Introduction
Endometrial cancer (EC) is the most frequently
occurring gynecologic malignant tumor and its incidence has been
increasing in recent years (1).
Obesity, diabetes and insulin resistance are clear risk factors
that promote the development of the more frequent type I EC
(2,3). Furthermore, obesity is associated with
an increased risk of EC fatality; obese women with EC have a
6.25-fold increased risk of succumbing to this disease compared
with non-obese counterparts (4).
The insulin-like growth factor (IGF) system is
associated with cell proliferation, obesity, diabetes and
hyperinsulinemia (5). IGF signaling
proteins are also important in the occurrence and development of
tumors (6). Indeed, the expression
levels of IGF-1, IGF-2 and IGF-1 receptor (IGF-1R) were shown to be
significantly higher in EC than in the normal endometrium (7). IGF-1 and IGF-2 are mitogenic
polypeptides of the IGF family and exert important roles in cell
growth and differentiation. The biological actions of IGF proteins
are mediated by IGF-1R, a transmembrane tyrosine kinase that is
structurally associated with the insulin receptor (8–10).
IGF-1R binds to the corresponding ligands, IGF-1, IGF-2 and
insulin, inducing autophosphorylation. This, in turn, results in
activation of distinct signaling pathways, including the
phosphatidylinositol 3-kinase-AKT/mammalian target of rapamycin
(mTOR) signaling pathway, eventually promoting cell proliferation
and suppressing apoptosis (11).
IGF-1, IGF-2, IGF-binding protein-1 (IGFBP-1) and
IGFBP-3 are expressed in normal and malignant endometrial tissues.
IGFBPs bind to IGF proteins, and are involved with the regulation
of cell proliferation, as well as the expression of IGFs. In the
human endometrium, IGFBP-1 is the predominant IGFBP. IGFBP-1 is
mainly synthesized in the liver; however, in premenopausal women,
late secretory endometrial basal cells also secrete IGFBP-1. In
obese and hyperinsulinemic patients, reduced levels of IGFBP-1 have
been observed (12,13). Notably, the expression levels of
IGF-1, IGF-2 and IGF-1R were observed to be significantly higher in
stage III and IV endometrial carcinoma tissues than in stage I or
II EC, and normal or hyperplastic endometrial tissue (14). In IGF-2- and IGF-1R-positive tumor
cells, IGF-1R-specific repressor significantly reduced cell
proliferation (14). In addition,
regardless of the IGF-2 expression status, IGF-1 and IGF-1R
expression levels were found to be positively correlated. These
previous studies suggest that IGF-1, IGF-2 and IGF-1R expression
levels are associated with the development of endometrial
adenocarcinoma, highlighting the crucial role of IGF-1R function in
EC and the importance of altered IGF-1R gene expression in
the development of the malignant phenotype (15–17).
Metformin is a safe, oral, antihyperglycemic agent
of the biguanides family and is widely used in the treatment of
type II diabetes, particularly in obese patients. Metformin is
commonly considered as an insulin sensitizer as it enhances
signaling through the insulin receptor, resulting in an decrease in
insulin resistance and subsequent reduction in circulating insulin
levels (18). Recent studies have
reported that metformin use is associated with a significant
reduction in the incidence of cancer (18,19). A
preliminary study suggested that metformin inhibits cancer cell
growth by activating adenosine monophosphate protein kinase (AMPK),
inactivating mTOR and eventually reducing the activity of the mTOR
effector S6K1 (20).
In a previous study, IGF-1 and IGF-2 were
demonstrated to promote EC cell proliferation, while metformin
inhibited this proliferation (20).
However, the effects of metformin on the IGF signaling pathway were
unclear. Therefore, the aim of the present study was to investigate
the regulatory mechanisms through which metformin affects the IGF
signaling pathway in EC cells, and to determine the effect of
metformin administered with an IGF-1R inhibitor on cell
proliferation and apoptosis.
Materials and methods
Cell lines and reagents
The Ishikawa (IK, well-differentiated) and HEC-1B
(moderately differentiated) human EC cell lines, provided by
Professor LH Wei (Peking University People’s Hospital, Beijing,
China), were maintained in phenol red-free Dulbecco’s modified
Eagle’s medium (DMEM)/F12 with 10% fetal bovine serum (FBS) at 37°C
in an atmosphere containing 5% CO2. The cell cultures
were routinely passaged every 3–5 days. Metformin and PPP (an
IGF-1R inhibitor) were purchased from Sigma-Aldrich (St. Louis, MO,
USA). IGF-1 and IGF-2 were purchased from Sigma-Aldrich and R&D
Systems (Minneapolis, MN), respectively. Compound C (an AMPK
inhibitor) was obtained from Calbiochem (Merck Millipore,
Billerica, MA, USA). Metformin was diluted in phosphate-buffered
saline (PBS) as a stock solution at a concentration of 100 mM.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The IK and HEC-1B cells were plated at a density of
2×105 cells/well in six-well plates for 24 h and were
then treated with metformin (1, 10 or 100 μM) in the presence or
absence of compound C (1 μM) in phenol red-free DMEM/F12 containing
3% steroid-stripped FBS, produced using dextran-coated charcoal
(DCC-FBS) for 72 h. Total RNA was extracted from cells with TRIzol
reagent (Invitrogen Life Technologies, Carlsbad, CA, USA) according
to the manufacturer’s instructions. RNA was subjected to DNase I
digestion to prevent possible genomic DNA contamination and then
reverse-transcribed with oligo-dT primers and M-MLV Reverse
Transcriptase (Promega Corporation, Madison, WI, USA). qPCR was
conducted using SYBR Green sequence detection reagents (Takara Bio,
Inc., Shiga, Japan) in a 20 μl reaction volume containing 1 μl
cDNA, 10 μl mix, 0.4 μl Rox and 1 μl of each primer (5 μM stock).
