1. Introduction
Prostate cancer (PCa) is the most common malignancy
and the second leading cause of cancer-related mortality in males
in western countries. Advanced and metastatic stages of the disease
were present in 35% of 1,589 patients with PCa diagnosed by autopsy
(1). Among those patients with
localized cancer who are able to receive radical prostatectomy
(RP), ~35% will develop a recurrence (metastatic disease) within
the 10 years following surgery (2,3). For
those who present at initial diagnosis or progress with advanced or
metastatic disease, androgen deprivation therapy (ADT) can be
effective. However, the median duration of response to ADT is
limited to between 8 months and 3 years (4), and these patients will eventually
become castration resistant. Another effective treatment for
castration-resistant PCa is chemotherapy, although the median
duration of response is only 10.3 months (5). Therefore, there is a clear and urgent
need to develop additional systemic interventions for patients with
progressive PCa.
2. Role of angiogenesis in prostate
cancer
Angiogenic switch
Angiogenesis plays a critical role in PCa
progression and metastasis. Without neovascularization, tumor
growth will stop at a diameter of 2–3 mm (6). Once tumor cells are able to make their
own new blood vessels, they can further expand and metastasize in a
process termed the ‘angiogenic switch’ (7). The angiogenic switch in tumors is due
to a shift in the balance towards neovascularization, when
pro-angiogenic factors outweigh anti-angiogenic factors (8). Cancer cells and other cells in tumor
tissue, such as macrophages and fibroblasts, can secrete
pro-angiogenic factors, including vascular endothelial growth
factor (VEGF), basic fibroblast growth factor and interleukin 8,
which promote the formation of new blood vessels, causing an
increase in microvessel density (MVD) in cancer tissue.
MVD
MVD has been found to be greater in PCa than in
benign prostatic hyperplasia (BPH) and normal tissue (9,10). It
has been reported that MVD increases with increasing Gleason score
in Pca, particularly in the poorly differentiated type (11). The MVD was also significantly
correlated with cancer-specific survival in 221 patients with PCa
followed for a median of 15 years (12). In other studies, mean MVD was found
to correlate with increasing Gleason score and disease progression
(from extraprostatic extension to metastasis) in PCa (13,14).
Weidner et al showed that MVD was significantly higher in
PCa samples from patients with metastatic disease, compared with
those from patients without metastatic disease (11). Results from another study indicated
that PCa angiogenesis correlated with progression after RP in
patients with a Gleason score >7 (15). These data highlight the important
role of angiogenesis in the progression and metastasis of PCa. MVD
counts have also been shown to potentially predict tumor
progression from high-grade intraepithelial neoplasia to PCa, as
well as outcome in patients undergoing RP (16).
VEGF and angiogenesis in PCa
As tumors undergo an angiogenic switch, many
pro-angiogenic growth factors are secreted. Among these, VEGF is
the most significant and also the most prominent regulator of
physiological angiogenesis. Cells in tumor tissue, including cancer
cells, fibroblasts and macrophages, secrete VEGF to stimulate new
vessel formation in response to hypoxia (17,18).
Clinical studies comparing PCa with BPH revealed
that VEGF expression was correlated with increased levels of
angiogenesis, and that this was at least partly mediated by VEGF
(10). In PCa, serum levels of the
humoral ligand, VEGF, have been found to be significantly higher in
patients with metastatic disease, compared with those patients with
localized disease or healthy controls (19). Plasma VEGF levels have also been
found to be an independent prognostic factor in males with
metastatic PCa (20). Peyromaure
et al compared 17 patients who developed bone metastases
after RP with 23 patients who remained disease free and found that
the expression of VEGF was significantly higher in the former group
(21). Levels of VEGF in serum,
plasma or urine are correlated with patient outcome in both
localized as well as disseminated PCa (22). In a study of 50 patients with
locally advanced disease treated with radical radiotherapy, Green
et al reported a correlation between higher VEGF expression
and worse disease-specific survival (23). In addition, levels of the VEGFR
cognate receptor were found to be associated with a poorer grade of
tumor differentiation and prognosis in PCa (24). Given these findings, angiogenesis
inhibition has been targeted as a strategy to treat PCa. However,
it has become increasingly apparent that current anti-angiogenic
therapy targeting VEGF elicits only modest effects in clinical
settings.
