HIF1, this suggests that PKM2 participates in a positive feedback
loop of HIF1 transcription in cancer cells (65). However, another recent study showed
no evidence of a shift from PKM1 to PKM2 expression during
tumorigenesis. Researchers found that PKM2 was the prominent
isoform in all analyzed cancer samples and cell lines but PKM2 was
also found in matched control tissues (66). Therefore, whether a shift from PKM1
to PKM2 expression occurs during tumorigenesis remains to be
clarified.
In addition to mediating HIFs, mTOR directly
upregulates the basic glycolytic processes, from glucose uptake to
lactate formation (56,58,67).
When glucose uptake goes beyond that required by cancer cells, it
results in pyruvate being reduced to lactate which is excreted into
the extracellular space.
The oncogene Myc, frequently overexpressed in
human cancers, is a master transcription factor which regulates
over 15% of human genes, including cell cycle, metabolism (glucose,
glutamine, protein and lipid), ribosome biogenesis, RNA (miRNA,
tRNA and rRNA), mitochondrial biogenesis, apoptosis and
transformation. Myc stimulates the Warburg effect in cancers in two
aspects (46,48). Firstly, Myc upregulates the
expression of glucose transporter GLUT and LDH-A
(1); and secondly, Myc promotes
glutamine metabolism, including glutamine uptake and glutaminolysis
to provide energy for use by cells (39). The upregulation of the glutamine
metabolism by c-Myc is related to microRNA-23a/b (miR-23a/b), which
targets GLS. c-Myc stimulates the glutamine metabolism and cell
proliferation by repressing miR-23a/b (68).
Compared with Ras, which regulates glycolysis via
the PI3K-Akt pathway, increase of the glutamine metabolism by Myc
does not depend on the PI3K-Akt pathway. Using the PI3K-Akt pathway
inhibitors suppressed the glucose metabolism, but not the glutamine
metabolism, suggesting that the oncogenes Ras and Myc cooperatively
maintain transforming cells via the two key nutrients glucose and
glutamine, complementing each other (39). In order to engage in replicative
division, a cell must duplicate its genome, proteins and lipids and
assemble the components into daughter cells; in short, it becomes a
factory for macromolecular biosynthesis. These activities require
that cells take up extracellular nutrients such as glucose and
glutamine and allocate them into metabolic pathways that convert
them into biosynthetic precursors. Many intermediates from
glycolysis, truncated TCA and glutaminolysis are used in cell
biosynthesis during the cancer cell metabolic switch to glycolysis
by Ras and Myc (40,57).
In addition, HIF1 also binds to a DNA motif in the
promoter of Myc and enhances the transcription of Myc
(Fig. 1). HIF1 cooperates with
c-Myc to promote aerobic glycolysis by induction of hexokinase 2
(HK2), converting glucose to glucose 6-phosphate, and PDK1, a
negative regulator of pyruvate dehydrogenase (PDH) (69).
Tumor suppressor p53, one of the most common gene
mutations in human cancers, is a transcription factor and widely
regulates diverse biological functions, including the cellular
energy metabolism. It plays a pivotal role in balancing between
glycolysis and OXPHOS (70,71). p53 combined with HIF1 and c-Myc has
been described as the ‘triad’ of transcription factors responsible
for the glycolytic phenotype in cancer (Fig. 1) (1,69,72).