The primer sequences were as follows: IGFBP-1 forward:
5′-CTATGATGGCTCGAAGGCTC-3′; IGFBP-1 reverse:
5′-TTCTTGTTGCAGTTTGGCAG-3′; IGF-1R forward:
5′-AAGGCTGTGACCCTCACCAT-3′; IGF-1R reverse:
5′-CGATGCTGAAAGAACGTCCAA-3′; glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) forward: 5′-CAGTCAGCCGCATCTTCTTTT-3′, GAPDH
reverse: 5′-GTGACCAGGCGCCCAATAC-3′; GAPDH forward:
5′-CTCTCTGCTCCTCCTGTTCG-3′, GAPDH reverse:
5′-TTGATTTTGGAGGGATCTCG-3′. The PCR cycling conditions were as
follows: 95°C for 30 sec followed by 40 cycles of two steps at 95°C
for 5 sec and 60°C for 31 sec. Fluorescent signals were detected
using an ABI 7500 instrument (Applied Biosystems, Foster City, CA,
USA) and the accumulation of PCR product was measured in real-time
as the increase in SYBR green fluorescence. qPCR was performed in
triplicate for each sample. The obtained IGF-1R and
IGFBP-1 mRNA levels were calculated by normalizing the
threshold cycle (Ct) of IGF-1R and IGFBP-1 to the Ct
of GAPDH. The relative levels of IGF-1R and IGFBP-1 mRNA
were calculated by normalizing the threshold cycle (Ct) to the Ct
of GAPDH, which served as a control, using the following formula:
2-ΔΔCt. The relative levels of mRNA were then expressed
as a ratio, compared with that of the control (metformin in the
absence or presence of compound C).
Western immunoblotting
The IK and HEC-1B cells were plated at a density of
2×105 cells/well in six-well plates for 24 h and were
then treated with metformin (1, 10 or 100 μM) in the presence or
absence of compound C (1 μM) in phenol red-free DMEM/F12 containing
3% DCC-FBS for 72 h to observe the changes in IGF-1R and IGFBP-1
protein levels. Cell lysates were prepared using RIPA buffer
containing 1% NP40, 0.5 sodium deoxycholate and 0.1% sodium dodecyl
sulfate (SDS). Subsequently, 20 μg of each protein extract was
subjected to SDS-polyacrylamide gel electrophoresis in 10% gels and
subsequently electrotransferred to nitrocellulose membranes. The
membranes were blocked with 5% non-fat dry milk and 0.1% Tween-20
for 1 h at room temperature (RT) with constant agitation, and then
incubated with primary monoclonal rabbit anti-human GAPDH (1:1,000;
Cell Signaling Technology, Danvers, MA, USA), monoclonal rabbit
anti-human IGFBP1 (1:1,1000; Cell Signaling Technology) and
monoclonal rabbit anti-human IGF-1R (1:1,1000; Cell Signaling
Technology) antibodies overnight at 4°C. Subsequent to washing
three times for 5 min each with PBS and Tween-20 (PBST), the
membranes were incubated with secondary polyclonal goat anti-rabbit
horseradish peroxidase-linked antibody (1:2,000; Cell Signaling
Technology) for 2 h. The membranes were then washed again three
times for 5 min each with PBST, and bands were visualized by
enhanced chemiluminescence using the Pierce ECL Plus Western
Blotting Substrates (32132, 32134) according to the manufacturer’s
instructions (Pierce Biotechnology, Inc., Rockford, IL, USA).
Subsequent to development, the membranes were stripped and reprobed
using antibodies against GAPDH (1:1,000; Cell Signaling
Technologies) to confirm equal loading. The relative protein
expression levels was normalized to the GAPDH expression levels and
are expressed as the ratio of treated versus untreated cells.
Protein bands, including those of GAPDH, were quantified by
densitometry with the Quantity One imaging program (Bio-Rad,
Hercules, CA, USA).
Cell proliferation assays
Cell proliferation assays were performed using a
5-bromodeoxyuridine (BrdU)-enzyme-linked immunosorbent assay
(ELISA) kit (Roche Diagnostics GmbH, Mannheim, Germany). The IK and
HEC-1B cells were plated into 96-well plates at 8×103 or
1×104 cells/well, respectively. At 24 h after plating,
the cells were serum-starved for an additional 24 h and were then
treated with increasing concentrations of metformin (0.1, 1, 10 or
100 μM) in the absence or presence of PPP (0.5 or 1 μM) for 72 h.
