Introduction
Lung cancer is the most common malignancy and the
leading cause of cancer-related mortality worldwide (1). Despite significant progress in
surgical techniques and other conventional therapeutic modalities,
such as chemotherapy and radiotherapy, most patients diagnosed with
lung cancer succumb to the disease in a short period (2–4).
Consequently, understanding the molecule mechanisms underlying the
oncogenesis of lung cancer is crucially important for the
development of more effective therapy of lung cancer (5–7).
The recent discovery of the cohesin complex in yeast
has aided the further understanding of the molecular basis
underlying genome instability, which has been recognized as a
hallmark of human carcinomas (8).
The cohesin complex, evolutionarily conserved from yeast to humans,
comprises four subunits: a pair of SMC (structural maintenance of
chromosomes) proteins, namely SMC1A and SMC3, and two non-SMC
proteins, RAD21/SCC1 and STAG/SCC3/SA. SMC1A and SMC3 are composed
of two coiled domains and interact with each other via their hinge
domain to form an antiparallel heterodimer. Their head domains
interact with RAD21, creating a ring-like structure (9). By trapping DNA within the ring-like
structure, cohesin is associated with chromosomes, holding pairs of
sister chromatids from the time of replication in S phase until
their separation in anaphase to ensure faithful chromosome
segregation during mitosis (10–12).
It has been shown that the cohesin complex participates in a number
of aspects of DNA repair, cell cycle, gene expression regulation
and genomic imprinting, contributing to genome stability (13–15).
Additionally, studies have demonstrated that the dysfunction of
cohesin and cohesin regulatory genes makes them strong candidates
for promoting genome instability and cancer development (16–18).
Previous studies have demonstrated that an increased
risk of lung cancer is associated with deficiencies in DNA repair
capacity, including in the DNA base excision repair genes XRCC1,
PARP-1 and ERCC4 (19). The SMC1A
gene maps to Xp11.22-p11.21 and consists of 25 exons, encoding a
core component of the cohesin complex. In addition to the canonical
role in sister chromotid cohesion, SMC1A is also known as a
substrate of ATM/ATR protein kinases activated by specific DNA
damage signaling, thereby playing a critical role in the regulation
of gene expression and DNA repair (20–23).
In recent years, the downregulated expression of SMC1A and other
cohesin-related genes (NIPBL, SMC3, SCC3) caused by somatic
mutations has been detected in colorectal cancers characterized by
chromosome instability (CIN) (24).
Moreover, researchers have reported that knockdown of SMC1A by RNA
interference (RNAi) resulted in chromatid cohesion defects,
mis-segregation and CIN in vitro(24,25).
These findings imply that SMC1A may serve as a mutational target,
whose disruption leads to the onset of CIN and cancer development.
Apart from mutations, cohesin genes were found to be deregulated in
diverse carcinomas. RAD21 and SMC3 were found to be overexpressed
in breast and prostate cancer and colon carcinoma (26–28),
while SMC1A and RAD21 were found to be downregulated in acute
myeloid leukemia and oral squamous cancer (29,30).
However, to date, the functional roles of SMC1A in human pulmonary
carcinomas have not been demonstrated.
The RNAi technique, a powerful tool for carrying out
loss-of-function assays, is a novel alternative to gene inhibition
and provides a new approach for studying cancer gene therapy
(31,32). Applications of RNAi for mammalian
cells have emerged. In this study, we adopted a lentiviral
vector-mediated RNAi system to achieve highly stable silencing of
SMC1A. The safety of lentiviral vectors has been recognized in the
scientific community (33).
In the present study, we constructed an
SMC1A-specific small interfering RNA (siRNA)-lentiviral vector that
is capable of effectively inhibiting the expression of the SMC1A
gene in human lung adenocarcinoma A549 and H1299 cells and
systemically investigated the impacts of SMC1A silencing on the
growth and invasive ability of the cancer cells in vitro.
Furthermore, we determined the effects of SMC1A knockdown on the
cell cycle distribution and apoptosis of A549 and H1299 cells. As
result, we found that SMC1A is a novel oncogeme, which modulates
the proliferation and migration capabilities of lung cancer cells
via G1/S phase cell cycle arrest and apoptosis.
Materials and methods
Cell culture
The human lung adenocarcinoma cell lines A549 and
H1299 (Cell Bank of Chinese Academy of Sciences, Shanghai, China)
and human embryonic kidney (HEK) 293T cell line (American Type
Culture Collection, ATCC, Manassas, VA, USA) were maintained in
DMEM (Hyclone, Logan, UT, USA) with 10% FBS (Hyclone) and
penicillin/streptomycin at 37°C in humidified atmosphere of 5%
CO2.
