Introduction
Previous epidemiological studies have indicated that
dietary intake of cruciferous vegetables may protect against
multiple cancers and their protective effects are partially
credited to chemopreventive phytochemicals, particularly
isothiocyanates (ITCs) (1,2). Sulforaphane (4-methylsulfinylbutyl
ICT; SFN), a significant type of ITC, is formed by the hydrolysis
of its glucosinolate precursor, glucoraphanin, which is highly
enriched in cruciferous vegetables. As a chemopreventive compound
that interferes with various cancers, SFN targets the different
stages of carcinogenesis, including initiation, promotion and
progression (3). During cancer
initiation, SFN may enhance the detoxification and removal of
carcinogens by inducing phase II enzymes, and blocking carcinogen
activation via the inhibition of phase I enzymes. Furthermore, SFN
may block the promotion and progression of carcinogenesis by
regulating various signaling pathways to suppress cell growth and
induce cell death (4).
With respect to phase I enzyme metabolism, SFN has
been demonstrated to decrease the enzyme activities of cytochromes
P450 (CYPs) 1A1 and 2B1/2 in rat hepatocytes, as well as CYP3A4 in
human hepatocytes (5). Zhou et
al (6) further revealed that
SFN is an antagonist of human pregnane X receptor (hPXR) and
inhibits hPXR-mediated CYP3A4 expression in human primary
hepatocytes. The majority of SFN studies have focused on phase II
enzyme induction via the Keap1-nuclear factor erythroid 2-related
factor 2 (Nrf2)-antioxidant response element (ARE) signaling
pathways. SFN has been demonstrated to induce phase II enzymes,
namely glutathione-S-transferase (GST), nicotinamide adenine
dinucleotide phosphate quinine oxidoreductase and uridine
5′-diphospho (UDP)-glucuronosyltransferase (UGT) (3). UGT, the representative member of the
phase II enzyme family, conjugates endogenous and exogenous
substrates with a β-glucuronic acid moiety. Following
glucuronidation, these compounds are more water soluble and easily
excreted (7). UGT1A1 is a major
isoform of the UGT1A family and is the only isoform with bilirubin
glucuronidating activity in humans. In addition to bilirubin
glucuronidation, numerous therapeutic agents and mutagenic
xenobiotics are also substrates of UGT1A1 (8). UGT1A8 and UGT1A10 have been
demonstrated to be specifically expressed in the human intestine,
and are significant in carcinogen metabolism, as is UGT1A1, which
is expressed in the epithelium of colorectal tissues (9).
Autophagy is a protein degradation pathway by which
macromolecules and organelles are transported to lysosomes for
degradation. This process has been implicated in physiological
conditions, such as protein turnover, and in certain diseases, such
as neurodegenerative disorders, infectious diseases and cancer
(10,11). Due to the cancer-associated changes
in autophagy induction, numerous studies focus on the
pharmacological manipulation of autophagy for cancer prevention or
therapy. Various autophagy activators and inhibitors of autophagy
are currently under investigation to combat cancer, and autophagy
inhibition in particular is considered to be a promising strategy
for cancer treatment (12).
Previous studies have also revealed that SFN may induce autophagy
in human prostate (13), colon
(14), and breast (15) cancer cells and that SFN-induced
apoptosis was prevented by its induction of autophagy.
Consequently, in these studies, the cells were co-treated with the
autophagy inhibitor, 3-methyladenine (3-MA) or bafilomycin A1 to
potentiate SFN-induced apoptosis.
The above-mentioned studies of SFN predominantly
focus on cell growth arrest or cell death, which were induced by
co-treatment with SFN and autophagy modulators; the studies aimed
to promote the chemotherapeutic effects in cancer cases. However,
the aim of the current study is to elucidate the chemopreventive
properties of SFN, which may be exploited to decrease the incidence
of cancer. As described, SFN may reduce the exposure of cells to
carcinogens via the induction of phase II enzymes and inhibition of
phase I enzymes at the cancer initiation stage. Therefore, the
present study investigated whether the addition of autophagy
modulators exerts changes on phase II enzymes, and evaluated the
mechanism of changes in expression. In the present study, the
expression of UGT1A1, UGT1A8, and UGT1A10 (three representative
members of the phase II enzyme family) and transcription factors,
Nrf2 and hPXR, was analyzed in cells co-treated with SFN and
rapamycin (an activator of autophagy). In addition, the expression
of the phase I enzyme, CYP3A4 (an hPXR target gene) was assayed in
cells co-treated with SFN and rapamycin. Experiments were performed
in the human colon cancer cell line, Caco-2, an effective model for
investigating UGT expression and the hPXR signaling pathway
(16–18).
Materials and methods
Reagents
SFN (D,L-Sulforaphane) and 3-MA were purchased from
Sigma-Aldrich (St. Louis, MO, USA). Rapamycin was purchased from
Cell Signaling Technology (Beverly, MA, USA). Dulbecco’s modified
Eagle’s medium (DMEM), and fetal bovine serum (FBS) were acquired
from Gibco (Rockville, MD, USA) and cell counting kit-8 (CCK-8) was
acquired from Dojindo Laboratories (Kumamoto, Japan). The primary
rabbit anti-human polyclonal antibody against
microtubule-associated protein 1 light chain 3 (LC3) was purchased
from Cell Signaling Technology and the rabbit anti-human polyclonal
anti-Nrf2 primary antibody was manufactured by Santa Cruz
Biotechnology (Santa Cruz, CA, USA). The mouse anti-human
monoclonal anti-β-actin primary antibody and fluorescein
isothiocyanate (FITC)-conjugated monoclonal goat anti-rabbit
secondary antibodies were purchased from ZSGB-Biotechnology, Co.,
Ltd. (Beijing, China).
Cell culture
The human colon Caco-2 adenocarcinoma cells
(Shanghai Institutes for Biological Science, Chinese Academy of
Sciences; Shanghai, China) were maintained as monolayers in DMEM
supplemented with 20% non-heat-inactivated FBS, 100 U/ml penicillin
and 100 μg/ml streptomycin at 37°C in a humidified atmosphere of 5%
CO2. The cells used in the experiments were in the
logarithmic growth phase. A stock solution of SFN was initially
prepared in dimethyl sulfoxide (DMSO) and further diluted to the
required concentration in the culture medium. An equal volume of
DMSO was also added to the control cells (DMSO-treated control; 0
μM SFN). The final DMSO concentration in the culture medium was
maintained at <0.1%.