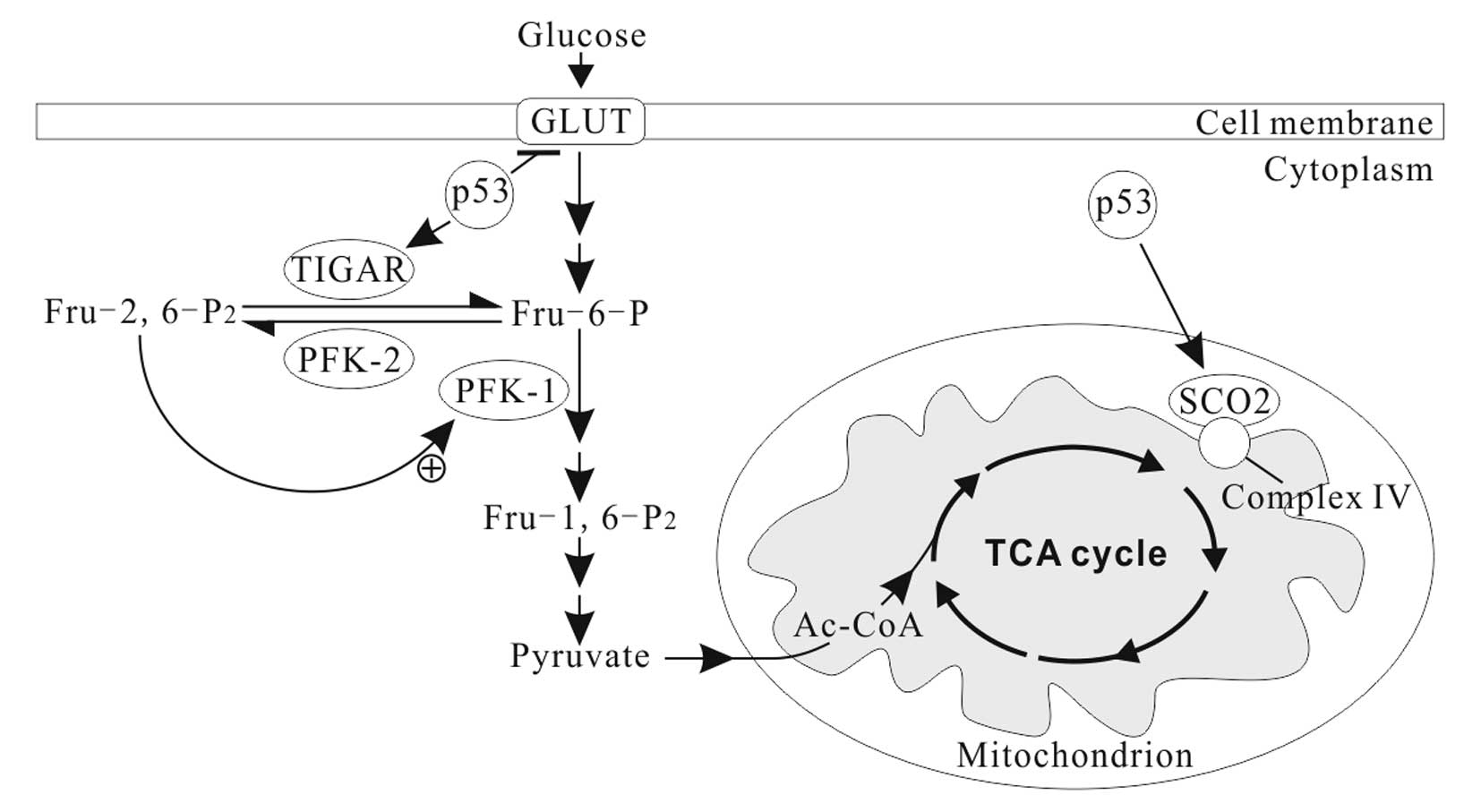
In normal conditions, p53 downregulates the
expression of GLUT1, GLUT4 and HK2, and upregulates
the expression of TP53-induced glycolysis and apoptosis regulator
(TIGAR) and synthesis of cytochrome c oxidase 2
(SCO2) (Fig. 2) and
apoptosis-inducing factor (AIF) (73). Thus, the basic effects of p53 on the
cellular energy metabolism are to inhibit glycolysis and promote
OXPHOS (70,73). TIGAR, an enzyme that decreases the
levels of fructose-2, 6-bisphosphate (Fru-2, 6-P2) by
dephosphorylating, inhibits glycolytic activity (74). Fru-2, 6-P2 is an
important allosteric effector (+) of PFK1, one of key regulatory
enzymes of glycolysis (Fig. 2).
SCO2 facilitates the assembly of cytochrome c oxidase complex in
the mitochondrial ETC complex IV. AIF is essential for ETC complex
I function (75). p53 deficiency
leads to reduced SCO2 and AIF activity, resulting in mitochondrial
OXPHOS impairment (76).
Moreover, p53 is also a potent negative regulator of
HIF1α (Fig. 1). Activation of p53
blocks the accumulation of HIF1α in normoxia and hypoxia (77), and inhibits HIF1 by inducing
microRNA-107 (78).