Construction of SMC1A short hairpin
(shRNA)-expressing lentivirus
To permit robust inducible RNAi-mediated SMC1A
silencing, shRNA lentiviral vector was constructed. The RNAi was
designed based on a 21-nt SMC1A (NM_006306)-targeting sequence
(5′-TAGGAGGTTCTTCTGAGTACA-3′) of oligonucleotides and negative
control sequence (5′-TTCTCCGAACGTGTCACGT-3′). The sequences were
annealed and ligated into the NheI/PacI- (NEB,
Ipswich, MA, USA) linearized pFH1UGW vector (Shanghai Hollybio Co.
Ltd., Shanghai, China). The lentiviral-based shRNA-expressing
vectors were confirmed by DNA sequencing.
Lentivirus infection
Recombinant lentiviral vectors and packaging vectors
were cotransfected into 293T cells using Lipofectamine 2000
(Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s
instructions for the generation of recombinant lentiviruses [SMC1A
shRNA (Lv-shSMC1A) and negative control shRNA (Lv-shCon)].
Supernatants containing lentiviruses expressing Lv-shSMC1A and
Lv-shCon were harvested 72 h after transfection. Lentiviruses were
purified using ultracentrifugation. A549 and H1299 cells were
infected with the lentiviruses at a multiplicity of infection (MOI)
of 30. Uninfected A549 and H1299 cells were used as controls.
Quantitative real-time PCR
Quantitative real-time PCR was carried out using a
previously described method (34,35).
In brief, total RNA was extracted from A549 and H1299 cells 96 h
after infection using the RNeasy Midi kit (Promega, Madison, WI,
USA). cDNA was synthesized with SuperScriptII reverse transcriptase
(Invitrogen). A mixture containing 1 μg total RNA, 0.5
μg oligo-dT primer (Shanghai Sangon, Shanghai, China) and
nuclease-free water in a total volume of 15 μl was heated at
70°C for 5 min and then cooled on ice for another 5 min. The
mixture was supplemented with 2 μl 10X buffer and 200 units
Super-Script II reverse transcriptase to a final volume of 20
μl, followed by incubation at 42°C for 60 min. Real-time
quantitative PCR analysis was performed using SYBR-Green Master mix
kit on DNA Engine Opticon™ system (MJ Research, Waltham, MA, USA).
Each PCR mixture, containing 10 μl 2X SYBR-Green Master mix
(Takara, Dalian, China), 1 μl sense and antisense primers (5
μmol/μl) and 1 μl of cDNA (10 ng), was run for
45 cycles with denaturation at 95°C for 15 sec, annealing at 60°C
for 30 sec and extension at 72°C for 30 sec in a total volume of 20
μl. For relative quantification, 2-ΔΔCt was
calculated and used as an indication of the relative expression
levels by subtracting CT values of the control gene from the CT
values of SMC1A (36). The primer
sequences for PCR amplification of the SMC1A gene were 5′-AAGTGAGGA
GGAGGAGGAG-3′ and 5′-ACTTTCTTCAGGGTCTTG TTC-3′. β-actin was applied
as an internal control. The primer sequences for β-actin were
5′-GTGGACATCCGCAAAGAC-3′ and 5′-AAAGGGTGTAACGCAACTA-3′.
Western blot analysis
Western blotting was performed using our previously
described method with modifications (34,35).
In brief, A549 and H1299 cells were collected and lysed with
precooled lysis buffer after 96 h of infection. Total protein was
extracted from the cells and determined by the BCA method. Protein
(20 μg) was loaded onto a 10% SDS-PAGE gel. The gel was run
at 30 mA for 2 h and transferred to polyvinylidene difluoride
membrane (Millipore, Billerica, MA, USA). The resulting membrane
was blocked in 5% non-fat dry milk blocking buffer and then probed
with goat anti-SMC1A (1:1,000 dilution; Sigma, St. Louis, MO, USA;
Cat. no. SAB4300451) and mouse anti-GAPDH (1:6,000; Santa Cruz
Biotechnology, Inc., Sana Cruz, CA, USA) overnight at 4°C. The
protein level of GAPDH was used as a control and detected by an
anti-GAPDH antibody. The membrane was washed three times with
Tris-buffered saline Tween-20 (TBST), followed by incubation for 2
h with anti-mouse IgG at a 1:5,000 dilution (Santa Cruz
Biotechnology, Inc.). The membrane was developed using enhanced
chemiluminescence (Amersham, UK). Bands on the developed films were
quantified with an ImageQuant densitometric scanner (Molecular
Dynamics, Sunny-Vale, CA, USA).
Methylthiazol tetrazolium (MTT)
assay
The MTT assay was performed using a previously
described method (33). Briefly,
exponentially growing cells were inoculated into 96-well plates
with 2×103 A549 cells or 6×104 H1299 cells
per well. After incubation for 24, 48, 72, 96 and 120 h, 10
μl sterile MTT (5 mg/ml) was added into each well. Following
incubation at 37°C for 4 h, the reaction was stopped by adding 100
μl dimethyl sulfoxide. The formazan production was detected
by measurement of the spectrometric absorbance at 595 nm. The
values obtained are proportional to the amount of viable cells.
Colony formation assay
The colony formation assay was performed using a
previously described method (34).