3. Role of the RhoA/Rho kinase pathway in
angiogenesis
RhoA and its effector Rho kinase
(ROCK)
RhoA and its activation
RhoA is a small guanosine triphosphate hydrolase
(GTPase) belonging to the Ras homology (Rho) family. The Rho family
of GTPases comprise at least 23 members (25,26),
which serve as key regulators of extracellular stimulus-mediated
signaling networks involved in a diversity of cellular processes
including motility, mitosis, proliferation and apoptosis (27). RhoA promotes actin stress fiber
formation and focal adhesion assembly.
Rho GTPases function as molecular switches, cycling
between an active GTP-bound conformation and an inactive guanosine
diphosphate (GDP)-bound conformation (28,29).
When binding with GTP, they interact with downstream effectors to
propagate signal transduction (30). Intrinsic phosphatase activity
hydrolyzes GTP to GDP, deactivating RhoA function, and this process
is accelerated by interaction with GTPase-activating proteins.
Conversely, interaction with guanine-nucleotide exchange factors
facilitates the exchange of GDP to GTP, which restores the
activation of RhoA. The relative affinity difference of the
effector molecules between the GTP- and GDP-bound states of the Rho
GTPase can be as much as 100-fold, resulting in a highly-specific
interaction only with the GTP-bound activated state (Fig. 1). In addition, Rho proteins are also
regulated by guanine nucleotide dissociation inhibitors (GDIs),
which can inhibit both the exchange of GDP to GTP and the
hydrolysis of bound GTP. In the majority of cases, Rho proteins are
post-translationally modified at their C-termini by prenylation of
a conserved cysteine, and this facilitates their attachment to cell
membranes.
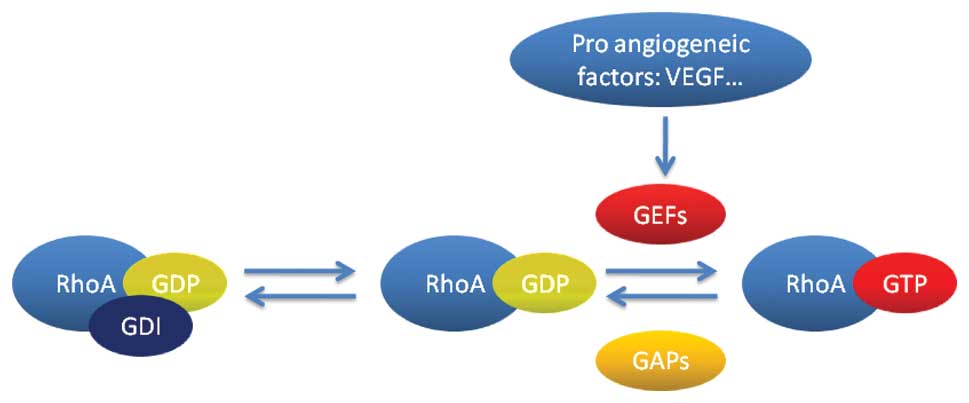 | Figure 1Regulation of RhoA. RhoA functions as
a molecular switch, cycling between an active GTP-bound
conformation and an inactive GDP-bound conformation. Intrinsic
phosphatase activity hydrolyzes GTP to GDP, deactivating RhoA
function, and this process is accelerated by interaction with GAPs.
Conversely, interaction with GEFs facilitates the exchange of GDP
to GTP, which restores the activation of RhoA. RhoA is also
regulated by GDIs, which can inhibit the exchange of GDP to GTP.
Pro-angiogenic factors may activate RhoA by GEFs. GTP, guanosine
triphosphate; GDP, guanosine diphosphate; GAPs, GTPase-activating
proteins; GEFs, guanine-nucleotide exchange factors; GDIs, guanine
nucleotide dissociation inhibitors; VEGF, vascular endothelial
growth factor. |
ROCK and its function
Rho kinase (ROCK) is a serine/threonine kinase with
a molecular mass of ~160 kDa, which has been shown to be the
principle downstream target of RhoA. There are two ROCK isoforms:
ROCK1 (ROCKβ or p160 ROCK) and ROCK2 (ROCKα or Rho kinase). ROCK1
and ROCK2 show an overall homology of 65% in their amino acid
sequence and 92% in their kinase domains. Both ROCK1 and ROCK2 are
expressed in vascular endothelial cells (ECs) (31,32).