The effects of metformin and PPP treatment were calculated as the
percentage of control cell growth obtained in PBS- or DMSO-treated
cells grown in the same 96-well plates. Assays were performed under
serum-free conditions. DNA synthesis was monitored as determined by
the incorporation of BrdU into DNA, which was detected by
immunoassay according to the manufacturer’s instructions (Roche
Diagnostics GmbH). Briefly, following incubation, the cells were
incubated again with 10 μl/well BrdU labeling solution for an
additional 2 h at 37°C. The labeling medium was removed, 200
μl/well FixDenat was added and the cells were incubated for 30 min
at 20°C. Subsequently, the FixDenat solution was removed completely
and the cells were incubated with 100 μl/well anti-BrdU POD working
solution for 90 min at 20°C. The antibody conjugate was removed and
the cells were rinsed three times with washing solution. Following
removal of the washing solution, 100 μl/well substrate solution was
added and the cells were incubated at 20°C for 20 min, followed by
incubation with 25 μl 1 M H2SO4 for 1 min on
a shaker at 100 × g. The absorbance of the samples was measured
using the Fluostar Optima ELISA reader (BMG Labtech GmbH,
Ortenberg, Germany) at 450 nm (reference wavelength, 690 nm). Each
experiment was performed in triplicate and repeated three times to
assess the consistency of the results. The BrdU assay results were
compared using MTT assays and the validity of the findings was
confirmed (data not shown).
Apoptosis assay using TdT-mediated dUTP
nick end labeling (TUNEL)
The apoptotic cells were detected in situ
using a Roche TUNEL kit (Roche Diagnostics GmbH). TUNEL was
conducted according to the manufacturer’s instructions to visualize
the 3’-OH ends of DNA fragments in apoptotic cells. Subsequent to
xylene dewaxing, the sections were rinsed three times in distilled
water for 5 min and then dipped in methanol containing 0.3%
H2O2 at RT for 30 min to inhibit endogenous
peroxidase activity. Following rinsing in PBS three times at RT for
5 min, the sections were treated with proteinase K (Sigma-Aldrich
Chemie GmbH, Mannheim, Germany) at 37°C for 8 min. The sections
were then rinsed again in PBS three times at RT for 5 min, soaked
in TdT buffer for 10 min and then incubated at 37°C for 60 min in a
moist chamber with 50 μl TdT buffer. Subsequent to rinsing in PBS