In brief, A549 and H1299 cells infected with Lv-shSMC1A or Lv-shCon
and uninfected cells (Con) were seeded in six-well plates
(2×102 cells/well of A549, 5×104 cells/well
of H1299) and cultured at 37°C with 5% CO2 for 8 days.
The cell colonies were washed twice with PBS, fixed in 4%
paraformaldehyde for 15 min and stained with Giemsa for 30 min.
Individual colonies with >50 cells were counted under a
fluorescence microscope.
Cell migration assay
The cell migration assay was performed using our
previously described method (34).
In brief, A549 and H1299 cells infected with Lv-shSMC1A or Lv-shCon
for 96 h and uninfected cells (Con) were harvested and their
ability to migrate in vitro was determined using a Transwell
chamber (Corning, NY, USA). Cells were seeded into the upper
chamber (3.0×104 cells/well of A549, 8.0×104
cells/well of H1299) in 100 μl serum-free medium. Medium (1
ml) containing 20% FBS was added to the lower chamber as a
chemo-attractant. After incubation for 24 h at 37°C in 5%
CO2, cells that invaded to the lower surface of the
filter were fixed in 4% paraformaldehyde and stained with crystal
purple. Cell numbers were counted in five random fields (×100) per
filter and detected by the spectrometric absorbance at 570 nm.
Fluorescence-activated cell sorting
(FACS) analysis
FACS flow cytometry analysis of cell cycle and
apoptosis was performed using our previously described method
(34). In brief, A549 and H1299
cells were seeded in six-well plates (A540, 1.5×106
cells/well; H1299, 2×106 cells/well). After 48 h, cells
were collected, washed with PBS and fixed with 75% cold ethanol.
The cells were then incubated for >24 h at 4°C. After washing
the cells with PBS, propidium iodide (PI) was added to the cell
suspension and the analysis of cell cycle distribution was
performed by FACScan (Becton-Dickinson, Franklin Lakes, NJ,
USA).
Statistical analysis
Data are expressed as mean ± SD. Student’s t-test
was performed to evaluate inter-group differences. P<0.05 was
considered to indicate a statistically significant result. All
statistical analyses were performed with SPSS 10.0 software (SPSS,
Inc., Chicago, IL, USA).
Results
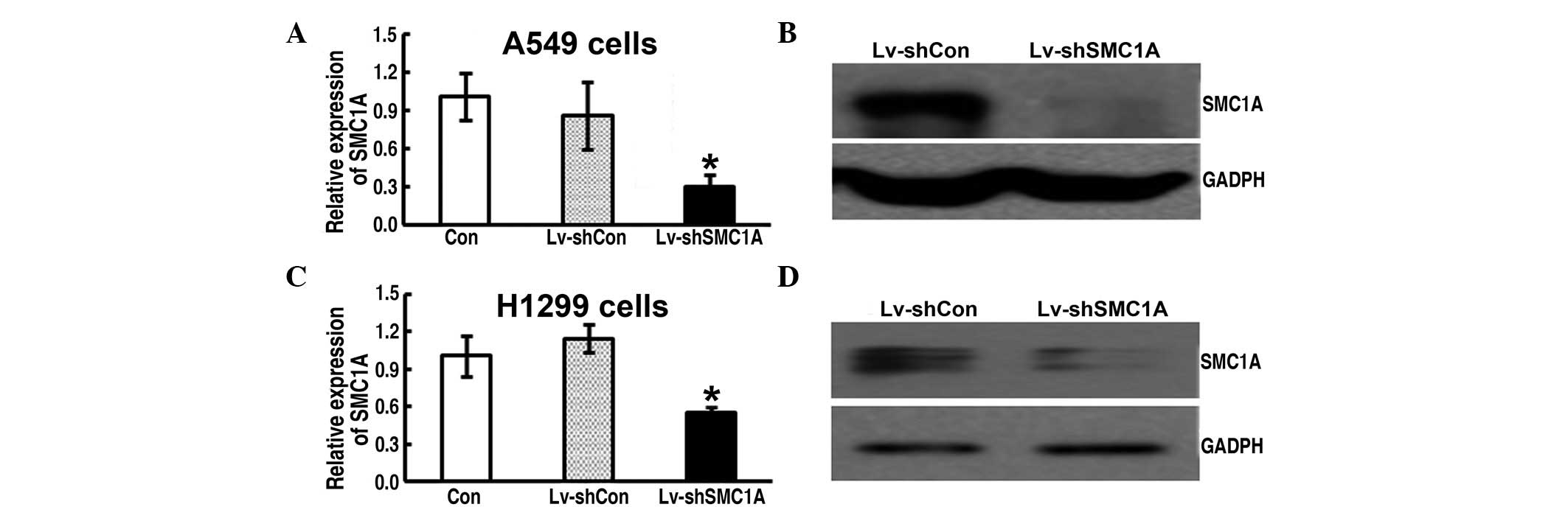
Efficacy of lentivirus-mediated RNAi
targeting of SMC1A
To determine the silencing effect of
lentivirus-mediated SMC1A RNAi on SMC1A expression in A549 and
H1299 cells, real-time PCR and western blot analysis were performed
after 72 h of infection. The expression level of SMC1A mRNA of the
Lv-shSMC1A-infected cells was significantly lower than that of the
parent (Con) and Lv-shCon-infected cells (Fig. 1A and C). Moreover, the western blot
assay further showed that SMC1A protein levels were significantly
decreased in Lv-shSMC1A-infected cells compared with those of
Lv-shCon-infected cells (Fig. 1B and
D). Therefore, this indicates the high efficacy of
lentivirus-mediated SMC1A shRNA on SMC1A expression in lung cancer
cells.