When bound and activated by RhoA, ROCK translocates
from the cytoplasm to the cell membrane, where it increases
phosphorylation of the myosin light chain (MLC) of myosin II. This
is achieved either by direct phosphorylation, or by phosphorylation
of the regulatory myosin-binding subunit of myosin phosphatase
(also known as the phosphatase-targeting subunit), which inhibits
the phosphatase activity of this molecule (33). This, in turn, enhances actin binding
and the actin-induced adenosine triphosphatase activity of myosin,
facilitating the interaction of myosin with F-actin, and ultimately
cell contractility. ROCK proteins can also phosphorylate cofilin
indirectly via LIM kinase, and this facilitates the organization of
F-actin into stress fibers and re-arrangement of the actin
cytoskeleton (30).
RhoA/ROCK pathway and cell
motility
In eukaryotes, organization and reorganization of
the cytoskeleton underpins cellular morphology and motility. The
actin cytoskeleton is composed of actin filaments and numerous
specialized actin-binding proteins. Actin filaments are created by
the simple polymerization of actin monomers, regulated dynamically
by numerous upstream signaling pathways, notably Rho GTPases
(30). A coordinated regulation of
the actin network is essential to produce directed cell
movement.
During cell movement, RhoA is active at the trailing
edge of the cell to promote retraction, while Rac, another member
of the Rho family, is active at the leading edge, promoting
protrusion. Active RhoA has also been shown to localize at the
leading edge of migrating cells (34–36),
indicating that RhoA not only acts in retraction, but also
regulates protrusion at the front of the cell. Notably, increases
in RhoA activity were found to be correlated with increases in
protrusion rates, and were synchronous with cell-edge advancement
(36). These data highlight the
important role of RhoA in cell movement.
Association between the RhoA/ROCK pathway
and angiogenesis
Mechanism of angiogenesis
Angiogenesis is a five-step process involving a
complex series of events. Firstly, an increase in the permeability
of the basement membrane allows a new capillary to sprout. Next,
ECs activated by angiogenic factors migrate through the basement
membrane into the extracellular matrix, towards the angiogenic
stimulus. The leading front of migrating cells is driven by
enhanced proliferation of ECs. This is then followed by
re-organization of ECs to form tubules with a central lumen,
together with the recruitment of periendothelial cells (pericytes)
and vascular smooth muscle cells for new capillary stabilization
(37). The RhoA/ROCK pathway plays
a role in each of these key steps (Fig.
2).
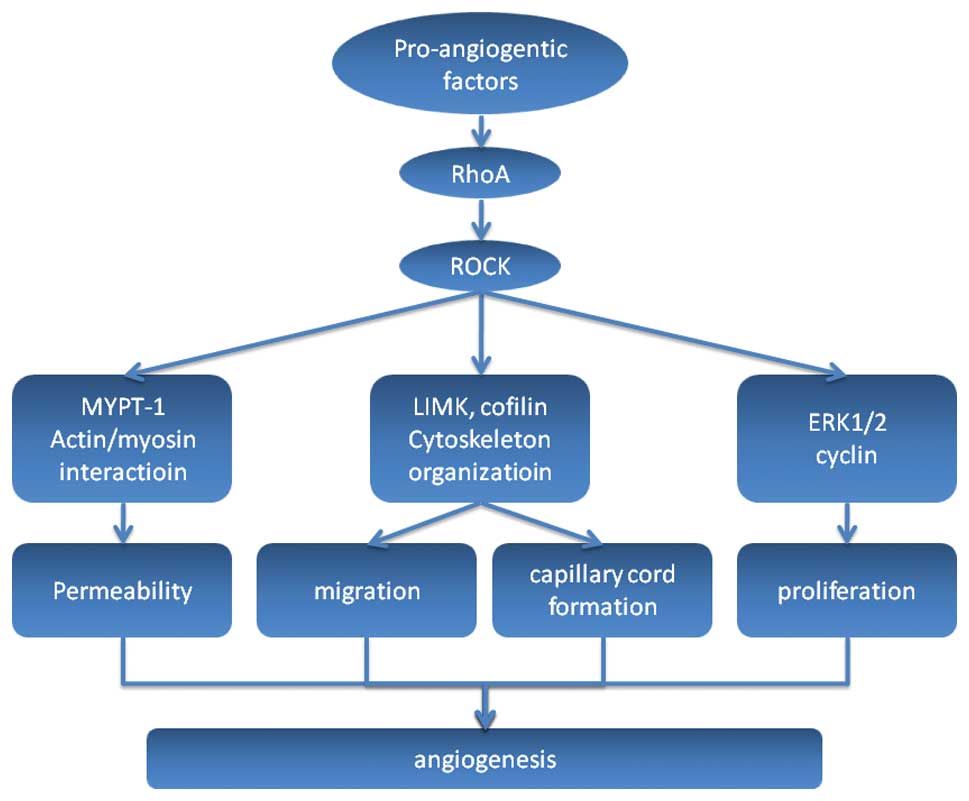 | Figure 2Illustration of the RhoA/ROCK pathway
in angiogenesis. Pro-angiogenic factors can activate the RhoA/ROCK
pathway then subsequently activate the downstream molecules that
take part in the multisteps of angiogenesis. Firstly, MYPT-1 can be
activated by ROCK and cause myosin light chain phosphorylation
followed by actin-mediated EC contraction, which leads to an
increase in the permeability of the basement membrane allowing a
new capillary to sprout. Other molecules involved in cytoskeletal