three times at RT for 5 min, the sections were placed in 50 μl
fluorescein isothiocyanate (Roche Diagnostics GmbH) and then
incubated at 37°C for 40 min. Following a further three 5-min
rinses in PBS, the sections were dipped in 3,3′-diaminobenzidine
(Roche Diagnostics GmbH) at RT for 3 min and the reaction was
observed under a microscope (Olympus IMT-2; Olympus Corporation,
Tokyo, Japan). The reactions were terminated with distilled water
and the nuclei were counterstained with hematoxylin buffer. Normal
nuclei were stained blue by DAPI, and apoptotic nuclei were stained
green using TUNEL. The number of apoptotic cells was then
calculated as a percentage of the total cells.
Statistical analysis
All data are presented as the mean ± standard error
of the mean. The data were analyzed by one-way analysis of variance
using SPSS software (version 13.0; SPSS, Inc., Chicago, IL, USA)
and P<0.05 was considered to indicate a statistically
significant difference.
Results
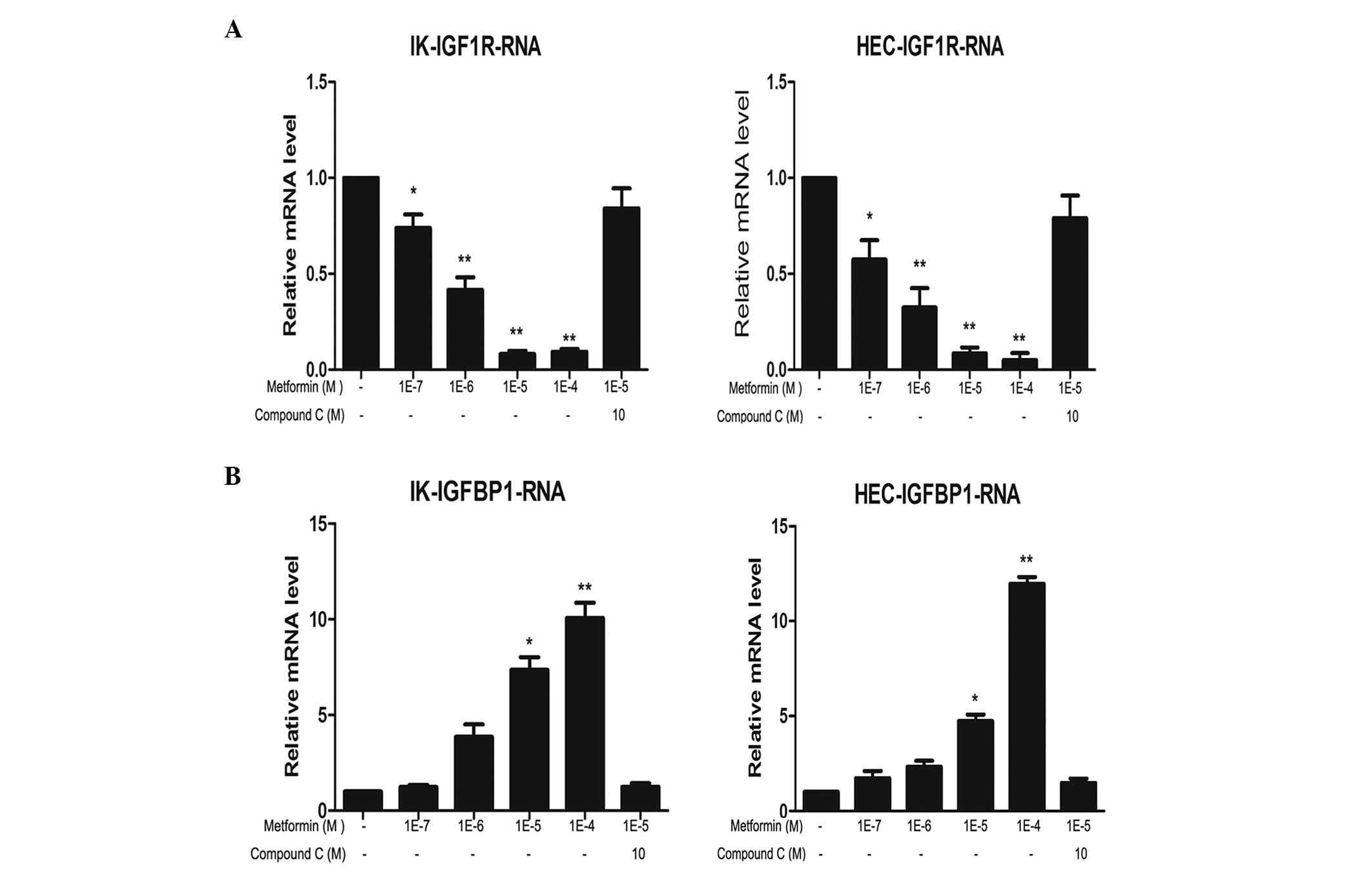
Metformin downregulates IGF-1R mRNA and
protein levels, and compound C reverses this effect
The expression levels of IGF-1R mRNA and protein in
IK and HEC-1B cells following treatment with metformin and/or
compound C were analyzed. Metformin markedly reduced IGF-1R mRNA
and protein expression levels in a concentration-dependent manner
in the two cell lines. The most evident effect was observed
following 100 μM metformin treatment. This inhibitory effect was
partially reversed by compound C treatment (Figs. 1 and 2).
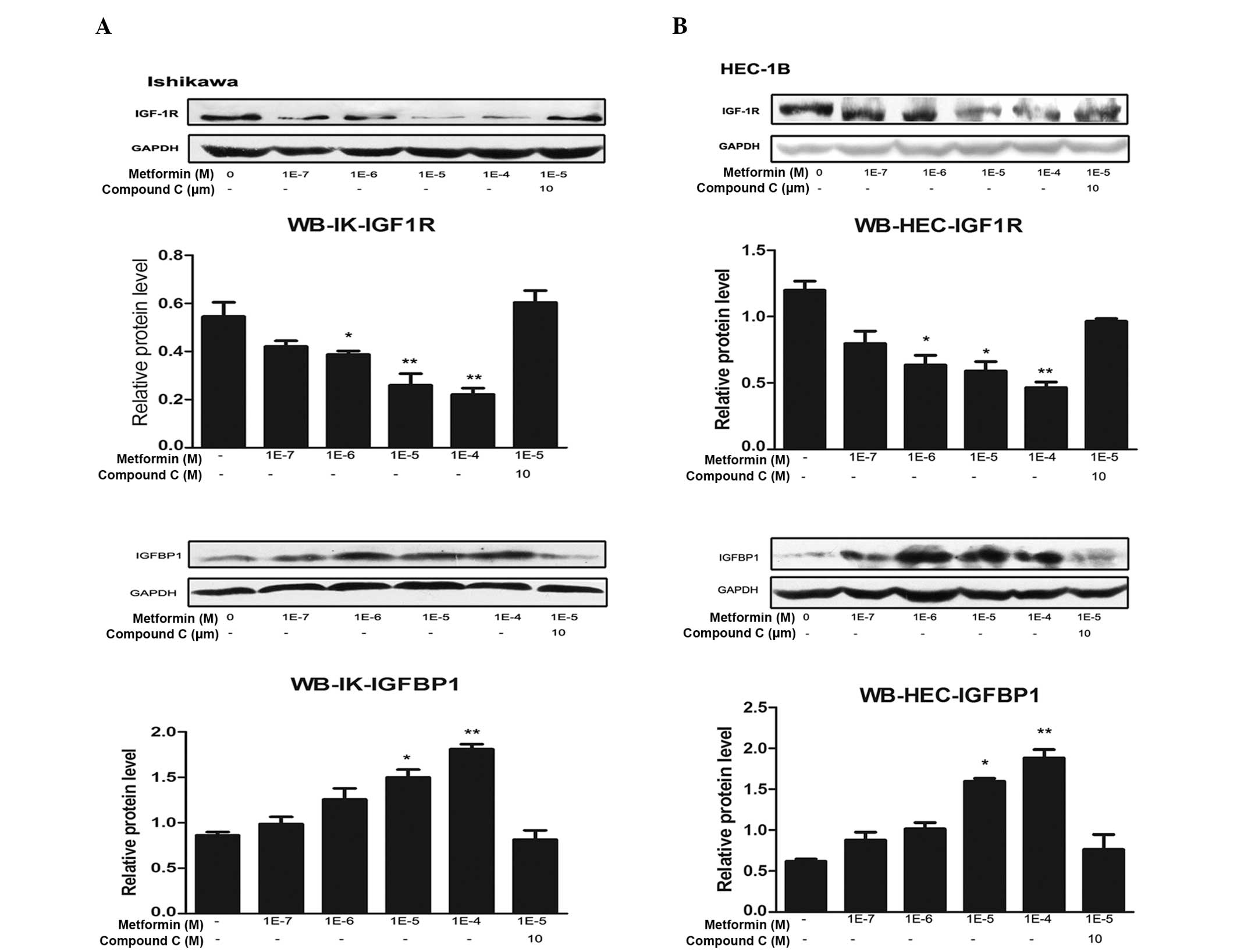 | Figure 2Metformin inhibits IGF-1R protein
expression and promotes IGFBP-1 protein expression through the AMPK
signaling pathway. (A) IK and (B) HEC-1B endometrial cancer cells
were plated at a density of 2×105 cells per well in
six-well plates. After 24 h, the cells were treated with the
indicated concentrations of metformin for 72 h in the presence or
absence of compound C (an AMPK inhibitor). Protein was then
extracted, and total protein was immunoblotted using specific
antibodies for IGF-1R and IGFBP-1. GAPDH served as a loading
control. All blots signify three independent experiments. Protein
bands, including those of GAPDH, were quantified by densitometry
with the Quantity One imaging program
(Bio-Rad).*P<0.05 vs. untreated cells and
**P<0.01 vs. untreated cells, by one-way analysis of
variance. IGF-1R, insulin-like growth factor 1 receptor; IGFBP-1,