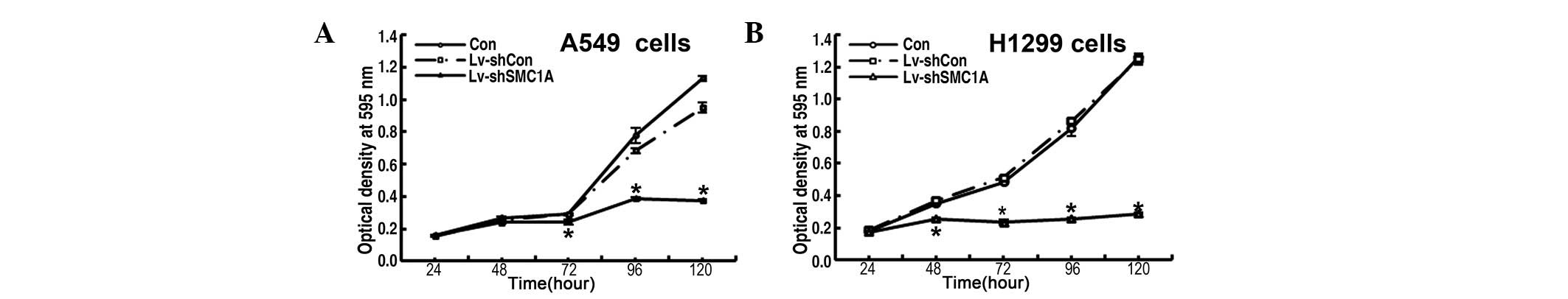
Impact of downregulation of SMC1A
expression on cell growth in vitro
To explore the functional role of SMC1A in the
proliferation of lung cancer cells, the growth dynamics of parent
or Lv-shCon and Lv-shSMC1A-infected A549 and H1299 cells was
determined by MTT and colony formation assays, respectively. The
MTT assay showed that, during the 120-h incubation period, the
growth of Lv-shCon-infected cells did not differ from that of the
uninfected parent cells and showed strong proliferation, whereas
the growth of Lv-shSMC1A-infected cells was markedly slower than
that of the parent or Lv-shCon-infected cells at 48, 72, 96 and 120
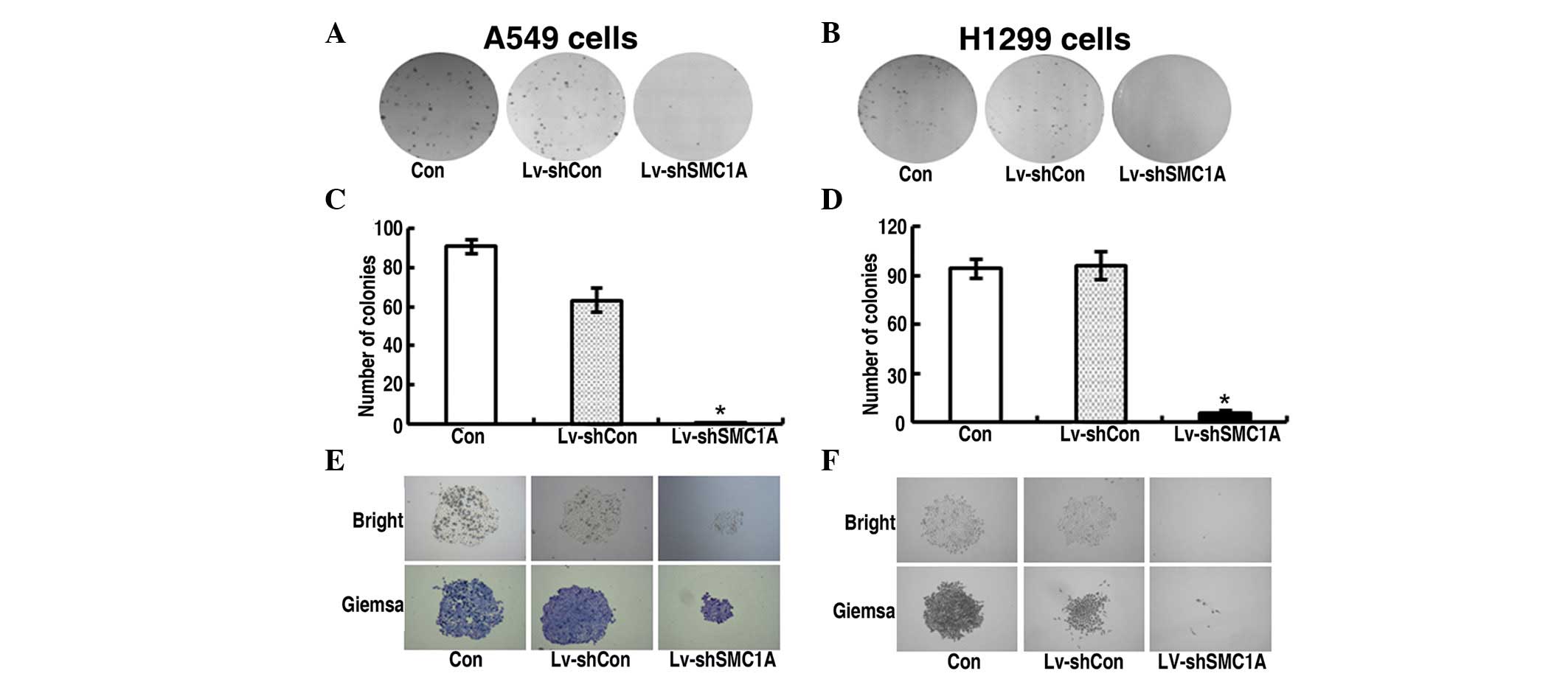
h (Fig. 2). Quantitative analysis
of colonies revealed that after incubation for 8 days, the number
of colonies of Lv-shSMC1A-infected cells was lower than that of the
parent and Lv-shCon-infected cells (P<0.01) (Fig. 3C and D). Therefore, the low
viability and colony-forming efficiency of Lv-shSMC1A-infected A549
and H1299 cells demonstrated that downregulation of SMC1A
expression inhibits the growth of lung cancer cells in
vitro.
Impact of downregulation of SMC1A
expression on cell invasion
To determine the role of SMC1A in lung cancer
invasion, we tested the invasive ability of parent and Lv-shCon- or
