Introduction
At present, no studies have analyzed the total
incidence of yolk sac tumors (YSTs), however, it has been reported
that YSTs most commonly occur in the pediatric testis (1). Pediatric germ cell tumors account for
60–75% of pediatric testicular tumors, mostly as YSTs. The
occurrence of the tumor in the cerebellar hemisphere is extremely
rare and few cases have been reported in the literature (2). Endodermal sinus tumors, also known as
YSTs, belong to an inferior class of germ cell tumors (GCTs) with a
poor prognosis. The optimal treatment is the surgical resection of
the tumor, followed by adjuvant chemotherapy (including bleomycin,
etoposide and cisplatin) (3),
however, the results are poor. Approximately 80–90% of YSTs arise
in the reproductive organs, but may also occur in the extragonadal
regions (1,4–12).
There have been several previously reported intracranial cases, the
majority of which were observed in the pineal region. However, pure
primary endodermal sinus tumors that occur in the cerebellar
hemisphere are extremely rare (1).
The current study presents the case of a three-year-old male with a
cerebellar YST, which initially presented as a medulloblastoma.
Follow-up was continued for six months. Patient provided written
informed consent.
Case report
A three-year-old male presented to The First
Affiliated Hospital of Nanchang University (Nanchang, China) with a
headache that had persisted for one month, and then worsened for
the last 10 days. This was accompanied by vomiting and gait
disturbance. The remainder of the patient’s physical examination
and medical, family and surgical histories were unremarkable. At
the time of presentation, routine laboratory tests, including a
routine blood examination and coagulation indices, were within the
normal ranges. Serum tumor markers, including β-human chorionic
gonadotropin and α-fetoprotein (AFP), were not measured, as a
diagnosis of GCT was not suspected at this stage.
For further investigation, the patient was referred
to the Department of Radiology for brain magnetic resonance imaging
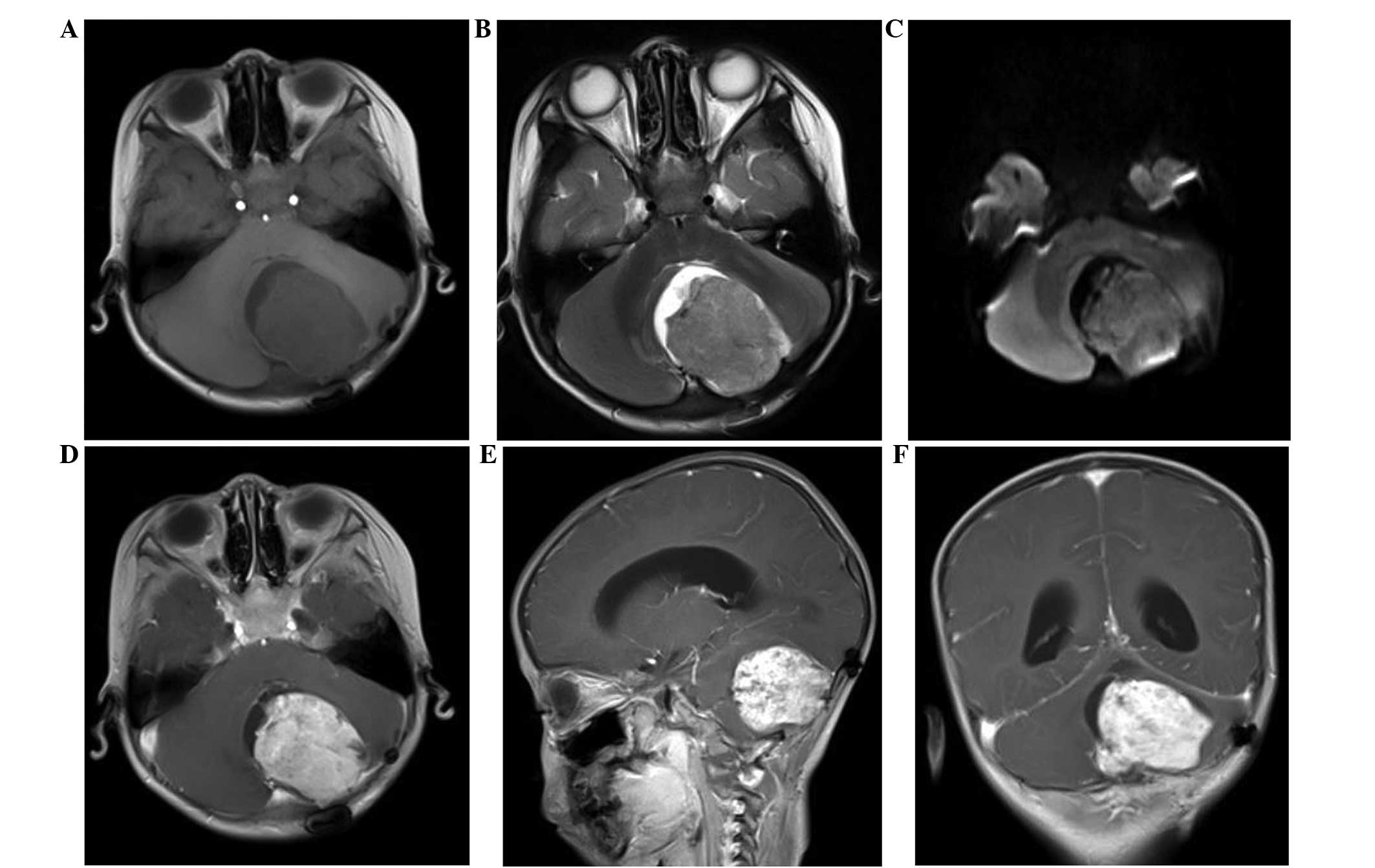
(MRI). The imaging revealed an abnormal signal mass in the left
cerebellar hemisphere (Fig. 1), but
no tumorous lesions were identified at other sites. The MRI clearly
revealed the tumors, which showed relatively homogeneous uniform
signal intensity on T1-weighted imaging, with patchy areas of a