insulin-like growth factor binding protein 1; AMPK, adenosine
monophosphate protein kinase; IK, Ishikawa; WB, western
blotting. |
Metformin upregulates IGFBP-1 mRNA and
protein levels, and compound C reverses this effect
The effects of metformin and compound C on IGFBP-1
expression levels in IK and HEC-1B cells were analyzed. Metformin
markedly increased IGFBP-1 mRNA and protein expression levels in a
concentration-dependent manner in the two cell lines. Similar to
IGF-1R, the most marked effect was detected following 100 μM
metformin treatment. This increase was partially reversed by
compound C treatment (Figs. 1 and
2).
PPP, an IGF-1R inhibitor, suppresses
cancer cell proliferation and enhances the antiproliferative
effects of metformin
The effects of metformin with or without PPP on the
proliferation of IK and HEC-1B EC cells were examined. As shown in
Fig. 3, 0.5 and 1 μM PPP
significantly inhibited the proliferation of EC cells (P<0.01).
In addition, using BrdU incorporation assays, PPP was found to
enhance the inhibitory effects of metformin on cell proliferation.
The greatest effect was observed when using 1 μM PPP combined with
10 μM metformin.
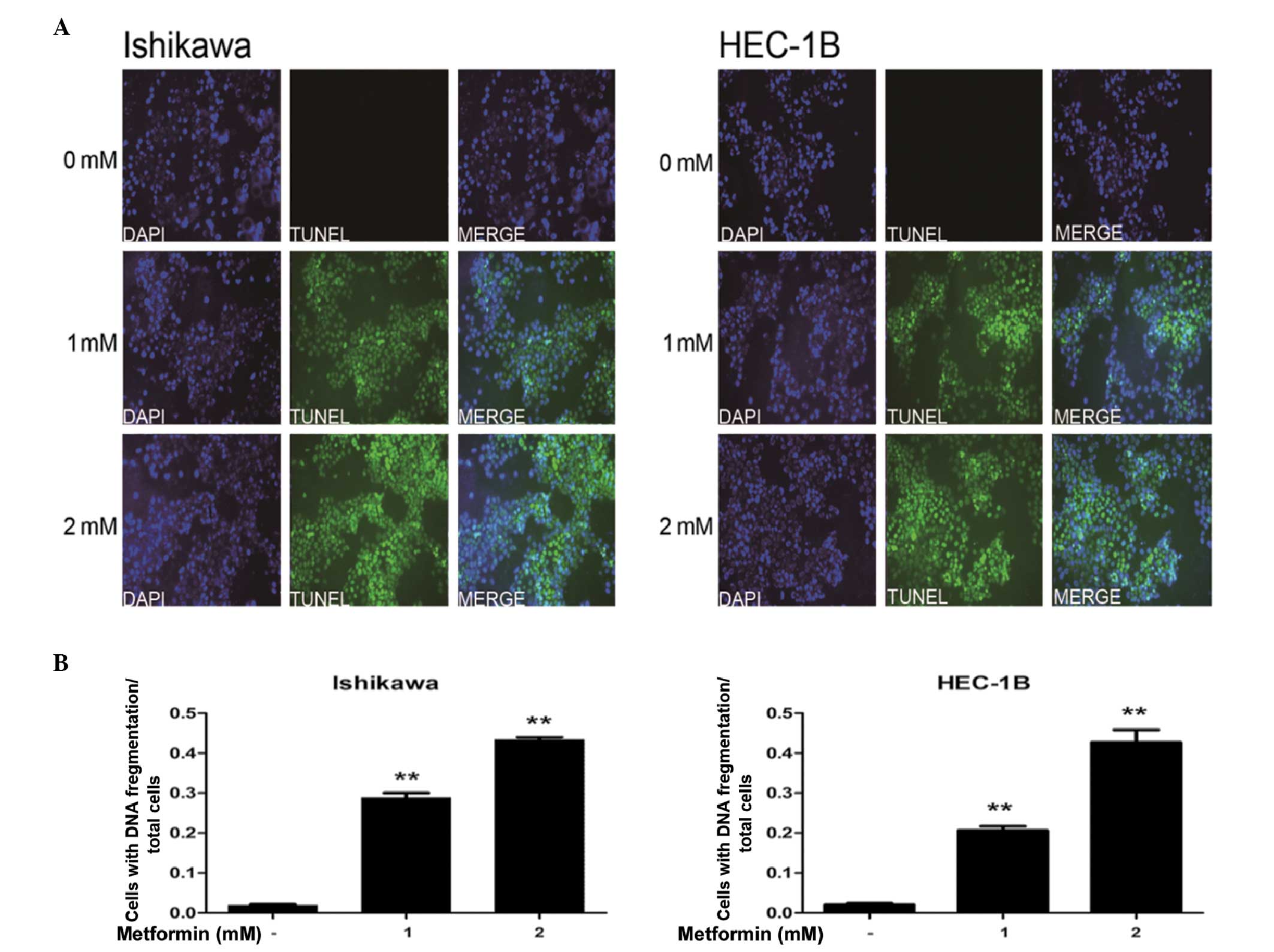
At high concentrations, metformin induces
apoptosis in EC cells
The effects of high-concentration metformin on
apoptosis in EC cells were examined using TUNEL assays. The data
demonstrated that incubation of IK and HEC-1B cells with 1 or 2 mM
metformin significantly increased the rate of apoptosis, compared
with that of the control (P<0.01; Fig. 4).
Discussion
EC is associated with obesity and diabetes (21,22).
IGF signaling proteins are expressed in endometrial tissue and are
the predominant regulatory factors of steroid hormones. The IGF
system has been shown to be associated with cell proliferation,
obesity, diabetes, hyperinsulinemia and EC. IGF proteins and
associated signaling molecules are involved in the pathogenesis of
numerous types of malignant tumor, including EC (5,6,23).
IGF-1 and IGF-2 promote mitogenic signaling and
exert antiapoptotic effects. Subsequent to binding to IGF-1R, IGF-1
and IGF-2 regulate IGF-1R tyrosine kinase activity to stimulate
cell growth (5,20). Certain studies have observed that
IGF-2 and IGF-1R are highly expressed in EC tissues compared with
normal endometrial tissues (7,13,14,24).
In the present study, an IGF-1R antagonist was demonstrated to