The p53-deficient cells produced
significantly higher levels of lactate, indicating a shift from
OXPHOS to glycolysis in energy production. For example, glycolysis
contributing to the total amount of ATP is 40% in human colon
cancer cell line HCT116 with wild-type (+/+) p53, rising to
66% in homozygous (−/−) p53 (71). Inactivation of p53 leads to favor
aerobic glycolysis is in many aspects, including increase of
glucose uptake and HIF-1α, and decrease of TIGAR, SCO2 and
AIF expression. Moreover, mutant p53 also increases
the activity of two other glycolytic enzymes, HK2 and
phosphoglycerate mutase (PGM), catalyzing 3-phosphoglycerate (3PG)
to 2-phosphoglycerate (2PG) (71,73,79).
Conclusion
Cancers have different metabolic phenotypes of
energy for several reasons. First, cancers are heterogeneous
diseases and their genetic heterogeneity determines metabolic
heterogeneity (15). Even from a
single cancer, its constituent cells are also heterogeneous and
reflect differences in metabolic phenotype from one cell to
another. The different subclones within a cancer benefit each other
in the metabolism and form a metabolic symbiont (26). This phenomenon is not
cancer-specific, and cancer cells also use other physiological
mechanisms to support their rapid growth. Second, cancer cells
continuously reprogram to adapt to environmental pressures and
alteration of growth conditions. The result is that the ratio
between glycolysis and OXPHOS to yield total ATP, the ratio between
glucose and glutamine to yield total ATP, or the ratio between
glucose/glutamine and fatty acid to yield total ATP, are
continuously changing. The result of these changes is that the
unfavorable environment provides a selective advantage to cancer
cells.
Acknowledgements
This study was in part supported by a
grant from the Ministry of Education in China (Grant No.
20110092110043). Dr Peng Gao is acknowledged for the artwork of the
figures.
References
|
1.
|
WH KoppenolPL BoundsCV DangOtto Warburg’s
contributions to current concepts of cancer metabolismNat Rev
Cancer113253372011
|
|
2.
|
G KroemerJ PouyssegurTumor cell
metabolism: cancer’s Achilles’ heelCancer Cell134724822008
|
|
3.
|
CE GriguerCR OlivaGY GillespieGlucose
metabolism heterogeneity in human and mouse malignant glioma cell
linesJ Neurooncol74123133200510.1007/s11060-004-6404-616193382
|
|
4.
|
VR FantinJ St-PierreP LederAttenuation of
LDH-A expression uncovers a link between glycolysis, mitochondrial
physiology, and tumor maintenanceCancer
Cell9425434200610.1016/j.ccr.2006.04.02316766262
|
|
5.
|
PP HsuDM SabatiniCancer cell metabolism:
Warburg and
beyondCell134703707200810.1016/j.cell.2008.08.02118775299
|
|
6.
|
HY LimQS HoJ LowM ChoolaniKP
WongRespiratory competent mitochondria in human ovarian and
peritoneal
cancerMitochondrion11437443201110.1016/j.mito.2010.12.01521211574
|
|
7.
|
DA ScottAD RichardsonFV FilippCA KnutzenGG
ChiangZA RonaiAL OstermanJW SmithComparative metabolic flux
profiling of melanoma cell lines: beyond the Warburg effectJ Biol
Chem2864262642634201110.1074/jbc.M111.28204621998308
|
|
8.
|
XL ZuM GuppyCancer metabolism: facts,
fantasy, and fictionBiochem Biophys Res
Commun313459465200410.1016/j.bbrc.2003.11.13614697210
|
|
9.
|
GA BrooksCell-cell and intracellular
lactate shuttlesJ
Physiol58755915600200910.1113/jphysiol.2009.17835019805739
|
|
10.
|
SN VaishnaviAG VlassenkoMM RundleAZ
SnyderMA MintunME RaichleRegional aerobic glycolysis in the human
brainProc Natl Acad Sci
USA1071775717762201010.1073/pnas.101045910720837536
|
|
11.
|
T PfeifferS SchusterS
BonhoefferCooperation and competition in the evolution of
ATP-producing
pathwaysScience292504507200110.1126/science.105807911283355
|
|
12.
|
A MiccheliA TomassiniC PuccettiM ValerioG
PelusoF TuccilloM CalvaniC ManettiF ContiMetabolic profiling by
13C-NMR spectroscopy: [1,2–13C2]glucose reveals a
heterogeneousmetabolism in human leukemia T
cellsBiochimie884374482006
|
|
13.