high T1 signal. A slightly increased signal intensity was observed
on the T2- and diffusion-weighted images, while the enhanced scan
with gadolinium suggested inhomogeneous enhancement. Mild
peritumoral edema was also observed around the tumor, and the
fourth ventricle was pushed to the right side and had become
narrowed. Due to these results, medulloblastoma was initially
diagnosed.
A resection of the left cerebellar tumor was
performed. The intraoperative findings revealed a well-defined
4.0×3.0×2.5-cm tumor, with a red and white appearance, an
inconsistently soft texture and a rich blood supply. The resected
tumor was a solid mass, and the cut surface exhibiteda friable,
white-to-tan appearance. The resected specimen was characterized
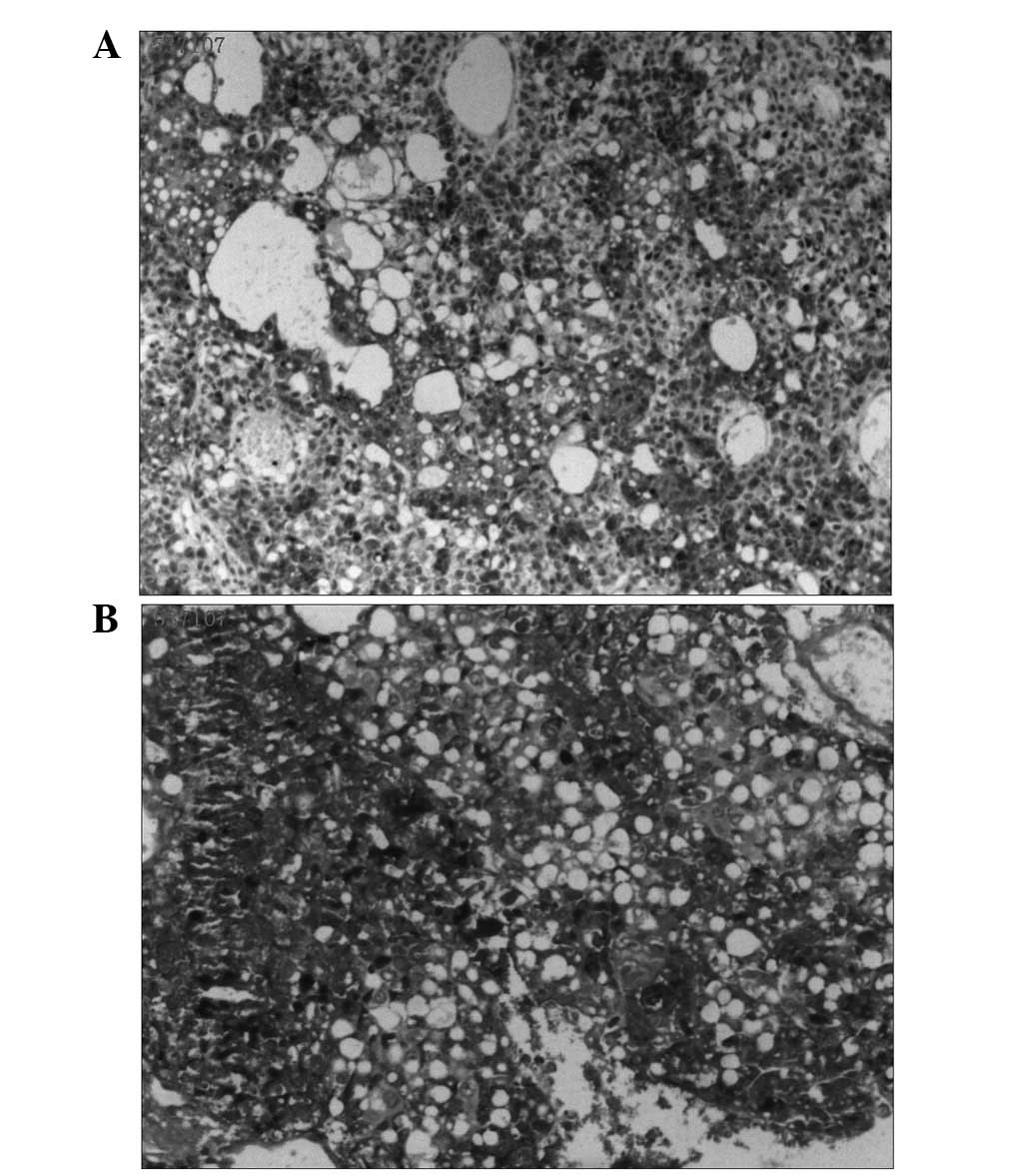
histologically by diffuse, malignant, neoplastic cells
proliferating in a microcystic or reticular pattern of growth
around the blood vessels or cavity. The neoplastic cells, forming
Schiller-Duval bodies, exhibited highly atypical, large nuclei,
with evident karyokinesis and eosinophilic bodies that partly
existed in the cytoplasm and partly in the extracellular matrix
(Fig. 2). Immunohistochemical
staining and periodic acid-Schiff (PAS) staining for AFP were
strongly positive (Fig. 3). The
histological features of the tumorous specimen indicated an
endodermal sinus tumor. The cut edge of the left cerebellar
hemisphere was not involved, and the vicinity of the tumor was free
of tumor cells. Therefore, the diagnosis of an endodermal sinus
tumor originating in the left cerebellar hemisphere was
determined.
Following surgery, the patient’s symptoms were
relieved for a while. However, subsequent to one month, MRI of the
brain revealed tumor recurrence in the same region (Fig. 4). After another month, the relapsing
mass had increased in size (Fig. 5)
and the patient’s conditioned had worsened, resulting in the
patient succumbing to the disease six months after the
diagnosis.
Discussion
Endodermal sinus tumors are rare malignant GCTs that
usually originate from the gonads and are rarely observed
extragonadally (4). Several case
studies have reported the occurrence of this entity in the vagina,
seminal vesicle, omentum, pancreas and stomach, as well as in the
sacrococcygeal and intracranial regions (1,5–12).
GCTs originating in the intracranial region almost always occur in
the pineal gland or suprasellar regions, therefore, a tumor arising