CCK-8 cytotoxicity assay
Caco-2 cells were seeded into flat-bottomed 96-well
culture plates at a density of 8×103 per well and
allowed to adhere for 24 h at 37°C. The cells were then treated
with 2.5, 5 and 10 mM of 3-MA, or 10 nM rapamycin, in the presence
or absence of 25 μM SFN for 24 h. One group of cells was treated
with only 25 μM SFN. Two control groups were included in the
experiments: One without drug treatment and one without cells. Cell
cytotoxicity was assessed using the CCK-8 kit according to the
manufacturer’s instructions. Cells were incubated in the culture
medium containing the CCK-8 reagent at 37°C for 2 h, and the
absorbance of the solution at a wavelength of 450 nm was determined
using a microplate spectrophotometer (Lambda 850; PerkinElmer,
Waltham, MA, USA). All experiments were performed in quadruplicate
wells. The rate of inhibition of cell viability was calculated
according to the following formula: Inhibition rate of cell
viability (%) = [1–A450 (sample)/A450 (control)] × 100.
Western blot analysis of LC3
expression
Caco-2 cells were treated with SFN at a range of
concentrations (0, 5, 15, 25 and 35 μM) for 24 h, and treated with
SFN at 25 μM for varying lengths of time (6, 12, 24 and 36 h). In
addition, the cells were treated with autophagy modulators alone
(2.5 mM 3-MA or 10 nM rapamycin) or in combination with 25 μM SFN
for 24 h. Cells were then washed twice in ice-cold
phosphate-buffered saline (PBS) and lysed in complete cell lysis
buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% Triton X-100,
0.25% sodium deoxycholate, 1 mM EDTA, 1 mM NaF, 1 mM
dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 1 mM activated
Na3VO4, 1 μg/ml aprotinin, 1 μg/ml leupeptin and 1 μg/ml pepstatin;
Beyotime Institute of Biotechnology, Beijing, China). The protein
concentration of the cell lysate was determined using the
bicinchoninic acid assay (Hyclone-Pierce, Logan, UT, USA). Proteins
were resolved by SDS-PAGE and transferred to polyvinylidene
difluoride (PVDF) membranes. The membranes were blocked in 5%
nonfat dry milk containing 0.1% Tween-20 at room temperature for 1
h, and then probed with the primary antibodies against LC3
(1:1,000) and β-actin (1:2,000) overnight at 4°C. Following
washing, the PVDF membranes were incubated with horseradish
peroxidase-conjugated goat anti-rabbit polyclonal secondary
antibody (1:5,000; Dako, Glostrup, Denmark). The bands were
detected using enhanced chemiluminescence and the intensities of
acquired bands were evaluated using a computerized image analysis
system (Kodak MI software; Carestream Health UK Ltd.,
Hertfordishire, UK) and normalized to β-actin (the endogenous
control).
Localization of LC3 and Nrf2 by
immunocytochemistry
Caco-2 cells were plated on sterile coverslips in a
6-well plate at a density of 1.5×105 cells per well. The
cells were divided into the six groups as described in the
cytotoxic assay subsection. Following fixing with 4%
paraformaldehyde, the cells were permeabilized with 0.3% Triton
X-100 for 5 min and blocked with 5% normal goat serum for 1 h at
25°C. Cells were subsequently incubated with the rabbit anti-LC3
(1:200) or rabbit anti-Nrf2 (1:100) primary antibodies overnight at
4°C. Following washing with PBS, the cells were immunostained with
FITC-conjugated goat anti-rabbit secondary antibody (1:100) for 1 h
at room temperature. The nuclei of the cells were stained with
4′,6-diamidino-2-phenylindole. Immunofluorescence images were
acquired using a Carl Zeiss LSM710 confocal laser microscope (Carl
Zeiss, Oberkochen, Germany; objective lens magnification, ×63).
Ultrastructures observed by transmission
electron microscopy
On entering the logarithmic phase, the Caco-2 cells
were divided into six treatment groups: 0 μM SFN (DMSO-treated
control); 25 μM SFN; 2.5 mM 3-MA; 10 nM rapamycin; 25 μM SFN plus
2.5 mM 3-MA; and 25 μM SFN plus 10 nM rapamycin. Following 24 h of
treatment, the cells were washed with PBS, collected by
centrifugation using a Minispin centrifuge (Eppendorf, Hamburg,
Germany) at 1,000 × g and fixed in ice-cold 2.5% electron
microscopy grade glutaraldehyde. The specimens were subsequently
rinsed with 0.1 M PBS, postfixed in 1% osmium tetroxide (Absin
Bioscience Inc., Beijing, China), dehydrated through a graded
series of ethanol solutions (30–90%) and processed for Epon™
embedding (Momentive Specialty Chemicals, Inc., Columbus, OH, USA).
Semi-thin sections (600–800 nm) were stained with toluidine blue
and representative areas were selected for ultra-thin sectioning.
Ultra-thin sections (50–70 nm) stained with uranyl acetate and lead
citrate were examined with a JEM-1011 electron microscope (Jeol
Ltd., Tokyo, Japan).
Quantitative reverse transcription
polymerase chain reaction (RT-qPCR)
Caco-2 cells were divided into the six
above-mentioned groups and total RNA was isolated from the cells
using TRIzol reagent (Invitrogen Life Technologies, Carlsbad, CA,
USA) according to the manufacturer’s instructions. Reverse
transcription was conducted with 1 μg RNA, oligo-dT15 primer and
Moloney murine leukemia virus reverse transcriptase (Toyobo, Osaka,
Japan) in a volume of 20 μl. The levels of UGT1A1, UGT1A8, UGT1A10,
hPXR and CYP3A4 transcripts were analyzed by RT-qPCR, with
SYBR® Green fluorescence (Toyobo) detected using the
LightCycler® system (LightCycler 2.0; Roche, Basel,
Switzerland). In all samples the levels of the reference gene,
β-actin served as an internal control for the normalization of RNA
loading and quality differences. The qPCR reaction conditions were
as follows: An initial step of 30 sec at 95°C, followed by 45
cycles for 5 sec at 95°C, 10 sec at 58°C and 15 sec at 72°C. The
experiments were performed in triplicate. Relative mRNA levels were
calculated as the ratio between the target mRNA level and the
corresponding internal control (β-actin).
Gene-specific primer sequences were acquired from
PrimerBank (http://pga.mgh.harvard.edu/primerbank/index.html)
and are shown in Table I. All
primers were synthesized by BioSune (Shanghai, China) following
BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) searches to
ensure their target specificity. Gene-specific amplifications were
identified by analyzing the melting curve data and visualizing
RT-qPCR products by agarose gel electrophoresis.
 | Table IPrimer sequences for quantitative
reverse transcription polymerase chain reaction. |
Table I
Primer sequences for quantitative
reverse transcription polymerase chain reaction.
| Gene name | Primer | Primer sequence,
5′→3′ |
|---|
| UGT1A1 | Forward |
TCCCACTTACTGCACAACAAG |
| Reverse |
GGTCCGTCAGCATGACATCA |
| UGT1A8 | Forward |
TTGATGCCTGTGCGTTAATTGT |
| Reverse |
GGCAACCTATTCCCCTGGC |
| UGT1A10 | Forward |
GCCCCGTTCCTTTATGTGTGT |
| Reverse |
ATCTTCCAGAGTGTACGAGGTT |
| β-actin | Forward |
CATGTACGTTGCTATCCAGGC |
| Reverse |
CTCCTTAATGTCACGCACGAT |
| hPXR | Forward |
TTGCCCATCGAGGACCAGAT |
| Reverse |
GTCTCCGCGTTGAACACTGT |
| CYP3A4 | Forward |
AAGTCGCCTCGAAGATACACA |
| Reverse |
AAGGAGAGAACACTGCTCGTG |
Statistical analysis
Statistical analysis was performed using SPSS
version 17.0 (SPSS, Inc., Chicago, IL, USA). Data are from a
minimum of three independent experiments and are expressed as the
mean ± standard error. P<0.05 was considered to indicate a
statistically significant difference.