|
MV BerridgePM HerstAS TanMetabolic
flexibility and cell hierarchy in metastatic
cancerMitochondrion10584588201010.1016/j.mito.2010.08.00220709626
|
|
14.
|
JL ChenJE LucasT SchroederS MoriJ WuJ
NevinsM DewhirstM WestJT ChiThe genomic analysis of lactic acidosis
and acidosis response in human cancersPLoS
Genet4e1000293200810.1371/journal.pgen.100029319057672
|
|
15.
|
A MarusykK PolyakTumor heterogeneity:
causes and consequencesBiochim Biophys
Acta1805105117201019931353
|
|
16.
|
K SuganumaH MiwaN ImaiM ShikamiM GotouM
GotoS MizunoM TakahashiH YamamotoA HiramatsuEnergy metabolism of
leukemia cells: glycolysis versus oxidative phosphorylationLeuk
Lymphoma5121122119201010.3109/10428194.2010.51296620860495
|
|
17.
|
R Moreno-SánchezS Rodríguez-EnríquezA
Marín-HernándezE SaavedraEnergy metabolism in tumor cellsFEBS
J274139314182007
|
|
18.
|
C JoseN BellanceR RossignolChoosing
between glycolysis and oxidative phosphorylation: a tumor’s
dilemma?Biochim Biophys Acta1807552561201120955683
|
|
19.
|
K SmolkováL Plecitá-HlavatáN BellanceG
BenardR RossignolP JežekWaves of gene regulation suppress and then
restore oxidative phosphorylation in cancer cellsInt J Biochem Cell
Biol43950968201120460169
|
|
20.
|
PM HerstMV BerridgeCell surface oxygen
consumption: a major contributor to cellular oxygen consumption in
glycolytic cancer cell linesBiochim Biophys
Acta1767170177200710.1016/j.bbabio.2006.11.01817266920
|
|
21.
|
S Rodríguez-EnríquezL Carreño-FuentesJC
Gallardo-PérezE SaavedraH QuezadaA VegaA Marín-HernándezV
Olín-SandovalME Torres-MárquezR Moreno-SánchezOxidative
phosphorylation is impaired by prolonged hypoxia in breast and
possibly in cervix carcinomaInt J Biochem Cell
Biol4217441751201020654728
|
|
22.
|
G BonuccelliA TsirigosD Whitaker-MenezesS
PavlidesRG PestellB ChiavarinaPG FrankN FlomenbergA HowellUE
Martinez-OutschoornKetones and lactate ‘fuel’ tumor growth and
metastasis: evidence that epithelial cancer cells use oxidative
mitochondrial metabolismCell Cycle9350635142010
|
|
23.
|
S PavlidesD Whitaker-MenezesR
Castello-CrosN FlomenbergAK WitkiewiczPG FrankMC CasimiroC WangP
FortinaS AddyaRG PestellUE Martinez-OutschoornF SotgiaMP LisantiThe
reverse Warburg effect: aerobic glycolysis in cancer associated
fibroblasts and the tumor stromaCell
Cycle839844001200910.4161/cc.8.23.1023819923890
|
|
24.
|
H KiarisI ChatzistamouCh KalofoutisH
KoutseliniCh PiperiA KalofoutisTumour-stroma interactions in
carcinogenesis: basic aspects and perspectivesMol Cell
Biochem261117122200410.1023/B:MCBI.0000028746.54447.6c15362494
|
|
25.
|
MI KoukourakisA GiatromanolakiAL HarrisE
SivridisComparison of metabolic pathways between cancer cells and
stromal cells in colorectal carcinomas: a metabolic survival role
for tumor-associated stromaCancer
Res66632637200610.1158/0008-5472.CAN-05-3260
|
|
26.
|
O FeronPyruvate into lactate and back:
from the Warburg effect to symbiotic energy fuel exchange in cancer
cellsRadiother
Oncol92329333200910.1016/j.radonc.2009.06.02519604589
|
|
27.
|
VC SandulacheTJ OwCR PickeringMJ
FrederickG ZhouI FoktM Davis-MalesevichW PriebeJN MyersGlucose, not
glutamine, is the dominant energy source required for proliferation
and survival of head and neck squamous carcinoma
cellsCancer11729262938201121692052
|
|
28.