in the cerebellar hemisphere is extremely rare (1).
The histogenesis of extragonadal YST remains
controversial. There are currently two theories that may explain
the occurrence of primary GCT at extragonadal sites; the first is
the aberrant differentiation of somatic cells, while the second is
the malignant transformation of remnant of germ cells along the
pathway of migration (6,7). The latter may be more feasible for
explaining the location of the YST in the present case.
The typical morphological structure of the tumor,
known as the Schiller-Duval body, is a glomerulus-like structure
composed of a monolayer of cubic or columnar neoplastic cells
wrapping around the capillaries, thin-walled blood sinus or small
venous blood vessels. This forms vessel-centered, sleeve-shaped
structures, similar to glomerulus-like structures (6,13).
In the present case, the patient had initially been
diagnosed with a medulloblastoma, however, the post-operative
pathology confirmed the lesion to be a YST, indicating that caution
must be taken when conducting initial examinations in order to
avoid misdiagnosis. Initially, the case was consistent with a
medulloblastoma with regard to the age distribution and
predilection site, and due to its rarity in the cerebellar
hemisphere, primary YST was not considered. Furthermore, the
clinical and imaging manifestations of YST are not specific. The
clinical symptoms are associated with the location of the tumor,
whereas the final diagnosis mainly depends on the pathology.
Notably, the present patient did not receive the pre-operative
tests associated with YST, particularly the test for serum AFP. To
the best of our knowledge, the serum AFP levels in YST patients are
likely to significantly increase when these types of tumors contain
YST elements. It has also been confirmed that an increased AFP
level in the serum and cerebrospinal fluid correlates with the
presence of a YST tumor (14). In
the current case, according to the microscopic pathology,
immunohistochemical staining and PAS staining of the tumor, a final
diagnosis of YST was determined. Studies have suggested that AFP,
glypican-3 and Sal-like protein 4 are sensitive diagnostic markers
for YST (6,13). In particular, AFP is useful not only
for immunohistochemical staining and pathological diagnosis, but
also for assessing the response to treatment and detecting
recurrence (1).