Results
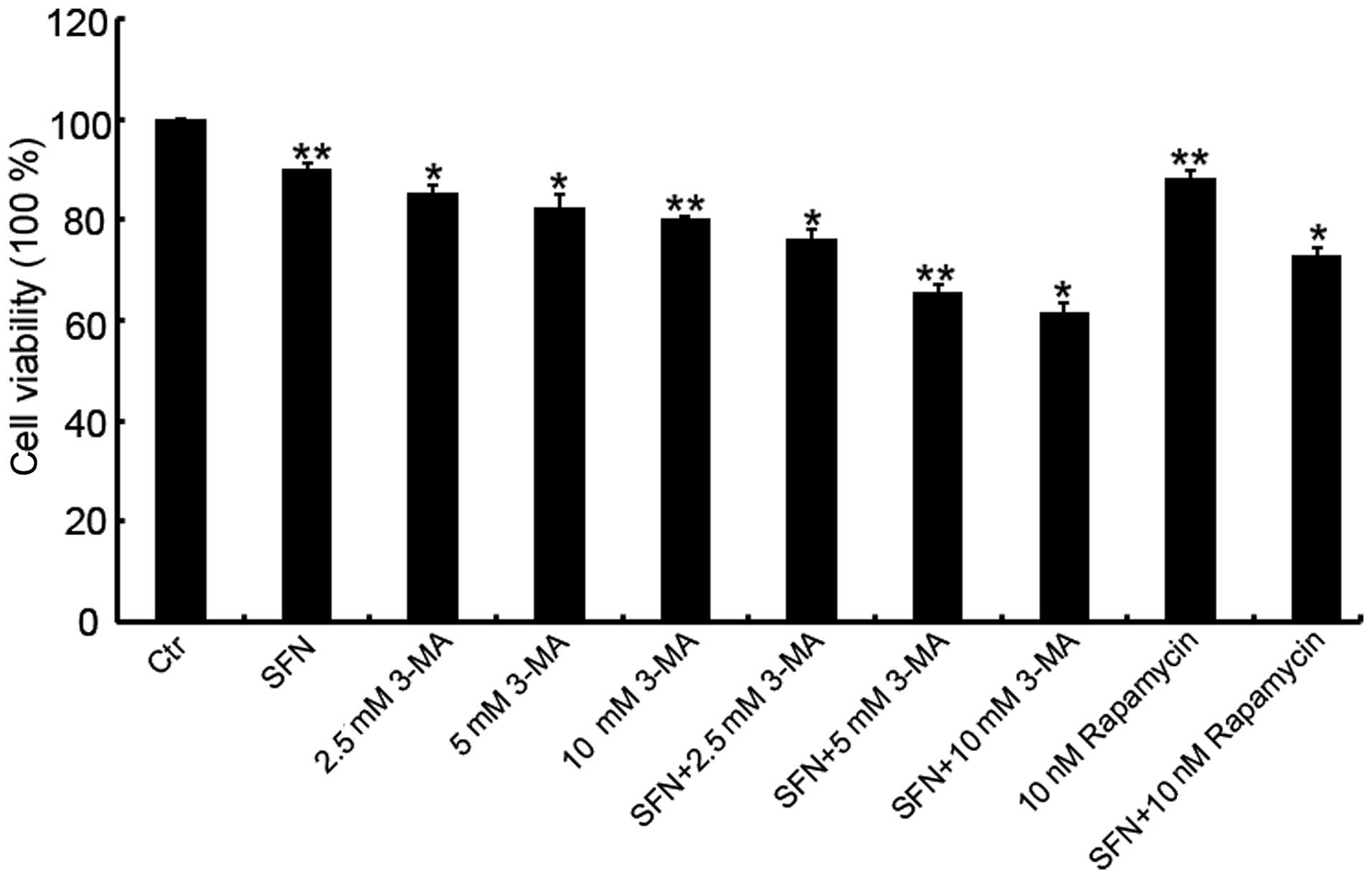
Probing the cytotoxicity of SFN and
autophagy modulators in Caco-2 cells
To limit the changes in viability due to therapeutic
agent-induced cytotoxicity, the appropriate therapeutic agent
concentration was selected to be used in future experiments by
assessing the Caco-2 cell cytotoxicity of SFN and 3-MA at various
concentrations with the CCK-8 assay. Compared with the cytotoxicity
of 25 μM SFN treatment alone, co-treatment with 3-MA exhibited a
higher cytotoxicity, and a dose-dependent increase in the
inhibition rate of cell viability was observed when 25 μM SFN was
combined with 2.5, 5 or 10 μM 3-MA (Fig. 1). To reduce the influence of
cytotoxicity on cell viability, a concentration of 2.5 μM 3-MA was
used in combination with 25 μM SFN in subsequent experiments due to
its low inhibition rate (23.9%).
The concentration of rapamycin (10 nM) that was used
in the experiments was selected based on the manufacturer’s
recommendation. The cytotoxicity of the rapamycin/SFN combination
treatment was further investigated and the results demonstrated
that this combination exhibited a relatively low inhibition rate
(26.9%; Fig. 1).
SFN, 3-MA and rapamycin modulate
autophagy
The process of autophagy commences with the
formation of double-membrane vacuoles (autophagosomes), which
enclose cytoplasmic material. Autophagosomes subsequently fuse with
late endosomes and/or lysosomes to mature into autolysosomes, where
the inner membrane and contents are degraded. To demonstrate the
autophagy-induction effect of SFN on colon cancer Caco-2 cells, and
investigate the appropriate concentration and duration of SFN
treatment, the autophagy induction was analyzed by examining the
conversion of LC3-I to LC3-II, which is the conventional approach
for monitoring autophagy induction. LC3 is an autophagosome marker
that exists in two forms: LC3-I, a 16-kDa cytosolic protein, and
LC3-II, a processed 14-kDa form localized in the outer and inner
membranes of the autophagosome (19). SFN and rapamycin are autophagy
activators while 3-MA inhibits autophagy at an early stage.