|
PE PorporatoS DhupRK DadhichT CopettiP
SonveauxAnticancer targets in the glycolytic metabolism of tumors:
a comprehensive reviewFront
Pharmacol249201110.3389/fphar.2011.0004921904528
|
|
29.
|
M BélangerI AllamanPJ MagistrettiBrain
energy metabolism: focus on astrocyte-neuron metabolic
cooperationCell Metab14724738201122152301
|
|
30.
|
SY LuntMG Vander HeidenAerobic glycolysis:
meeting the metabolic requirements of cell proliferationAnnu Rev
Cell Dev
Biol27441464201110.1146/annurev-cellbio-092910-15423721985671
|
|
31.
|
MG Vander HeidenLC CantleyCB
ThompsonUnderstanding the Warburg effect: the metabolic
requirements of cell
proliferationScience32410291033200919460998
|
|
32.
|
RA GatenbyRJ GilliesA microenvironmental
model of carcinogenesisNat Rev
Cancer85661200810.1038/nrc225518059462
|
|
33.
|
P VaupelMetabolic microenvironment of
tumor cells: a key factor in malignant progressionExp
Oncol32125127201021403604
|
|
34.
|
NC DenkoHypoxia, HIF1 and glucose
metabolism in the solid tumourNat Rev
Cancer8705713200810.1038/nrc246819143055
|
|
35.
|
V NogueiraY ParkCC ChenPZ XuML ChenI
TonicT UntermanN HayAkt determines replicative senescence and
oxidative or oncogenic premature senescence and sensitizes cells to
oxidative apoptosisCancer
Cell14458470200810.1016/j.ccr.2008.11.00319061837
|
|
36.
|
D ChandraKK SinghGenetic insights into
OXPHOS defect and its role in cancerBiochim Biophys
Acta1807620625201110.1016/j.bbabio.2010.10.02321074512
|
|
37.
|
KM OwensM KulawiecMM DesoukiA
VanniarajanKK SinghImpaired OXPHOS complex III in breast cancerPLoS
One6e23846201110.1371/journal.pone.002384621901141
|
|
38.
|
F López-RíosM Sánchez-AragóE
García-GarcíaAD OrtegaJR BerrenderoF Pozo-RodríguezA
López-EncuentraC BallestínJM CuezvaLoss of the mitochondrial
bioenergetic capacity underlies the glucose avidity of
carcinomasCancer Res6790139017200717909002
|
|
39.
|
DR WiseRJ DeBerardinisA MancusoN SayedXY
ZhangHK PfeifferI NissimE DaikhinM YudkoffSB McMahonCB ThompsonMyc
regulates a transcriptional program that stimulates mitochondrial
glutaminolysis and leads to glutamine addictionProc Natl Acad Sci
USA1051878218787200810.1073/pnas.081019910519033189
|
|
40.
|
RJ DeBerardinisT ChengQ’s next: the
diverse functions of glutamine in metabolism, cell biology and
cancerOncogene293133242010
|
|
41.
|
LJ ReitzerBM WiceD KennellEvidence that
glutamine, not sugar, is the major energy source for cultured HeLa
cellsJ Biol Chem254266926761979429309
|
|
42.
|
M GuppyP LeedmanX ZuV RussellContribution
by different fuels and metabolic pathways to the total ATP turnover
of proliferating MCF-7 breast cancer cellsBiochem
J364309315200211988105
|
|
43.
|
M YunevaN ZamboniP OefnerR SachidanandamY
LazebnikDeficiency in glutamine but not glucose induces
MYC-dependent apoptosis in human cellsJ Cell
Biol17893105200710.1083/jcb.20070309917606868
|
|
44.
|
C YangJ SudderthT DangRM BachooJG
McDonaldRJ DeBerardinisGlioblastoma cells require glutamate
dehydrogenase to survive impairments of glucose metabolism or Akt
signalingCancer
Res6979867993200910.1158/0008-5472.CAN-09-226619826036
|
|
45.
|
YH KoZ LinN FlomenbergRG PestellA HowellF
SotgiaMP LisantiUE Martinez-OutschoornGlutamine fuels a vicious
cycle of autophagy in the tumor stroma and oxidative mitochondrial
metabolism in epithelial cancer cells: Implications for preventing
chemotherapy resistanceCancer Biol
Ther1210851097201110.4161/cbt.12.12.18671
|
|
46.