Patients diagnosed with YST generally have a poor
prognosis (15,16), and in the present case, the infant
succumbed to the disease. This may be attributed to the fact that
only surgical treatment was administered as opposed to combined
chemotherapy, as chemotherapy is known to be an extremely effective
treatment, which may improve quality of life and prolong survival
time (17).
In conclusion, the current study presents an
extremely rare case of primary YST originating in the left
cerebellar hemisphere. Extragonadal YST is aggressive and difficult
to diagnose, and further investigations are required to define its
pre-operative diagnosis and to further optimize the treatment
regimen.
References
|
1
|
Cheon HC, Jung S, Moon KS, et al: Primary
endodermal sinus tumor of the cerebellar hemisphere: a case report
with review of the literature. J Neurooncol. 77:173–176. 2006.
|
|
2
|
Zhao J, Chen C, Zhang H, Shen J, Zhang H,
Lin X, et al: Evaluation of cloned cells, animal model, and ATRA
sensitivity of human testicular yolk sac tumor. J Trans Med.
10:462012.
|
|
3
|
Guida M, Pignata S, Palumbo AR, Miele G,
Marra ML, Visconti F and Zullo F: Laparoscopic treatment of a Yolk
Sac Tumor: case report and literature review. Transl Med UniSa.
7:1–5. 2013.
|
|
4
|
McKenney JK, Heerema-McKenney A and Rouse
RV: Extragonadal germ cell tumors: a review with emphasis on
pathologic features, clinical prognostic variables, and
differential diagnostic considerations. Adv Anat Pathol. 14:69–92.
2007.
|
|
5
|
Khanchel-Lakhoua F, Koubâa-Mahjoub W,
Jouini R, et al: Sacrococcygeal yolk sac tumor: an uncommon site.
APSP J Case Rep. 3:172012.
|
|
6
|
Yao XD, Hong YP, Ye DW and Wang CF:
Primary yolk sac tumor of seminal vesicle: a case report and
literature review. World J Surg Oncol. 10:1892012.
|
|
7
|
Harano K, Ando M, Sasajima Y, et al:
Primary yolk sac tumor of the omentum: a case report and literature
review. Case Rep Oncol. 5:671–675. 2012.
|
|
8
|
Magni E, Sonzogni A and Zampino MG:
Primary pure gastric yolk sac tumor. Rare Tumors. 2:e102010.
|
|
9
|
Kim YS, Kim SH, Seong JK, et al: Gastric
yolk sac tumor: a case report and review of the literature. Korean
J Intern Med. 24:143–146. 2009.
|
|
10
|
Dhanasekharan A, Cherian AG, Emmanuel P
and Patel K: Endodermal sinus tumor of the vagina in a child. J
Obstet Gynaecol India. 62(Suppl 1): S81–S82. 2012.
|
|
11
|
Chauhan S, Nigam JS, Singh P, et al:
Endodermal sinus tumor of vagina in infants. Rare Tumors. 5:83–84.
2013.
|
|
12
|
Zhang B, Gao S, Chen Y and Wu Y: Primary
yolk sac tumor arising in the pancreas with hepatic metastasis: a
case report. Korean J Radiol. 11:472–475. 2010.
|
|
13
|
Talerman A: Germ cell tumors of the ovary.
Curr Opin Obstet Gynecol. 9:44–47. 1997.
|
|
14
|
Arita N, Bitoh S, Ushio Y, et al: Primary
pineal endodermal sinus tumor with elevated serum and CSF
alphafetoprotein levels. Case report. J Neurosurg. 53:244–248.
1980.
|
|
15
|
Jennings MT, Gelman R and Hochberg F:
Intracranial germ-cell tumors: natural history and pathogenesis. J
Neurosurg. 63:155–167. 1985.
|
|
16
|
Sano K: Pathogenesis of intracranial germ
cell tumors reconsidered. J Neurosurg. 90:258–264. 1999.
|
|
17
|
Mann JR, Raafat F, Robinson K, et al:
UKCCSG’s germ cell tumour (GCT) studies: improving outcome for
children with malignant extracranial non-gonadal tumours -
carboplatin, etoposide, and bleomycin are effective and less toxic
than previous regimens. United Kingdom Children’s Cancer Study
Group. Med Pediatr Oncol. 30:217–227. 1998.
|