inhibit the growth of EC cells. Thus, IGF-1R antagonists may be
useful as secondary therapy drugs in EC patients.
Metformin significantly inhibits cell proliferation
in EC cells. This effect may be associated with the increased
expression levels of IGFBP1 in these cells. IGF-1 and IGF-2 promote
cell proliferation in EC cells. Since IGFBP1 binds to IGFs, this
reduces the levels of IGFs in the circulation. Therefore, metformin
eventually reduces the levels of IGF-1 and IGF-2 in the serum, and
reduces the role of IGF-1 and IGF-2 in promoting cell proliferation
and inhibiting apoptosis (18,20).
The effects of metformin on apoptosis remain
controversial. Cantrell et al (17) revealed that metformin induces
apoptosis in EC, but only at high concentrations. Chen et al
(25) demonstrated that apoptosis
in metformin-treated cells was significantly higher compared with
that in untreated cells, but the concentration-dependent effects of
metformin on apoptosis were not observed. By contrast, Quentin
et al (26) observed that
metformin treatment does not induce apoptosis. The results of the
present study are consistent with those of Cantrell et al
(18), in that only high
concentrations of metformin, not physiological concentrations,
induced apoptosis in the EC cells.
Increasing clinical evidence suggests a potential
correlation between biomarkers associated with the IGF1R signaling
pathway and clinical benefits from IGF1R-targeted therapies. High
IGF1R expression levels and elevated circulating IGF1 levels have
been demonstrated to be correlated with improved response to
IGF1R-targeted therapies in clinical trials of malignant tumors
(27–29). In the present study, metformin
combined with the IGF-1R receptor inhibitor PPP was found to
markedly inhibit EC cell proliferation to a greater extent than
either agent alone. This may be associated with the suppression of
the IGF signaling pathway negative feedback mechanism. Metformin
may be considered to simultaneously target multiple protein kinases
in cancer cells, such as AMPK, S6K1, human epidermal growth factor
receptor 1 (HER1), HER2 and Src (30–35).
However, the majority of studies in the field have employed a
simplified signal model, in which metformin functions as a general
inhibitor of cancer cell growth by activating AMPK, inactivating
mTOR and reducing the activity of the mTOR effector, S6K1 (36,37).