Lv-shSMC1A-infected A549 and H1299 cells using the Transwell
chamber assay 96 h after infection. As shown in Fig. 4, the invasive ability of
Lv-shCon-infected cells did not significantly differ from that of
the parent cells and showed strong invasiveness. However, the
invasive ability of Lv-shSMC1A-infected cells was markedly lower
than that of the parent and Lv-shCon-infected cells (Fig. 4C–F). Therefore, this indicates that
the downregulation of SMC1A expression mitigates the invasion of
lung cancer cells in vitro.
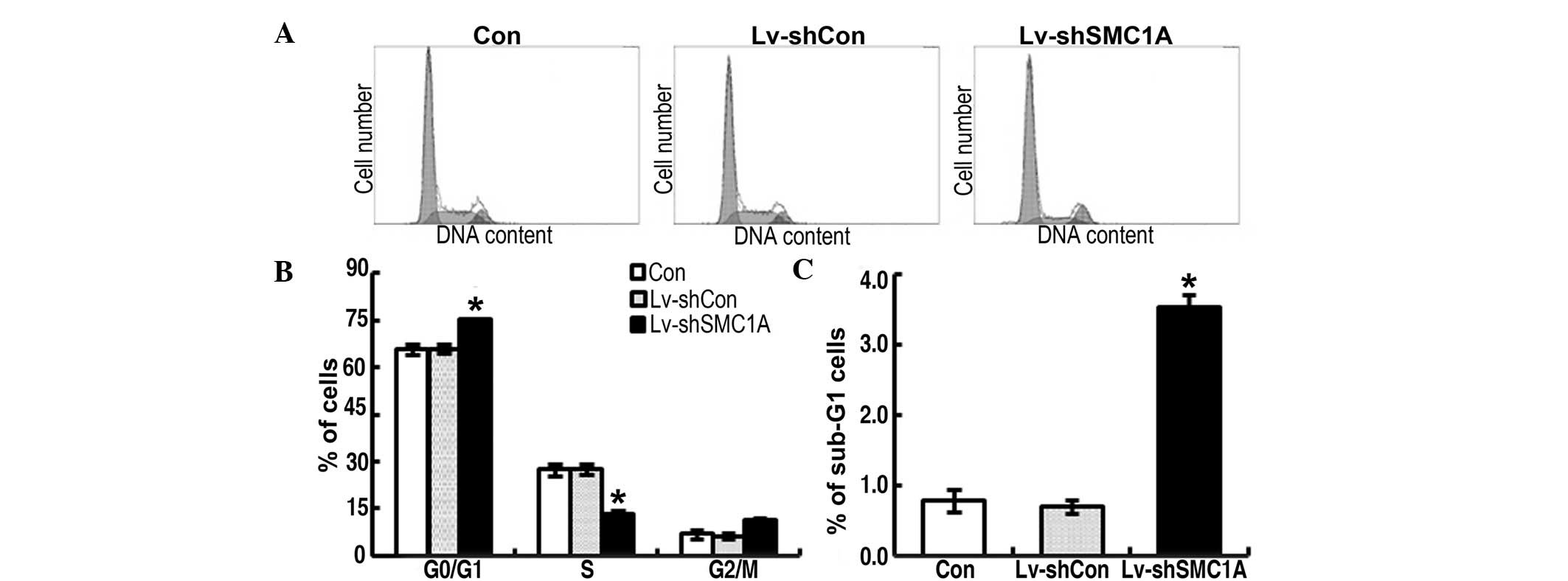
Impact of downregulated SMC1A expression
on cell cycle distribution in vitro
To explore the potential mechanism underlying the
action of SMC1A in the growth of A549 and H1299 cells, the cell
cycling patterns of parent, Lv-shConand Lv-shSMC1A-infected cancer
cells were determined by FACS flow cytometric analysis 96 h after
infection. As shown in Fig. 5B and
Fig. 6B, there was no evident
difference in the frequency at G2 stage of each group of cells. The
frequency of Lv-shCon-infected cells at G1 and S stage did not
significantly differ from its parent cells. At G1 stage, the
frequency of Lv-shSMC1A-infected cells was significantly higher
than that of its parent or Lv-shCon-infected cells. By contrast, at
S stage, the frequency of Lv-shSMC1A-infected cells was lower than
that of the controls (P<0.01) (Figs.