organization, such as LIMK and cofilin, are activated and cause ECs
to migrate into the extracellular matrix towards the angiogenic
stimulus. The leading front of migrating cells is driven by
enhanced proliferation of ECs, in which ERK1/2 and cyclin may play
a role when activated by ROCK. This is followed by re-organization
of ECs to form tubules with a central lumen, which finally
reorganize to result in new capillary stabilization. ROCK, Rho
kinase; MYPT-1, myosin phosphatase target subunit 1; ECs,
endothelial cells; ERK1/2, extracellular signal-regulated kinase
1/2. |
Permeability
The endothelium is a semi-permeable barrier that
lines the vasculature, comprising ECs that are connected to each
other by interendothelial junctions, consisting of protein
complexes organized as tight junctions and adherent junctions. The
latter are in the majority (38),
and are composed of vascular endothelial (VE) cadherin that
associates homotypically with VE-cadherin on adjacent cells.
VE-cadherin binds to the actin cytoskeleton. Actin-mediated EC
contraction occurs as a result of MLC phosphorylation, and this can
cause dysfunction of the endothelial barrier by inducing the
formation of small gaps between neighboring cells (39). RhoA, through its downstream effector
ROCK, plays a role in endothelial barrier dysfunction by
potentiating MLC phosphorylation via inhibition of MLC phosphatase
activity. Studies have also confirmed that RhoA contributes to
VEGF-induced hyperpermeability in the endothelium (40).
Migration
The formation of stress fibers and cellular
contraction is essential for EC migration, and these processes are
mediated by Rho GTPases (41). van
Nieuw Amerongen et al demonstrated in vitro that VEGF
induces the activation of RhoA and this increase in RhoA activity
is necessary for VEGF-induced reorganization of the F-actin
cytoskeleton. This process can be inhibited by transfection of ECs
with a RhoA dominant-negative mutant vector or by a RhoA inhibitor
C3 (42). Zhao et al showed
that increased expression of RhoA in human umbilical vein ECs
significantly enhanced cytoskeletal reorganization of transfected
cells, cell migration and angiogenic capacity, which suggests that
RhoA plays a key part in these processes in vitro (43).
Proliferation
Several lines of evidence suggest that Rho proteins
play an important role in normal and cancerous cell growth
processes, including G1 phase cell cycle progression and
mitogenesis (44). Cytokinesis is a
step in mitogenesis which is critical within the cell cycle. In
eukaryotic cells, cytokinesis requires an actin and myosin
contractile ring, which constricts and cleaves the cell, forming
two daughter cells. Inhibition of Rho GTPases prevents the assembly
of this contractile ring in a variety of mammalian cells.
Expression of constitutively activated Rho GTPases also blocks
cytokinesis, suggesting that cycling between the active and
inactive forms is required for its function (45).
The role of RhoA signaling in cell survival has been
evaluated in several non-EC cell types. Results showed that
inhibition of Rho signaling leads to apoptosis via alterations in
cell adhesion and the induction of p53 and other pro-apoptotic
proteins, or via ceramide upregulation leading to caspase cleavage
and subsequent activation (46,47).