Therefore, the levels LC3-II expression positively correlate with
their degrees of autophagy induction, however, an enhancement of
LC3 expression may also indicate inhibition of the autophagic
process at a later stage.
As shown in Fig. 2A,
autophagy was induced by SFN, as a dose-dependent increase in
LC3-II levels was observed in Caco-2 cells that were treated with
SFN ranging from 0 μM (DMSO-treated control) to 25 μM; however, the
levels of LC3-II protein diminished when the concentration was
increased to 35 μM. The lysates of Caco-2 cells were subsequently
subjected to western blot analysis following treatment with 25 μM
SFN or DMSO for 6, 12, 24 and 36 h; the LC3-II levels increased in
a time-dependent manner, in the presence of 25 μM SFN, from 0–36 h
(Fig. 2B). These results indicated
that SFN induced autophagy in a dose- and time-dependent manner in
Caco-2 cells.
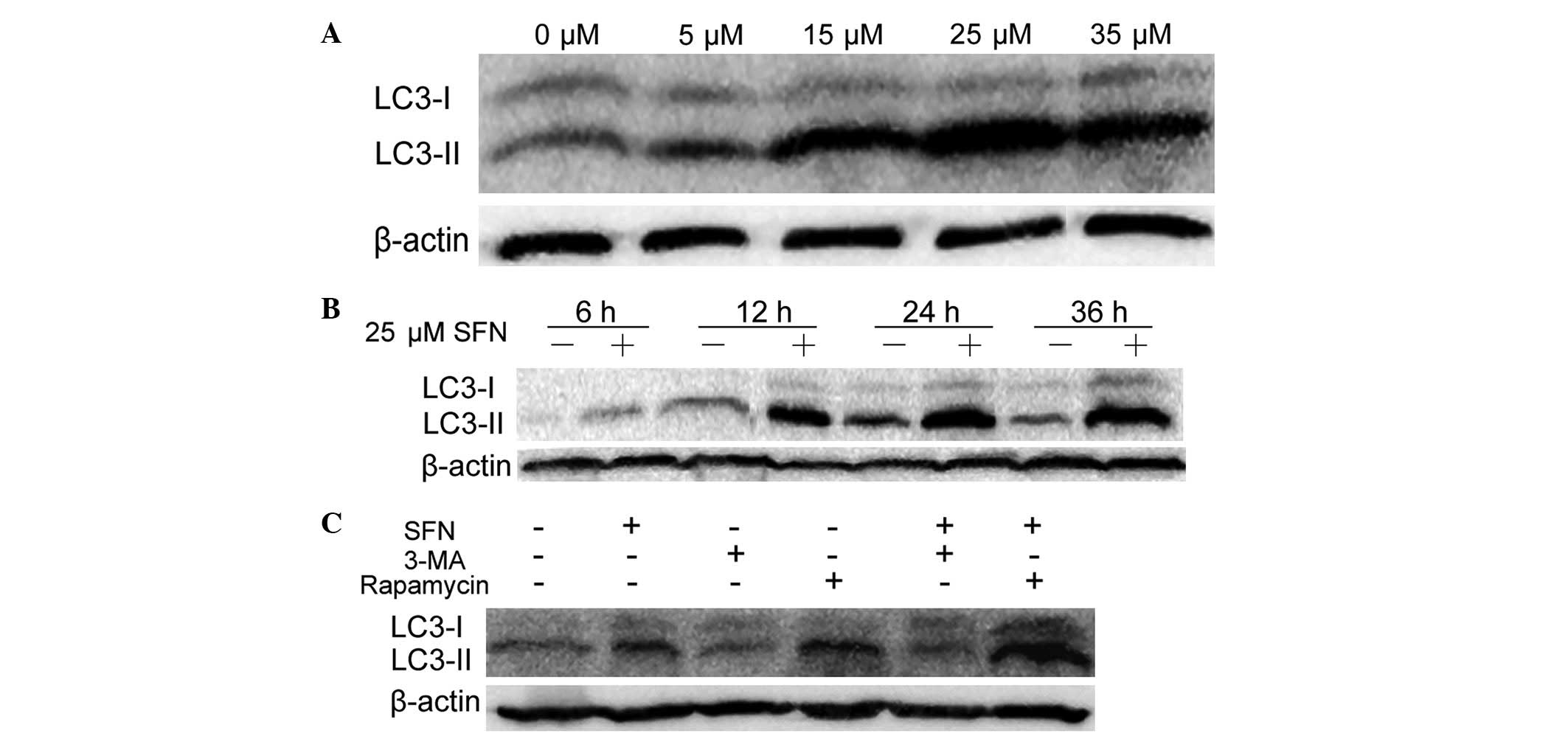 | Figure 2Western blot analysis of LC3-I and -II
using lysates from Caco-2 cells following treatment with SFN or SFN
combined with autophagy modulators. (A) Caco-2 cells treated with
0, 5, 15, 25 and 35 μM SFN for 24 h. (B) Caco-2 cells were treated
with 0 or 25 μM SFN for 6, 12, 24 and 36 h. (C) Caco-2 cells were
treated with 0 μM SFN (dimethyl sulfoxide-treated control), 25 μM
SFN, 2.5 mM 3-MA or 10 nM rapamycin in the presence or absence of
25 μM SFN for 24 h. The gel shown is representative of three
independent experiments. LC3, protein 1 light chain 3; SFN,
sulforaphane; 3-MA, 3-methyladenine. |
3-MA and rapamycin are known autophagy modulators;
3-MA has been demonstrated to inhibit autophagy at an early stage,
causing a reduction in autophagosome formation. Rapamycin is an
autophagy activator that inhibits the mammalian target of rapamycin
complex (11). To investigate the
effects of 3-MA and rapamycin on autophagy in Caco-2 cells, their
impact on LC3 conversion was analyzed. As shown in Fig. 2C, compared with the DMSO-treated
control, 3-MA reduced LC3-II protein expression levels while
rapamycin enhanced its expression levels. Compared with cells
treated only with SFN, the 3-MA/SFN combination treatment reduced
LC3-II expression levels and the rapamycin/SFN combination
treatment enhanced the expression levels. These results are
consistent with the above-mentioned inhibitory and activation
effects of 3-MA and rapamycin, respectively on autophagy
regulation.