5B and 6B). These results
suggest that the downregulation of SMC1A expression resulted in
cell cycle arrest at the G1/S transition in A549 and H1299 cells,
which contributed to the inhibition of SMC1A cell growth.
Impact of downregulated SMC1A expression
on apoptosis in vitro
To detect the apoptosis, sub-G1 phase cells were
measured. Such cells are usually considered to be the result of
apoptotic DNA fragmentation: during apoptosis, the DNA is degraded
by cellular endonucleases. Therefore, nuclei of apoptotic cells
contain less DNA than nuclei of healthy G0/G1 cells, resulting in a
sub-G1 peak in the fluorescent histogram that may be used to
determine the relative amount of apoptotic cells (37,38).
As shown in Figs. 5C and 6C, there was no marked difference in the
cell population at sub-G1 phase between parent and
Lv-shCon-infected cells, whereas Lv-shSMC1A-infected cells
exhibited a significantly higher proportion in sub-G1 phase than
that of parent or Lv-shCon-infected cells. This suggest that the
downregulation of SMC1A expression may trigger apoptosis in lung
cancer cells, contributing to the suppression of SMC1A cell
growth.
Discussion
Lung cancer is well established as a highly
heterogeneous disease, with a multitude of cellular components and
patterns of gene expression that affect tumor development (39). An in-depth understanding of the
molecular mechanisms underlying cancer proliferation is critical
for the development of optimal therapeutic modalities. Moreover,
there is evidence to suggest that therapeutic drugs specifically
targeting tumor-related molecules are expected to be highly
specific to malignant cells and have minimal adverse reactions due
to their actions through well-defined mechanisms. Cohesin is
emerging as the master regulator of genome stability and its
related genes have been found to be highly relevant to diverse
human malignancies. In the present study, we determined the
expression levels of SMC1A expression in lung adenocarcinoma A549
and H1299 cell lines using quantitative real-time PCR assay and
western blot analysis, and observed clear expression of SMC1A in
lung cancer cells. Consequently, this led to a hypothesis that, as
an indispensible subunit of the cohesin complex, SMC1A may play a
functional role in the biological behavior of lung cancer.
We adopted a lentiviral vector-mediated RNAi system
to further determine the roles of SMC1A in the growth and invasive
ability of lung cancer cells. Using a constructed lentivirus
expressing SMC1A-specific shRNA, we infected A549 and H1299 cells
to silence endogenous SMC1A and investigated the impact of SMC1A
knockdown on the lung cancer development in vitro. We found
that downregulation of SMC1A expression greatly impaired the
proliferation and colony-forming ability of A549 and H1299 cells.
Furthermore, our study also showed that SMC1A knockdown may greatly
reduce the migration capacity of the lung cancer cecolls, as
evidenced by the Transwell chamber invasion assay. Notably, we
observed that SMC1A knockdown caused cell cycle arrest at the G1/S
transition of A549 and H1299 cells, as evidenced by the
accumulation of G1 phase cells and decrease in S phase. In
addition, SMC1A silencing induced apoptosis, as characterized by
the prominent presence of sub-G1 apoptotic cancer cells.
Collectively, these findings are the first report that SMC1A is a
novel regulator of proliferation in lung cancer.
The hallmarks of cancer involve several critical
biological capabilities acquired during cell proliferation and the
invasion-metastasis cascade of malignant tumors. Genome instability
has been found to foster these multiple hallmarks and generates the
genetic diversity that expedites their acquisition (40). Recently, cohesion defects are
emerging as critical factors of genome instability that involve
defects in DNA repair, cell cycle checkpoints and epigenetic
processes (41). Studies have
revealed that, apart from its role in sister chromatid cohesion,
cohesin is also key in various aspects of DNA damage response, cell
cycle and gene expression regulation (13–15).