Studies have shown that the ROCK inhibitors, fasudil and Y-27632,
not only inhibit VEGF-induced cell proliferation, but also reverse
the protective effect of VEGF on apoptosis, which results in a
decrease in viability of VEGF-stimulated ECs (48,49).
Data obtained with these inhibitors have revealed the important
role of the RhoA/ROCK pathway in EC proliferation and cell
viability.
Morphogenesis
Cultured ECs can undergo marked changes in shape and
tube formation that closely imitate pre-capillary cord formation
in vivo (50). In
vitro angiogenesis assays found that the mean tube length of
the capillary-like tubular structures formed by ECs was reduced by
transfection of a RhoA dominant-negative mutant vector, the RhoA
inhibitor C3, or the ROCK inhibitor Y-27632 (42). In another study, Zhao et al
demonstrated that overexpression of RhoA increased the tube length
in transfected ECs (43).
4. RhoA/Rho kinase pathway and angiogenesis
in prostate cancer
As discussed previously, the RhoA/ROCK pathway
participates in the process of angiogenesis in numerous types of
cancer, including PCa. Tumor blood vessels always exhibit abnormal
structure and function. A study investigating the ECs of mice
carrying the transgenic adenocarcinoma of the mouse prostate
transgene revealed that the aberrant mechanosensing of
extracellular matrix cues and resulting abnormal responses in these
cells correlated with a constitutively high level of baseline
activity of Rho GTPase and its downstream effector, ROCK (51). These findings highlighted the
important role of the RhoA/ROCK pathway in the angiogenesis of PCa.
A number of other studies have also demonstrated that RhoA/ROCK
pathway inhibitors decrease angiogenesis and cell growth in PCa
(52–54).
In an in vitro study, Y-27632 inhibited
metastatic growth of highly invasive PC3 cells in immunocompromised
mice (52). Another ROCK inhibitor,
Wf-536, greatly enhanced the in vitro inhibition of EC
migration, vacuolation, lumen and cord formation, and VEGF- and
hepatocyte growth factor-stimulated endothelial sprout formation,
when combined with the matrix metalloproteinase inhibitor,
marimastat (53). Early treatment
with a combination of Wf-536 plus marimastat, with or without
paclitaxel, of immunocompromised mice bearing xenotransplants of
PC3 cells was associated with significant inhibition of tumor
growth and increased tumor necrosis (53). Certain potential anti-angiogenic
medications, such as anacardic acid, have been found to inhibit
human prostate tumor xenograft angiogenesis by targeting the Rho
GTPase signaling pathway (54).
5. Implications for therapy
VEGF antibody bevacizumab has been approved for
numerous cancer therapies, such as for colon, lung and kidney
cancer. One of its main side effects is hypertension, while ROCK
inhibitors have vasodilation effects. For example, fasudil has
shown favorable effects in patients with angina (55) and pulmonary hypertension (56) in clinical trials and has been
approved for cerebral vasospasm by the inhibition of vessel
contraction. Their effects on anti-angiogenesis have also gradually
been discovered, as we have reviewed above. It may be feasible to
combine the ROCK inhibitor with VEGF antibody in order to enforce
their anti-angiogenesis effects while reducing the side
effects.
6. Conclusion
Inhibition of angiogenesis has been targeted as a
strategy to treat PCa. Until now, the main focus has been on the
VEGF pathway. However, it has become increasingly apparent that
current anti-angiogenic therapy elicits only modest effects in
clinical settings. The RhoA/ROCK pathway plays a crucial role in
cancer angiogenesis and should also be a potential target for
anti-angiogenic therapy. Additional studies are required to
elucidate the molecular mechanisms of the RhoA/ROCK pathway in PCa
angiogenesis, and the potential value of modulating these
mechanisms for the treatment of PCa.
Acknowledgements
This study was supported by grant from the Program
of International Science and Technology Cooperation (grant no.
2012DFG31440) of the Ministry of Science and Technology, China.