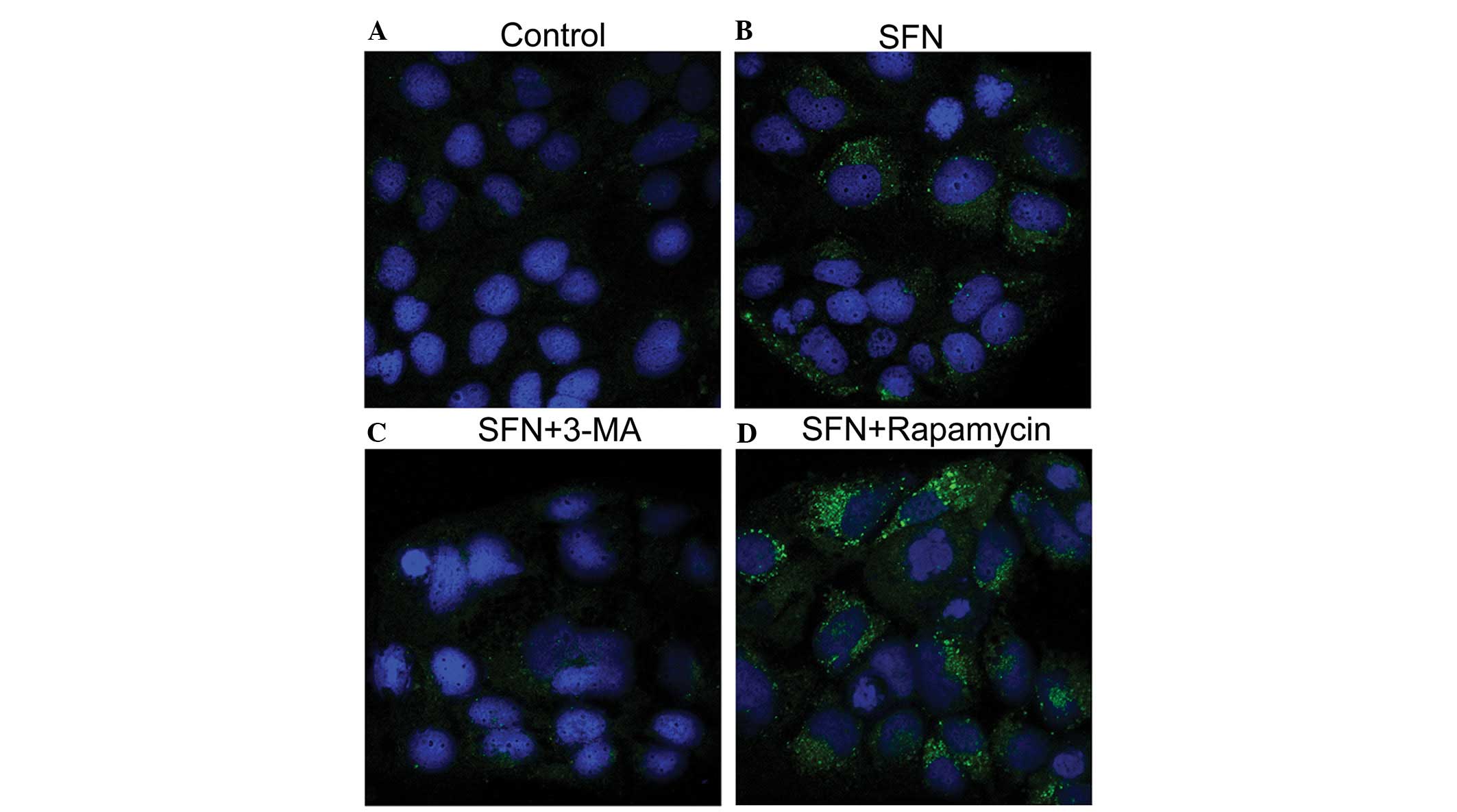
The intracellular localization of LC3 was visualized
by immunofluorescence microscopy to further establish that
autophagy is modulated by the combination treatment of Caco-2 cells
with SFN and 3-MA or rapamycin. As shown in Fig. 3, the DMSO-treated control Caco-2
cells exhibited weak and diffusely distributed LC3-associated green
fluorescence, whereas SFN treatment altered the distribution, with
coarse dots and punctate staining observed. This LC3-positive
punctate staining (LC3-II) is a typical feature of LC3 distribution
within autophagosomes (11).
Furthermore, this punctate staining was diminished by the addition
of 3-MA, while an increased quantity of LC3-positive punctates were
observed as a result of SFN/rapamycin combination treatment. These
results are largely consistent with the levels of LC3-II protein
that were observed by western blot analysis (Fig. 2C).
In addition, the ultrastructures of the cells were
examined by transmission electron microscopy (Fig. 4). Numerous membranous vacuoles,
autophagosomes and autolysosomes (indicated by the arrows),
containing residual digested material, appeared in the cytoplasm of
the SFN-, rapamycin- and SFN/rapamycin-treated cells, while
relatively few corresponding structures were observed in the
cytoplasm of the DMSO-treated control cells or in the groups of
cells that were treated with 3-MA alone or SFN/3-MA in combination
(data not shown). The observation of autophagic vacuoles further
demonstrates the effects of SFN and rapamycin on autophagy.
Rapamycin modulates the induction of