|
CV DangA LeP GaoMYC-induced cancer cell
energy metabolism and therapeutic opportunitiesClin Cancer
Res1564796483200910.1158/1078-0432.CCR-09-088919861459
|
|
47.
|
M YunevaFinding an ‘Achilles’ heel’ of
cancer: the role of glucose and glutamine metabolism in the
survival of transformed cellsCell Cycle7208320892008
|
|
48.
|
J ZhengFeatures of energy metabolism and
clinical application in cancer growthChin J Cell
Biol33115811652011
|
|
49.
|
AR MullenWW WheatonES JinPH ChenLB
SullivanT ChengY YangWM LinehanNS ChandelRJ DeBerardinisReductive
carboxylation supports growth in tumour cells with defective
mitochondriaNature481385388201222101431
|
|
50.
|
M BuzzaiDE BauerRG JonesRJ DeberardinisG
HatzivassiliouRL ElstromCB ThompsonThe glucose dependence of
Akt-transformed cells can be reversed by pharmacologic activation
of fatty acid
beta-oxidationOncogene2441654173200510.1038/sj.onc.120862215806154
|
|
51.
|
RL ElstromDE BauerM BuzzaiR KarnauskasMH
HarrisDR PlasH ZhuangRM CinalliA AlaviCM RudinCB ThompsonAkt
stimulates aerobic glycolysis in cancer cellsCancer
Res6438923899200410.1158/0008-5472.CAN-03-290415172999
|
|
52.
|
AJ LevineAM Puzio-KuterThe control of the
metabolic switch in cancers by oncogenes and tumor suppressor
genesScience33013401344201010.1126/science.119349421127244
|
|
53.
|
JP BayleyP DevileeThe Warburg effect in
2012Curr Opin Oncol246267201210.1097/CCO.0b013e32834deb9e
|
|
54.
|
Y HuW LuG ChenP WangZ ChenY ZhouM
OgasawaraD TrachoothamL FengH PelicanoK-ras(G12V) transformation
leads to mitochondrial dysfunction and a metabolic switch from
oxidative phosphorylation to glycolysisCell
Res22399412201210.1038/cr.2011.14521876558
|
|
55.
|
AJ MajmundarWJ WongMC
SimonHypoxia-inducible factors and the response to hypoxic
stressMol Cell40294309201010.1016/j.molcel.2010.09.02220965423
|
|
56.
|
K DüvelJL YeciesS MenonP RamanAI
LipovskyAL SouzaE TriantafellowQ MaR GorskiS CleaverActivation of a
metabolic gene regulatory network downstream of mTOR complex 1Mol
Cell39171183201020670887
|
|
57.
|
Y Pylayeva-GuptaE GrabockaD Bar-SagiRAS
oncogenes: weaving a tumorigenic webNat Rev
Cancer11761774201110.1038/nrc310621993244
|
|
58.
|
JL YeciesBD ManningmTOR links oncogenic
signaling to tumor cell metabolismJ Mol
Med89221228201110.1007/s00109-011-0726-621301797
|
|
59.
|
JJ LumT BuiM GruberJD GordanRJ
DeBerardinisKL CovelloMC SimonCB ThompsonThe transcription factor
HIF-1alpha plays a critical role in the growth factor-dependent
regulation of both aerobic and anaerobic glycolysisGenes
Dev2110371049200710.1101/gad.152910717437992
|
|
60.
|
AM WeljieFR JirikHypoxia-induced metabolic
shifts in cancer cells: moving beyond the Warburg effectInt J
Biochem Cell
Biol43981989201110.1016/j.biocel.2010.08.00920797448
|
|
61.
|
JA BertoutSA PatelMC SimonThe impact of O2
availability on human cancerNat Rev
Cancer8967975200810.1038/nrc254018987634
|
|
62.
|
Q SunX ChenJ MaH PengF WangX ZhaY WangY
JingH YangR ChenMammalian target of rapamycin up-regulation of
pyruvate kinase isoenzyme type M2 is critical for aerobic
glycolysis and tumor growthProc Natl Acad Sci
USA10841294134201110.1073/pnas.101476910821325052
|
|
63.
|
HR ChristofkMG Vander HeidenMH HarrisA
RamanathanRE GersztenR WeiMD FlemingSL SchreiberLC CantleyThe M2
splice isoform of pyruvate kinase is important for cancer
metabolism and tumour
growthNature452230233200810.1038/nature0673418337823
|
|
64.