SMC1A, an indispensible component of the versatile cohesin complex,
is implicated as an important molecular target in malignancies. Our
observation found that SMC1A facilitates important regulatory roles
in lung cancer cell proliferation and invasiveness. There is
evidence to suggest that several factors are implicated in the
genesis of lung cancer, including new fusion genes, new gene
expression, changing expression of p53, growth factors, cytokines
and chemokine receptors and STAT3 (signal transducer and activator
of transcription 3) (39,42–45).
However, to date, the issue of whether and how SMC1A interacts with
other regulators is poorly understood, and further investigation is
warranted to elucidate the detailed mechanisms underlying the
action of SMC1A.
In conclusion, our findings strongly suggest the
significance of the cohesin gene SMC1A in modulating the growth and
invasiveness of lung cancer and indicate that downregulation of
SMC1A expression induces growth suppression of human pulmonary
adenocarcinoma A549 and H1299 cells via G1/S phase cell cycle
arrest and apoptosis pathways. Hence, this study extends our
knowledge of the oncogenesis of lung cancer, and indicates that
SMC1A may serve as a new molecular target.
Acknowledgements
This study was supported by grants
from the Science and Technology Services of Jilin Province
Scientific and Technological Project (#20070720, #200805120 and
#20090732) and the Natural Science Foundation of China (#30670301,
#30870354 and #81272472).
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Ellison LF and Gibbons L; Canadian Cancer
Survival Analysis Group: Five-year relative survival from prostate,
breast, colorectal and lung cancer. Health Rep. 13:23–34.
2001.PubMed/NCBI
|
|
3
|
Paleari L, Russo P, Cesario A and Granone
P: Factors predicting poor survival after resection of stage IA
non-small cell lung cancer. J Thorac Cardiovasc Surg. 136:241–242.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Miller YE: Pathogenesis of lung cancer:
100 year report. Am J Respir Cell Mol Biol. 33:216–223.
2005.PubMed/NCBI
|
|
5
|
Normanno N, Bianco C, De Luca A, Maiello
MR and Salomon DS: Target-based agents against ErbB receptors and
their ligands: a novel approach to cancer treatment. Endocr Relat
Cancer. 10:1–21. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sun S, Schiller JH, Spinola M and Minna
JD: New molecularly targeted therapies for lung cancer. J Clin
Invest. 117:2740–2750. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lynch TJ, Bell DW, Sordella R, et al:
Activating mutations in the epidermal growth factor receptor
underlying responsiveness of non-small-cell lung cancer to
gefitinib. N Engl J Med. 350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Negrini S, Gorgoulis VG and Halazonetis
TD: Genomic instability - an evolving hallmark of cancer. Nat Rev
Mol Cell Biol. 11:220–228. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Haering CH, Löwe J, Hochwagen A and
Nasmyth K: Molecular architecture of SMC proteins and the yeast
cohesin complex. Mol Cell. 9:773–788. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Michaelis C, Ciosk R and Nasmyth K:
Cohesins: chromosomal proteins that prevent premature separation of
sister chromatids. Cell. 91:35–45. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Guacci V, Koshland D and Strunnikov A: A
direct link between sister chromatid cohesion and chromosome
condensation revealed through the analysis of MCD1 in S.
cerevisiae. Cell. 91:47–57. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hauf S, Waizenegger IC and Peters JM:
Cohesin cleavage by separase required for anaphase and cytokinesis
in human cells. Science. 293:1320–1323. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nasmyth K and Haering CH: Cohesin: its
roles and mechanisms. Annu Rev Genet. 43:525–558. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jessberger R: Cohesin’s dual role in the
DNA damage response: repair and checkpoint activation. EMBO J.
28:2491–2493. 2009.
|
|
15
|
Watrin E and Peters JM: The cohesin
complex is required for the DNA damage-induced G2/M checkpoint in
mammalian cells. EMBO J. 28:2625–2635. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xu H, Yan M, Patra J, et al: Enhanced
RAD21 cohesin expression confers poor prognosis and resistance to
chemotherapy in high grade luminal, basal and HER2 breast cancers.
Breast Cancer Res. 13:R92011. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Homme C, Krug U, Tidow N, et al: Low SMC1A
protein expression predicts poor survival in acute myeloid
leukemia. Oncol Rep. 24:47–56. 2010.PubMed/NCBI
|
|
18
|
Barber TD, McManus K, Yuen KW, et al:
Chromatid cohesion defects may underlie chromosome instability in
human colorectal cancers. Proc Natl Acad Sci USA. 105:3443–3448.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Luo S, Sun M, Jiang R, Wang G and Zhang X:
Establishment of promary mouse lung adenocarcinoma cell culture.