UGT1A isoform expression by SFN
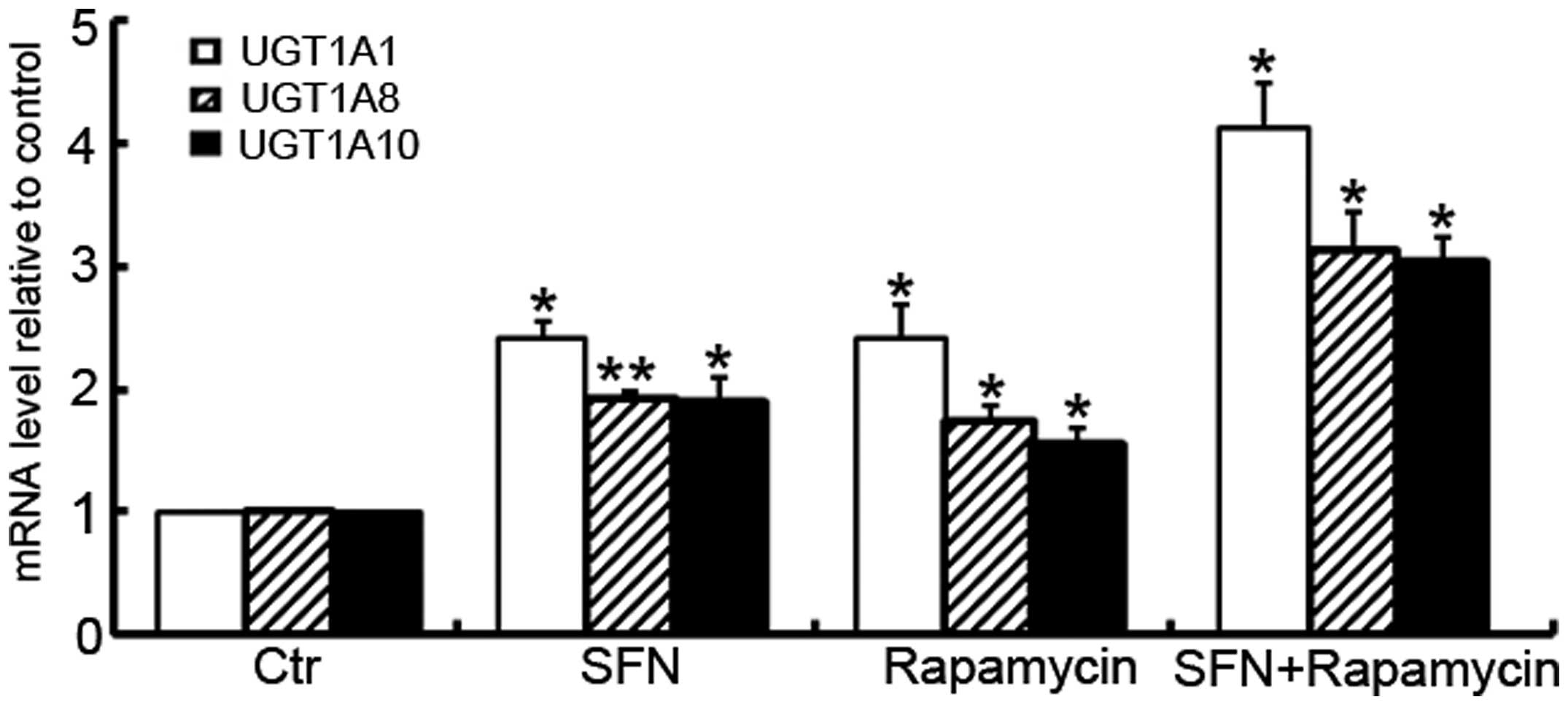
To further investigate the modulation of rapamycin
on UGT1A isoform expression by SFN, the mRNA levels of the UGT1A
isoforms for the SFN-, rapamycin- and SFN/rapamycin-treated cells
were analyzed using RT-qPCR. As shown in Fig. 5, similar trends were identified in
the induction of UGT1A isoforms, when Caco-2 cells were treated
with these small molecules. SFN-induced mRNA expression of UGT1A1,
UGT1A8 and UGT1A10 was further enhanced in the presence of
rapamycin (P=0.001, P=0.002 and P=0.003, respectively).
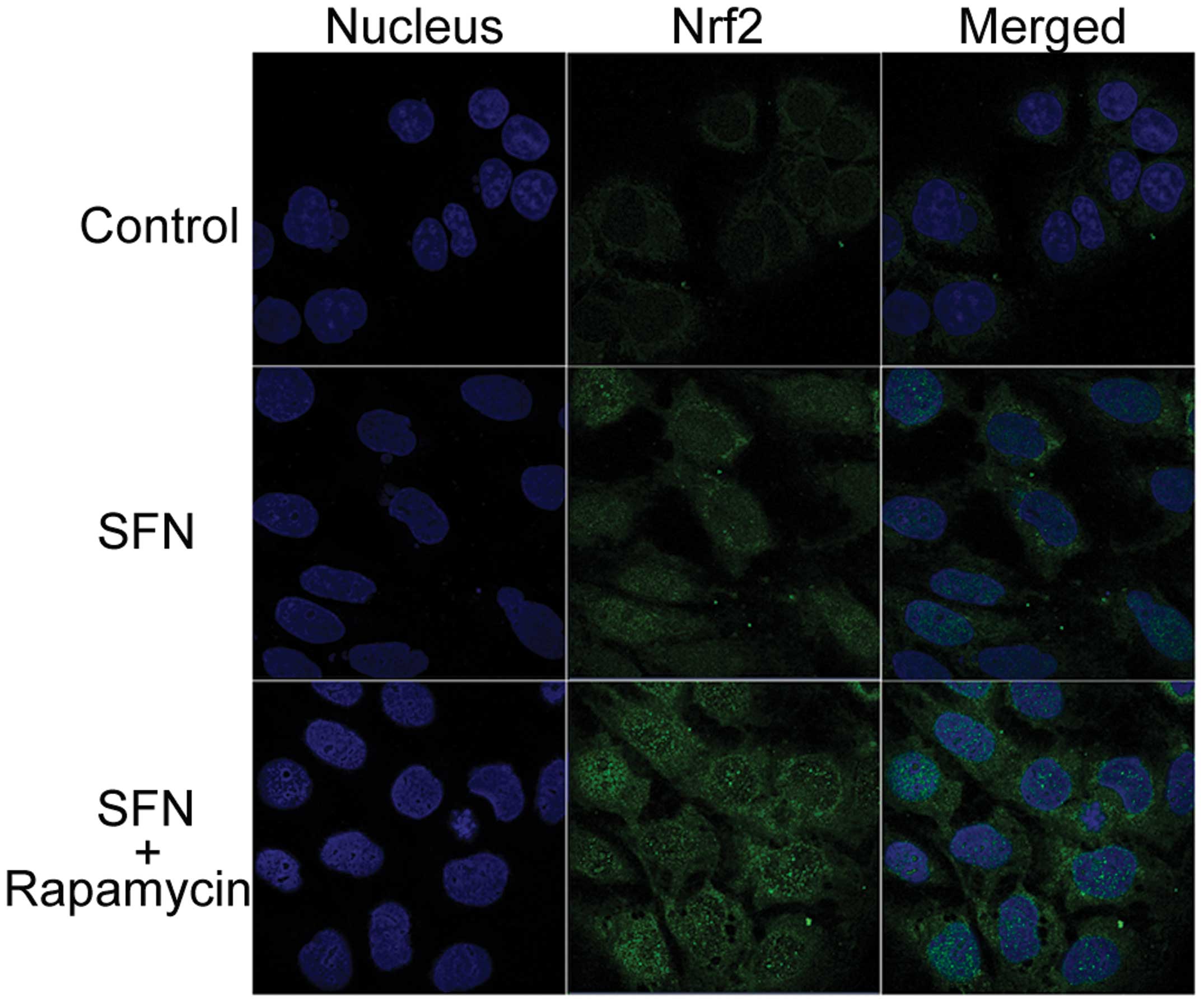
Nrf2 nuclear translocation in Caco-2
cells
Under basal conditions, Nrf2 is sequestered in the
cytoplasm by Keap1 and, therefore, is not involved in the induction
of phase II enzymes. The interaction between Nrf2 and Keap1 is
perturbed in response to chemical or oxidative stress, resulting in
the translocation of Nrf2 into the nucleus (20). Nrf2 may then bind to the AREs to
stimulate the transcription of phase II enzymes, including UGT1A
isoforms (21). Therefore, in the
present study, Nrf2 localization to the cytoplasm and nucleus of
Caco-2 cells was monitored by immunocytochemistry. As presented in
Fig. 6, a small quantity of Nrf2
protein (indicated by green fluorescence) was restricted to the
cytoplasm in the DMSO-treated control. Treatment with SFN alone
caused the accumulation of Nrf2 in the nuclear region, with
SFN/rapamycin combination treatment resulting in elevated Nrf2
protein staining, particularly increased Nrf2 nuclear staining.