References
|
1
|
Bubendorf L, Schöpfer A, Wagner U, et al:
Metastatic patterns of prostate cancer: an autopsy study of 1,589
patients. Hum Pathol. 31:578–583. 2000.
|
|
2
|
Roehl KA, Han M, Ramos CG, Antenor JA and
Catalona WJ: Cancer progression and survival rates following
anatomical radical retropubic prostatectomy in 3,478 consecutive
patients: long-term results. J Urol. 172:910–914. 2004.
|
|
3
|
Hull GW, Rabbani F, Abbas F, Wheeler TM,
Kattan MW and Scardino PT: Cancer control with radical
prostatectomy alone in 1,000 consecutive patients. J Urol.
167:528–534. 2002.
|
|
4
|
Daneshgari F and Crawford ED: Endocrine
therapy of advanced carcinoma of the prostate. Cancer.
71:1089–1097. 1993.
|
|
5
|
Eymard JC, Oudard S, Gravis G, et al:
Docetaxel reintroduction in patients with metastatic
castration-resistant docetaxel-sensitive prostate cancer: a
retrospective multicentre study. BJU Int. 106:974–978. 2010.
|
|
6
|
Folkman J, Cole P and Zimmerman S: Tumor
behavior in isolated perfused organs: in vitro growth and
metastases of biopsy material in rabbit thyroid and canine
intestinal segment. Ann Surg. 164:491–502. 1966.
|
|
7
|
Banerjee S, Dowsett M, Ashworth A and
Martin LA: Mechanisms of disease: angiogenesis and the management
of breast cancer. Nat Clin Pract Oncol. 4:536–550. 2007.
|
|
8
|
Bergers G and Benjamin LE: Tumorigenesis
and the angiogenic switch. Nat Rev Cancer. 3:401–410. 2003.
|
|
9
|
Bigler SA, Deering RE and Brawer MK:
Comparison of microscopic vascularity in benign and malignant
prostate tissue. Hum Pathol. 24:220–226. 1993.
|
|
10
|
Stefanou D, Batistatou A, Kamina S,
Arkoumani E, Papachristou DJ and Agnantis NJ: Expression of
vascular endothelial growth factor (VEGF) and association with
microvessel density in benign prostatic hyperplasia and prostate
cancer. In Vivo. 18:155–160. 2004.
|
|
11
|
Weidner N, Carroll PR, Flax J, Blumenfeld
W and Folkman J: Tumor angiogenesis correlates with metastasis in
invasive prostate carcinoma. Am J Pathol. 143:401–409. 1993.
|
|
12
|
Borre M, Offersen BV, Nerstrom B and
Overgaard J: Microvessel density predicts survival in prostate
cancer patients subjected to watchful waiting. Br J Cancer.
78:940–944. 1998.
|
|
13
|
Brawer MK, Deering RE, Brown M, Preston SD
and Bigler SA: Predictors of pathologic stage in prostatic
carcinoma. The role of neovascularity. Cancer. 73:678–687.
1994.
|
|
14
|
Bostwick DG, Wheeler TM, Blute M, et al:
Optimized microvessel density analysis improves prediction of
cancer stage from prostate needle biopsies. Urology. 48:47–57.
1996.
|
|
15
|
Silberman MA, Partin AW, Veltri RW and
Epstein JI: Tumor angiogenesis correlates with progression after
radical prostatectomy but not with pathologic stage in Gleason sum
5 to 7 adenocarcinoma of the prostate. Cancer. 79:772–779.
1997.
|
|
16
|
Bono AV, Celato N, Cova V, Salvadore M,
Chinetti S and Novario R: Microvessel density in prostate
carcinoma. Prostate Cancer Prostatic Dis. 5:123–127. 2002.
|
|
17
|
Byrne AM, Bouchier-Hayes DJ and Harmey JH:
Angiogenic and cell survival functions of vascular endothelial
growth factor (VEGF). J Cell Mol Med. 9:777–794. 2005.
|
|
18
|
Dvorak HF, Detmar M, Claffey KP, Nagy JA,
van de Water L and Senger DR: Vascular permeability factor/vascular
endothelial growth factor: an important mediator of angiogenesis in
malignancy and inflammation. Int Arch Allergy Immunol. 107:233–235.