Oncol Lett. 2:629–623. 2011.PubMed/NCBI
|
|
20
|
Kim ST, Xu B and Kastan MB: Involvement of
the cohesin protein, Smc1, in Atm-dependent and independent
responses to DNA damage. Genes Dev. 16:560–570. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yazdi PT, Wang Y, Zhao S, Patel N, Lee EY
and Qin J: SMC1 is a downstream effector in the ATM/NBS1 branch of
the human S-phase checkpoint. Genes Dev. 16:571–582. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kitagawa R, Bakkenist CJ, McKinnon PJ and
Kastan MB: Phosphorylation of SMC1 is a critical downstream event
in the ATM-NBS1-BRCA1 pathway. Genes Dev. 18:1423–1438. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sjögren C and Nasmyth K: Sister chromatid
cohesion is required for postreplicative double-strand break repair
in Saccharomyces cerevisiae. Curr Biol. 11:991–995.
2001.PubMed/NCBI
|
|
24
|
Barber TD, McManus K, Yuen KW, et al:
Chromatid cohesion defects may underlie chromosome instability in
human colorectal cancers. Proc Natl Acad Sci USA. 105:3443–3448.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Musio A, Montagna C, Mariani T, et al:
SMC1 involvement in fragile site expression. Hum Mol Genet.
14:525–533. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Porkka KP, Tammela TL, Vessella RL and
Visakorpi T: RAD21 and KIAA0196 at 8q24 are amplified and
overexpressed in prostate cancer. Genes Chromosomes Cancer.
39:1–10. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Oishi Y, Nagasaki K, Miyata S, et al:
Functional pathway characterized by gene expression analysis of
supraclavicular lymph node metastasis-positive breast cancer. J Hum
Genet. 52:271–279. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ghiselli G and Iozzo RV: Overexpression of
bamacan/SMC3 causes transformation. J Biol Chem. 275:20235–20238.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hömme C, Krug U, Tidow N, et al: Low SMC1A
protein expression predicts poor survival in acute myeloid
leukemia. Oncol Rep. 24:47–56. 2010.PubMed/NCBI
|
|
30
|
Yamamoto G, Irie T, Aida T, Nagoshi Y,
Tsuchiya R and Tachikawa T: Correlation of invasion and metastasis
of cancer cells, and expression of the RAD21 gene in oral squamous
cell carcinoma. Virchows Arch. 448:435–441. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kim DH, Behlke MA, Rose SD, Chang MS, Choi
S and Rossi JJ: Synthetic dsRNA Dicer substrates enhance RNAi
potency and efficacy. Nat Biotechnol. 23:222–226. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Guo W, Zhang Y, Chen T, et al: Efficacy of
RNAi targeting of pyruvate kinase M2 combined with cisplatin in a
lung cancer model. J Cancer Res Clin Oncol. 137:65–72. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Manilla P, Rebello T, Afable C, et al:
Regulatory considerations for novel gene therapy products: a review
of the process leading to the first clinical lentiviral vector. Hum
Gene Ther. 16:17–25. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gao N, Zhang XY, Jiang R, et al:
Lentivirus-mediated RNA interference and over-expression of CDK2AP1
cDNA regulate CDK2AP1 expression in human lung cancer A549 cells.
Chem Res Chin Univ. 27:445–449. 2011.
|
|
35
|
Sun M, Jiang R, Li JD, et al: MED19
promotes proliferation and tumorigenesis of lung cancer. Mol Cell
Biochem. 355:27–33. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
|
|
37
|
Elstein KH and Zucker RM: Comparison of
cellular and nuclear flow cytometric techniques for discriminating
apoptotic subpopulations. Exp Cell Res. 211:322–331. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Riccardi C and Nicoletti I: Analysis of
apoptosis by propidium iodide staining and flow cytometry. Nat
Protoc. 1:1458–1461. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen HY, Yu SL, Chen CH, et al: A
five-gene signature and clinical outcome in non-small-cell lung
cancer. N Engl J Med. 356:11–20. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mannini L and Musio A: The dark side of
cohesin: the carcinogenic point of view. Mutat Res. 728:81–87.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lin RK, Wu CY, Chang JW, et al:
Dysregulation of p53/Sp1 control leads to DNA methyltransferase-1
overexpression in lung cancer. Cancer Res. 70:5807–5817. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Altundag O, Altundag K, Morandi P and
Gunduz M: Cytokines and chemokines as predictive markers in
non-small cell lung cancer patients with brain metastases. Lung
Cancer. 47:291–292. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Shaozhang Z, Xiaomei L, Aiping Z, Jianbo
H, Xiangqun S and Qitao Y: Detection of EML4-ALK fusion genes in
non-small cell lung cancer patients with clinical features
associated with EGFR mutations. Genes Chromosomes Cancer.
51:925–932. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang CG, Wang R, Sun M, Li J, Gao N, Zhang
XY, Shi DL and Jin CY: In vivo antitumor effect of siRNA against
STAT3 on transplanted Lewis lung cancer in mice. Chem Res Chin
Univ. 24:330–337. 2008. View Article : Google Scholar
|