This indicated that the transcription factor, Nrf2 may be
significant in the induction of UGT1A1, UGT1A8 and UGT1A10
expression. Caco-2 cells that were treated with rapamycin alone
(data not shown) exhibited similar results to the DMSO-treated
control, as Nrf2 only exhibited a marginal quantity of cytosolic
fluorescence.
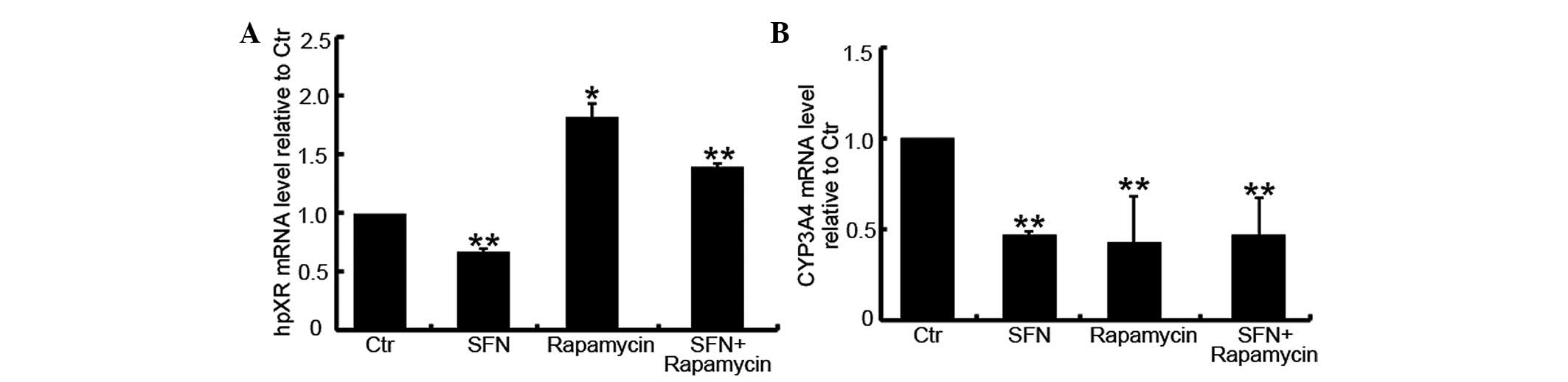
Modulation of hPXR mRNA expression by
rapamycin
As demonstrated by the findings of the present
study, the addition of rapamycin enhances the SFN-induced
expression of UGT1A isoforms. Rapamycin treatment alone was also
observed to induce UGT1A1 expression when compared with
DMSO-treated control cells. However, no apparent Nrf2 nuclear
translocation was observed in Caco-2 cells that were treated with
rapamycin. In addition to the activation of the Keap1-Nrf2-ARE
signaling pathway, the expression of UGT1A isoforms may also be
induced by the activation of hPXR (18,22).
Therefore, the levels of hPXR mRNA were investigated in Caco-2
cells treated with SFN, rapamycin or a combination of the two. As
shown in Fig. 7A, compared with the
DMSO-treated group, SFN inhibited the expression of hPXR mRNA
(P=0.024) while rapamycin enhanced its expression (P<0.001).
Furthermore, the SFN/rapamycin combination treatment group
exhibited an increased level of hPXR mRNA (P=0.008).
Modulation of CYP3A4 mRNA expression by
rapamycin
hPXR is a key transcription factor responsible for
the induction of CYP3A4 expression (23), and Zhou et al (6) reported that SFN may inhibit
hPXR-mediated CYP3A4 expression and CYP3A4-dependent drug
clearance. As rapamycin increases the mRNA expression of hPXR, the
present study evaluated whether this small molecule influences
CYP3A4 expression. As shown in Fig.
7B, compared with the DMSO-treated control group, SFN-,
rapamycin- and SFN/rapamycin-treated cells exhibited reduced levels
of CYP3A4 mRNA (P<0.001), however, no significant differences
were identified between these three groups (P>0.05). This
indicated that the induction of hPXR by rapamycin does not lead to
increased levels of CYP3A4 expression in Caco-2 cells.
Discussion
SFN has been demonstrated to activate autophagy in
human prostate (13), colon
(14), breast (15) and pancreatic carcinoma cells
(24). The present study
demonstrated that a dose-dependent increase in LC3-II expression
levels may be induced in Caco-2 cells by treatment with SFN at
concentrations ranging from 5 to 25 μM, and LC3-II levels may be
increased in a time-dependent manner by incubation with 25 μM SFN
for 6–36 h. In addition, the results indicate that SFN induces
autophagy in a dose- and time-dependent manner in Caco-2 cells.
Based on these results, 25 μM and 24 h were selected as the
concentration and duration of SFN treatment in the subsequent
experiments. To evaluate the potential role of autophagy in
SFN-mediated cancer chemoprevention, Caco-2 cells were treated with
SFN in combination with 3-MA or rapamycin. Initially, the
cytotoxicity of the co-treatment on Caco-2 cells was assessed using
a CCK-8 kit. In previous reports, 5 or 10 mM of 3-MA were used to
potentiate apoptosis, which is induced by chemoprevention or
chemotherapy agents (13,14,25,26).
Therefore, the cytotoxicity of 25 μM SFN combined with 2.5, 5 or 10
μM of 3-MA, as well as with 10 nM rapamycin (as recommended by the
manufacturer) was investigated. Based on the low level of
cytotoxicity observed in Caco-2 cells, concentrations of 2.5 μM
3-MA and 10 nM rapamycin were selected in combination with 25 μM
SFN for subsequent experiments. The ability of 3-MA and rapamycin
to inhibit or activate autophagy, respectively, in Caco-2 cells was
demonstrated by the conversion of LC3-I to LC3-II (as shown by
western blot and immunocytochemical analysis), and was also
established by the observed formation of autophagosomes and
autolysosomes by transmission electron microscopy.
In prostate, colon and breast cancer cells, the
addition of 3-MA or bafilomycin A1 potentiates SFN-induced
apoptotic cell death (15). In the
present study, co-treatment with 3-MA or rapamycin was observed to
aggravate the cytotoxicity of SFN on Caco-2 cells. In pancreatic
carcinoma (PC) cells (24), the
blockage of autophagy by chloroquine or the induction of autophagy
by rapamycin did not affect the cell viability of SFN-treated PC
cells, indicating that changes in autophagy do not influence
SFN-induced cell death in PC cells. This contradiction may be due
to the differences between cancer cell lines and autophagy
inhibitors that were analyzed.