Clinically, metformin may exert direct (insulin-independent)
antitumor effects via inhibition of the AMPK/mTOR/S6K1 signaling
pathway (37,38). However, the use of rapamycin and the
corresponding analogs in the clinic has revealed that the mTOR
signaling pathway is embedded in a network of signaling cross-talk
and feedback mechanisms, significantly reducing the effectiveness
in cancer treatment (39). If
metformin has the same role as rapamycin and its analogs as
inhibitor of mTOR, cancer cells may rapidly develop autoresistance
to metformin-induced tumoricidal effects due to the negative
feedback loop between mTORC1/S6K1 and IGF-1R/IRS-1 (40).
Therefore, IGF-1R axis inhibitors combined with
metformin may act synergistically to kill tumor cells, since
metformin delays and prevents feedback from the IGF-1R signaling
pathway. The present study provides a theoretical foundation and
new ideas which may provide a basis for further animal and
subclinical studies into demonstrating the feasibility of metformin
and IGF-1R axis inhibitor combination treatment in EC.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 81202070).
References
|
1
|
Kitchener H: Management of endometrial
cancer. Eur J Surg Oncol. 32:838–843. 2006.
|
|
2
|
Chia VM, Newcomb PA, Trentham-Dietz A and
Hampton JM: Obesity, diabetes, and other factors in relation to
survival after endometrial cancer diagnosis. Int J Gynecol Cancer.
17:441–446. 2007.
|
|
3
|
Calle EE, Rodriguez C, Walker-Thurmond K
and Thun MJ: Overweight, obesity, and mortality from cancer in a
prospectively studied cohort of U.S. adults. N Engl J Med.
348:1625–1638. 2003.
|
|
4
|
Steiner E, Plata K, Interthal C, et al:
Diabetes mellitus is a multivariate independent prognostic factor
in endometrial carcinoma: a clinicopathologic study on 313
patients. Eur J Gynaecol Oncol. 28:95–97. 2007.
|
|
5
|
Augustin LS, Dal Maso L, Franceschi S, et
al: Association between components of the insulin-like growth
factor system and endometrial cancer risk. Oncology. 67:54–59.
2004.
|
|
6
|
Schernhammer ES, Holly JM, Hunter DJ,
Pollak MN and Hankinson SE: Insulin-like growth factor-I, its
binding proteins (IGFBP-1 and IGFBP-3), and growth hormone and
breast cancer risk in The Nurses Health Study II. Endocr Relat
Cancer. 13:583–592. 2006.
|
|
7
|
Roy RN, Gerulath AH, Cecutti A and
Bhavnani BR: Discordant expression of insulin-like growth factors
and their receptor messenger ribonucleic acids in endometrial
carcinomas relative to normal endometrium. Mol Cell Endocrinol.
153:19–27. 1999.
|
|
8
|
LeRoith D, Werner H, Beitner-Johnson D and
Roberts CT Jr: Molecular and cellular aspects of the insulin-like
growth factor I receptor. Endocr Rev. 16:143–163. 1995.
|
|
9
|
Baserga R, Hongo A, Rubini M, Prisco M and
Valentinis B: The IGF-I receptor in cell growth, transformation and
apoptosis. Biochim Biophys Acta. 1332:F105–F126. 1997.
|
|
10
|
Werner H: The molecular basis of IGF-I
receptor gene expression in human cancer. Insulin-Like Growth
Factors. LeRoith D, Zumkeller W and Baxter RC: Landes Bioscience;
Austin, TX: pp. 346–356. 2003
|
|
11
|
Navarro M and Baserga R: Limited
redundancy of survival signals from the type 1 insulin-like growth
factor receptor. Endocrinology. 142:1073–1081. 2001.
|
|
12
|
Rutanen EM: Insulin-like growth factors in
endometrial function. Gynecol Endocrinol. 12:399–406. 1998.
|
|
13
|
Oh JC, Wu W, Tortolero-Luna G, et al:
Increased plasma levels of insulin-like growth factor 2 and
insulin-like growth factor binding protein 3 are associated with
endometrial cancer risk. Cancer Epidemiol Biomarkers Prev.
13:748–752. 2004.
|
|
14
|
Pavelić J, Radaković B and Pavelić K:
Insulin-like growth factor 2 and its receptors (IGF 1R and IGF
2R/mannose 6-phosphate) in endometrial adenocarcinoma. Gynecol
Oncol. 105:727–735. 2007.
|
|
15
|
McCampbell AS, Broaddus RR, Loose DS and
Davies PJ: Overexpression of the insulin-like growth factor I
receptor and activation of the AKT pathway in hyperplastic
endometrium. Clin Cancer Res. 12:6373–6378. 2006.
|
|
16
|
Pengchong H and Tao H: Expression of
IGF-1R, VEGF-C and D2-40 and their correlation with lymph node
metastasis in endometrial adenocarcinoma. Eur J Gynaecol Oncol.
32:660–664. 2011.
|
|
17
|
Werner H and Bruchim I: The insulin-like
growth factor-I receptor as an oncogene. Arch Physiol Biochem.