1995.
|
|
19
|
Duque JL, Loughlin KR, Adam RM, Kantoff
PW, Zurakowski D and Freeman MR: Plasma levels of vascular
endothelial growth factor are increased in patients with metastatic
prostate cancer. Urology. 54:523–527. 1999.
|
|
20
|
George DJ, Halabi S, Shepard TF, et al:
Cancer and Leukemia Group B 9480: Prognostic significance of plasma
vascular endothelial growth factor levels in patients with
hormone-refractory prostate cancer treated on Cancer and Leukemia
Group B 9480. Clin Cancer Res. 7:1932–1936. 2001.
|
|
21
|
Peyromaure M, Camparo P, Badoual C,
Descazeaud A and Dinh-Xuan AT: The expression of vascular
endothelial growth factor is associated with the risk of cancer
progression after radical prostatectomy. BJU Int. 99:1150–1153.
2007.
|
|
22
|
Bok RA, Halabi S, Fei DT, et al: Vascular
endothelial growth factor and basic fibroblast growth factor urine
levels as predictors of outcome in hormone-refractory prostate
cancer patients: a cancer and leukemia group B study. Cancer
research. 61:2533–2536. 2001.
|
|
23
|
Green MM, Hiley CT, Shanks JH, et al:
Expression of vascular endothelial growth factor (VEGF) in locally
invasive prostate cancer is prognostic for radiotherapy outcome.
Int J Radiat Oncol Biol Phys. 67:84–90. 2007.
|
|
24
|
Huss WJ, Hanrahan CF, Barrios RJ, Simons
JW and Greenberg NM: Angiogenesis and prostate cancer:
identification of a molecular progression switch. Cancer Res.
61:2736–2743. 2001.
|
|
25
|
Wennerberg K, Rossman KL and Der CJ: The
Ras superfamily at a glance. J Cell Sci. 118:843–846. 2005.
|
|
26
|
Bustelo XR, Sauzeau V and Berenjeno IM:
GTP-binding proteins of the Rho/Rac family: regulation, effectors
and functions in vivo. Bioessays. 29:356–370. 2007.
|
|
27
|
Etienne-Manneville S and Hall A: Rho
GTPases in cell biology. Nature. 420:629–635. 2002.
|
|
28
|
Bourne HR, Sanders DA and McCormick F: The
GTPase superfamily: conserved structure and molecular mechanism.
Nature. 349:117–127. 1991.
|
|
29
|
Bourne HR, Sanders DA and McCormick F: The
GTPase superfamily: a conserved switch for diverse cell functions.
Nature. 348:125–132. 1990.
|
|
30
|
Spiering D and Hodgson L: Dynamics of the
Rho-family small GTPases in actin regulation and motility. Cell Adh
Migr. 5:170–180. 2011.
|
|
31
|
Horowitz S, Binion DG, Nelson VM, et al:
Increased arginase activity and endothelial dysfunction in human
inflammatory bowel disease. Am J Physiol Gastrointest Liver
Physiol. 292:G1323–G1336. 2007.
|
|
32
|
Ming XF, Barandier C, Viswambharan H, et
al: Thrombin stimulates human endothelial arginase enzymatic
activity via RhoA/ROCK pathway: implications for atherosclerotic
endothelial dysfunction. Circulation. 110:3708–3714. 2004.
|
|
33
|
Somlyo AP and Somlyo AV: Ca2+
sensitivity of smooth muscle and nonmuscle myosin II: modulated by
G proteins, kinases and myosin phosphatase. Physiological reviews.
83:1325–1358. 2003.
|
|
34
|
Pertz O, Hodgson L, Klemke RL and Hahn KM:
Spatiotemporal dynamics of RhoA activity in migrating cells.
Nature. 440:1069–1072. 2006.
|
|
35
|
Kurokawa K and Matsuda M: Localized RhoA
activation as a requirement for the induction of membrane ruffling.
Mol Biol Cell. 16:4294–4303. 2005.
|
|
36
|
Machacek M, Hodgson L, Welch C, et al:
Coordination of Rho GTPase activities during cell protrusion.
Nature. 461:99–103. 2009.
|
|
37
|
Sakamoto S, Ryan AJ and Kyprianou N:
Targeting vasculature in urologic tumors: mechanistic and
therapeutic significance. J Cell Biochem. 103:691–708. 2008.
|
|
38
|
Mehta D and Malik AB: Signaling mechanisms
regulating endothelial permeability. Physiol Rev. 86:279–367.