|
CJ DavidM ChenM AssanahP CanollJL
ManleyHnRNP proteins controlled by c-Myc deregulate pyruvate kinase
mRNA splicing in
cancerNature463364368201010.1038/nature0869720010808
|
|
65.
|
W LuoH HuR ChangJ ZhongM KnabelR
O’MeallyRN ColeA PandeyGL SemenzaPyruvate kinase M2 is a
PHD3-stimulated coactivator for hypoxia-inducible factor
1Cell145732744201110.1016/j.cell.2011.03.05421620138
|
|
66.
|
K BluemleinNM GrüningRG FeichtingerH
LehrachB KoflerM RalserNo evidence for a shift in pyruvate kinase
PKM1 to PKM2 expression during
tumorigenesisOncotarget2393400201121789790
|
|
67.
|
RJ DeBerardinisJJ LumG HatzivassiliouCB
ThompsonThe biology of cancer: metabolic reprogramming fuels cell
growth and proliferationCell
Metab71120200810.1016/j.cmet.2007.10.00218177721
|
|
68.
|
P GaoI TchernyshyovTC ChangYS LeeK KitaT
OchiKI ZellerAM De MarzoJE Van EykJT MendellCV Dangc-Myc
suppression of miR-23a/b enhances mitochondrial glutaminase
expression and glutamine
metabolismNature458762765200910.1038/nature0782319219026
|
|
69.
|
CV DangJW KimP GaoJ YusteinThe interplay
between MYC and HIF in cancerNat Rev
Cancer85156200810.1038/nrc227418046334
|
|
70.
|
S MatobaJG KangWD PatinoA WraggM BoehmO
GavrilovaPJ HurleyF BunzPM Hwangp53 regulates mitochondrial
respirationScience31216501653200610.1126/science.112686316728594
|
|
71.
|
W MaHJ SungJY ParkS MatobaPM HwangA
pivotal role for p53: balancing aerobic respiration and glycolysisJ
Bioenerg Biomembr39243246200710.1007/s10863-007-9083-017551815
|
|
72.
|
SJ YeungJ PanMH LeeRoles of p53, MYC and
HIF-1 in regulating glycolysis - the seventh hallmark of cancerCell
Mol Life Sci6539813999200810.1007/s00018-008-8224-x18766298
|
|
73.
|
PY WangJ ZhuangPM Hwangp53: exercise
capacity and metabolismCurr Opin
Oncol247682201210.1097/CCO.0b013e32834de1d822123233
|
|
74.
|
K BensaadA TsurutaMA SelakMN VidalK
NakanoR BartronsE GottliebKH VousdenTIGAR, a p53-inducible
regulator of glycolysis and
apoptosisCell126107120200610.1016/j.cell.2006.05.03616839880
|
|
75.
|
N VahsenC CandéJJ BrièreP BénitN JozaN
LarochettePG MastroberardinoMO PequignotN CasaresV LazarAIF
deficiency compromises oxidative phosphorylationEMBO
J2346794689200410.1038/sj.emboj.760046115526035
|
|
76.
|
S ZhouS KachhapKK SinghMitochondrial
impairment in p53-deficient human cancer
cellsMutagenesis18287292200310.1093/mutage/18.3.28712714696
|
|
77.
|
J YangA AhmedE PoonN PerusingheA de Haven
BrandonG BoxM ValentiS EcclesK RouschopB WoutersM
AshcroftSmall-molecule activation of p53 blocks hypoxia-inducible
factor 1alpha and vascular endothelial growth factor expression in
vivo and leads to tumor cell apoptosis in normoxia and hypoxiaMol
Cell Biol2922432253200910.1128/MCB.00959-0819223463
|
|
78.
|
M YamakuchiCD LottermanC BaoRH HrubanB
KarimJT MendellD HusoCJ Lowensteinp53-induced microRNA-107 inhibits
HIF-1 and tumor angiogenesisProc Natl Acad Sci
USA10763346339201010.1073/pnas.091108210720308559
|
|
79.
|
H KondohME LleonartJ GilJ WangP DeganG
PetersD MartinezA CarneroD BeachGlycolytic enzymes can modulate
cellular life spanCancer Res65177185200515665293
|