115:58–71. 2009.
|
|
18
|
Cantrell LA, Zhou C, Mendivil A, Malloy
KM, Gehrig PA and Bae-Jump VL: Metformin is a potent inhibitor of
endometrial cancer cell proliferation - implications for a novel
treatment strategy. Gynecol Oncol. 116:92–98. 2010.
|
|
19
|
Rocha GZ, Dias MM, Ropelle ER, et al:
Metformin amplifies chemotherapy-induced AMPK activation and
antitumoral growth. Clin Cancer Res. 17:3993–4005. 2011.
|
|
20
|
Xie Y, Wang YL, Yu L, et al: Metformin
promotes progesterone receptor expression via inhibition of
mammalian target of rapamycin (mTOR) in endometrial cancer cells. J
Steroid Biochem Mol Biol. 126:113–120. 2011.
|
|
21
|
Parazzini F, La Vecchia C, Negri E, et al:
Diabetes and endometrial cancer: an Italian case-control study. Int
J Cancer. 81:539–542. 1999.
|
|
22
|
Grady D and Ernster VL: Endometrial
cancer. Cancer Epidemiology and Prevention. Schottenfeld D and
Fraumeni JF: 2nd edition. Oxford University Press; Oxford; pp.
1058–1089. 1996
|
|
23
|
Ouban A, Muraca P, Yeatman T and Coppola
D: Expression and distribution of insulin-like growth factor-1
receptor in human carcinomas. Hum Pathol. 34:803–808. 2003.
|
|
24
|
Petridou E, Koukoulomatis P, Alexe DM,
Voulgaris Z, Spanos E and Trichopoulos D: Endometrial cancer and
the IGF system: a case-control study in Greece. Oncology.
64:341–345. 2003.
|
|
25
|
Chen TW, Liang YN, Feng D, et al:
Metformin inhibits proliferation and promotes apoptosis of HER2
positive breast cancer cells by downregulating HSP90. J BUON.
18:51–56. 2013.
|
|
26
|
Quentin T, Steinmetz M, Poppe A and Thoms
S: Metformin differentially activates ER stress signaling pathways
without inducing apoptosis. Dis Model Mech. 5:259–269. 2012.
|
|
27
|
Olmos D, Martins AS, Jones RL, Alam S,
Scurr M and Judson IR: Targeting the Insulin-Like Growth Factor 1
Receptor in Ewing’s Sarcoma: Reality and Expectations. Sarcoma.
2011:4025082011.
|
|
28
|
Gualberto A, Hixon ML, Karp DD, et al:
Pre-treatment levels of circulating free IGF-1 identify NSCLC
patients who derive clinical benefit from figitumumab. Br J Cancer.
104:68–74. 2011.
|
|
29
|
de Bono JS, Attard G, Adjei A, et al:
Potential applications for circulating tumor cells expressing the
insulin-like growth factor-I receptor. Clin Cancer Res.
13:3611–3616. 2007.
|
|
30
|
Dowling RJ, Zakikhani M, Fantus IG, Pollak
M and Sonenberg N: Metformin inhibits mammalian target of
rapamycin-dependent translation initiation in breast cancer cells.
Cancer Res. 67:10804–10812. 2007.
|
|
31
|
Shell SA, Lyass L, Trusk PB, Pry KJ,
Wappel RL and Bacus SS: Activation of AMPK is necessary for killing
cancer cells and sparing cardiac cells. Cell Cycle. 7:1769–1775.
2008.
|
|
32
|
Vazquez-Martin A, Oliveras-Ferraros C, del
Barco S, Martin-Castillo B and Menendez JA: The antidiabetic drug
metformin: a pharmaceutical AMPK activator to overcome breast
cancer resistance to HER2 inhibitors while decreasing risk of
cardiomyopathy. Ann Oncol. 20:592–595. 2009.
|
|
33
|
Vazquez-Martin A, Oliveras-Ferraros C and
Menendez JA: The antidiabetic drug metformin suppresses HER2
(erbB-2) oncoprotein overexpression via inhibition of the mTOR
effector p70S6K1 in human breast carcinoma cells. Cell Cycle.
8:88–96. 2009.
|
|
34
|
Alimova IN, Liu B, Fan Z, et al: Metformin
inhibits breast cancer cell growth, colony formation and induces
cell cycle arrest in vitro. Cell Cycle. 8:909–915. 2009.
|
|
35
|
Liu B, Fan Z, Edgerton SM, et al:
Metformin induces unique biological and molecular responses in
triple negative breast cancer cells. Cell Cycle. 8:2031–2040.
2009.
|
|
36
|
Hadad SM, Fleming S and Thompson AM:
Targeting AMPK: a new therapeutic opportunity in breast cancer.
Crit Rev Oncol Hematol. 67:1–7. 2008.
|
|
37
|
Cazzaniga M, Bonanni B, Guerrieri-Gonzaga
A and Decensi A: Is it time to test metformin in breast cancer
clinical trials? Cancer Epidemiol Biomarkers Prev. 18:701–705.
2009.
|
|
38
|
Pollak M: Insulin and insulin-like growth
factor signalling in neoplasia. Nat Rev Cancer. 8:915–928.
2008.
|
|
39
|
Carracedo A, Baselga J and Pandolfi PP:
Deconstructing feedback-signaling networks to improve anticancer
therapy with mTORC1 inhibitors. Cell Cycle. 7:3805–3809. 2008.
|
|
40
|
Vazquez-Martin A, Oliveras-Ferraros C, Del
Barco S, Martin-Castillo B and Menendez JA: If mammalian target of
metformin indirectly is mammalian target of rapamycin, then the
insulin-like growth factor-1 receptor axis will audit the efficacy
of metformin in cancer clinical trials. J Clin Oncol. 27:e207–e209.
2009.
|