2006.
|
|
39
|
Dudek SM and Garcia JG: Cytoskeletal
regulation of pulmonary vascular permeability. J Appl Physiol
(1985). 91:1487–1500. 2001.
|
|
40
|
Sun H, Breslin JW, Zhu J, Yuan SY and Wu
MH: Rho and ROCK signaling in VEGF-induced microvascular
endothelial hyperpermeability. Microcirculation. 13:237–247.
2006.
|
|
41
|
Kiosses WB, Daniels RH, Otey C, Bokoch GM
and Schwartz MA: A role for p21-activated kinase in endothelial
cell migration. J Cell Biol. 147:831–844. 1999.
|
|
42
|
van Nieuw Amerongen GP, Koolwijk P,
Versteilen A and van Hinsbergh VW: Involvement of RhoA/Rho kinase
signaling in VEGF-induced endothelial cell migration and
angiogenesis in vitro. Aterioscler Thromb Vasc Biol. 23:211–217.
2002.
|
|
43
|
Zhao L, Xu G, Zhou J, et al: The effect of
RhoA on human umbilical vein endothelial cell migration and
angiogenesis in vitro. Oncol Rep. 15:1147–1152. 2006.
|
|
44
|
Van Aelst L and D’Souza-Schorey C: Rho
GTPases and signaling networks. Genes Dev. 11:2295–2322. 1997.
|
|
45
|
Glotzer M: Animal cell cytokinesis. Annu
Rev Cell Dev Biol. 17:351–386. 2001.
|
|
46
|
Bobak D, Moorman J, Guanzon A, Gilmer L
and Hahn C: Inactivation of the small GTPase Rho disrupts cellular
attachment and induces adhesion-dependent and adhesion-independent
apoptosis. Oncogene. 15:2179–2189. 1997.
|
|
47
|
Petrache I, Crow MT, Neuss M and Garcia
JG: Central involvement of Rho family GTPases in TNF-alpha-mediated
bovine pulmonary endothelial cell apoptosis. Biochem Biophys Res
Commun. 306:244–249. 2003.
|
|
48
|
Yin L, Morishige K, Takahashi T, et al:
Fasudil inhibits vascular endothelial growth factor-induced
angiogenesis in vitro and in vivo. Mol Cancer Ther. 6:1517–1525.
2007.
|
|
49
|
Bryan BA, Dennstedt E, Mitchell DC, et al:
RhoA/ROCK signaling is essential for multiple aspects of
VEGF-mediated angiogenesis. FASEB J. 24:3186–3195. 2010.
|
|
50
|
Montesano R, Orci L and Vassalli P: In
vitro rapid organization of endothelial cells into capillary-like
networks is promoted by collagen matrices. J Cell Biol.
97:1648–1652. 1983.
|
|
51
|
Ghosh K, Thodeti CK, Dudley AC, Mammoto A,
Klagsbrun M and Ingber DE: Tumor-derived endothelial cells exhibit
aberrant Rho-mediated mechanosensing and abnormal angiogenesis in
vitro. Proc Natl Acad Sci USA. 105:11305–11310. 2008.
|
|
52
|
Somlyo AV, Bradshaw D, Ramos S, Murphy C,
Myers CE and Somlyo AP: Rho-kinase inhibitor retards migration and
in vivo dissemination of human prostate cancer cells. Biochem
Biophys Res Commun. 269:652–659. 2000.
|
|
53
|
Somlyo AV, Phelps C, Dipierro C, et al:
Rho kinase and matrix metalloproteinase inhibitors cooperate to
inhibit angiogenesis and growth of human prostate cancer
xenotransplants. FASEB J. 17:223–234. 2003.
|
|
54
|
Wu Y, He L, Zhang L, et al: Anacardic acid
(6-pentadecylsalicylic acid) inhibits tumor angiogenesis by
targeting Src/FAK/Rho GTPases signaling pathway. J Pharmacol Exp
Ther. 339:403–411. 2011.
|
|
55
|
Shimokawa H, Hiramori K, Iinuma H, et al:
Anti-anginal effect of fasudil, a Rho-kinase inhibitor, in patients
with stable effort angina: a multicenter study. J Cardiovasc
Pharmacol. 40:751–761. 2002.
|
|
56
|
Fukumoto Y, Matoba T, Ito A, et al: Acute
vasodilator effects of a Rho-kinase inhibitor, fasudil, in patients
with severe pulmonary hypertension. Heart. 91:391–392. 2005.
|