The majority of previous studies regarding the
cancer chemopreventive properties of SFN focused on its
chemopreventive effects at the promotion and progression stages in
carcinogenesis, as well as attempting to determine whether the
regulation of autophagy (such as by co-treatment with autophagy
modulators) may promote SFN-induced apoptosis in cancer cells. The
current study focussed on elucidating the SFN-mediated
chemopreventive effects at the cancer initiation stage, and
investigated the role of autophagy in phase I and phase II enzyme
expression. In a previous study, SFN at hypotoxic levels (10–30 μM)
was observed to induce UGT1A in a dose-dependent manner, while 25
μM SFN treatment for 24 h enhanced UGT1A1, UGT1A8 and UGT1A10 mRNA
expression in Caco-2 cells (27).
To the best of our knowledge, the present study is the first to
report that rapamycin increases the mRNA expression level of
UGT1A1, and that co-treatment with rapamycin and SFN results in a
synergistic induction of UGT1A1, UGT1A8 and UGT1A10 expression at
the mRNA level. Conversely, 3-MA exhibited no influence on UGT1A
isoform expression levels, and the addition of 3-MA attenuated the
SFN-induced mRNA expression levels of the UGT1A isoforms.
The potential mechanisms by which UGT1A isoform
expression is regulated by autophagy modulators alone or in
combination with SFN was subsequently investigated. Studies have
demonstrated that several ligand-activated transcription factors
regulate the induction of UGTs, including Nrf2, aryl hydrocarbon
receptor, constitutive androstane receptor (CAR), peroxisome
proliferator-activated receptor-α and PXR (7,28). In
our previous study, SFN was observed to induce Nrf2 nuclear
translocation and activation in Caco-2 cells. The present study
demonstrated that co-treatment with SFN and rapamycin exhibited
higher Nrf2 intracellular expression levels and a more intense
nuclear staining than was observed with SFN treatment alone. This
indicates that the addition of rapamycin may enhance UGT1A isoform
expression levels through an increase in Nrf2 activation.
However, while rapamycin treatment alone enhanced
UGT1A1 expression levels in Caco-2 cells, it did not upregulate
Nrf2 intracellular expression levels or activate Nrf2 nuclear
translocation. This indicated that other transcription factors may
be participating in the rapamycin-mediated induction of UGT1A
isoforms. Certain reports have proposed that UGT1A1, UGT1A3, UGT1A4
and UGT1A6 contain functional PXR response elements and are
inducible via PXR-specific ligands (18,22,29).
Gardner-Stephen et al (18)
reported that UGT1A1 was markedly upregulated by the transfection
of hPXR variants in Caco-2 cells, and the results of UGT1A8 and
UGT1A10 were not consistent among replicates. Therefore, hPXR mRNA
transcription was examined in the present study using RT-qPCR.
Rapamycin was shown to upregulate hPXR mRNA levels, while SFN
suppressed its expression, which was consistent with a previous
report (6). These results indicated
that the induction of UGT1A1 gene expression by rapamycin is
partially attributed to signaling through the hPXR pathway, while
SFN induces UGT1A1 gene expression, including the expression of
UGT1A8 and UGT1A10, predominantly through the Nrf2 signaling
pathway and independently of hPXR regulation. In the present study,
the mRNA expression of UGT1A8 and UGT1A10 was found to be enhanced
by rapamycin treatment alone, however, this effect was not
significant when compared with the control group, which may have
been due to the upregulation of hPXR. Therefore, the combination of
SFN and rapamycin appears to synergistically induce UGT1A1 gene
expression in Caco-2 human colon cancer cells via the simultaneous
stimulation of the Nrf2 and hPXR signaling pathways. However, the
potential underlying mechanism of hPXR involvement in the
enhancement of UGT1A8 and UGT1A10 expression requires further
investigation.
CYP3A4 is the major CYP isoform that is present in
the human liver and small intestine. It is regulated by PXR and CAR
at the transcriptional level (30)
and contributes to the biotransformation of >50% of current
prescription medications (31).
CYP3A4 induction is a common mechanism of adverse drug-drug
interactions. In addition, certain procarcinogens require
CYP3A4-mediated metabolic activation and are converted into highly
reactive intermediates that promote carcinogenesis. For example,
CYP3A4 expression is the most important determinant of the hepatic
carcinogen, aflatoxin B1 (AFB1) conversion into the AFB,
8,9-epoxide, which binds to DNA to form an AFB1-DNA adduct
(32,33). Thus, SFN as an antagonist of hPXR
may reduce adverse therapeutic agent responses and enhance cancer
chemoprevention via the repression of hPXR-regulated CYP3A4. In the
current study, the addition of rapamycin did not influence
SFN-regulated CYP3A4 expression, however, it was observed to
enhance the expression of hPXR at the mRNA level. Therefore, with
regards to CYP3A4 expression, co-treatment with rapamycin may not
affect the chemopreventive property of SFN, which involves the
inhibition of CYP3A4. The results also indicate that in addition to
hPXR, other signaling pathways, which were inhibited by
co-treatment with rapamycin and SFN are involved in CYP3A4
expression. However, CYP3A4 is also particularly important for the
detoxification of xenobiotic chemicals, and the excessive
suppression of CYP3A4 expression may lead to negative impacts on
normal metabolism.
In conclusion, co-treatment with rapamycin may
enhance SFN-induced autophagy and UGT1A1, UGT1A8 and UGT1A10
expression levels. The underlying mechanism of the synergistic
induction of UGT1A isoforms may be associated with the combined
activation of Nrf2 and hPXR signaling pathways. Furthermore, the
upregulation of hPXR did not lead to CYP3A4 induction, which is
associated with adverse therapeutics agent responses and
procarcinogen activation. However, a limitation of the current
UGT1A isoform and CYP3A4 study is that these enzymes were only
analyzed with regard to their mRNA levels. Further information may
be obtained by analyzing the metabolic activity of enzymes in
Caco-2 cells following co-treatment with SFN and rapamycin. The
current study provides evidence supporting the potential use of an
autophagy activator for the enhancement of the chemopreventive
effects of SFN, particularly in the induction of phase II enzymes.
However, further studies are required to demonstrate the efficacy
of this strategy in healthy cells and in vivo.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 81372681).
References
|
1
|
Verhoeven DT, Goldbohm RA, van Poppel G,
Verhagen H and van den Brandt PA: Epidemiological studies on
brassica vegetables and cancer risk. Cancer Epidemiol Biomarkers
Prev. 5:733–748. 1996.
|
|
2
|
Gullett NP, Ruhul Amin AR, Bayraktar S,
Pezzuto JM, Shin DM, Khuri FR, Aggarwal BB, Surh YJ and Kucuk O:
Cancer prevention with natural compounds. Semin Oncol. 37:258–281.
2010.
|
|
3
|
Cheung KL and Kong AN: Molecular targets
of dietary phenethyl isothiocyanate and sulforaphane for cancer
chemoprevention. AAPS J. 12:87–97. 2010.
|
|
4
|
Clarke JD, Dashwood RH and Ho E:
Multi-targeted prevention of cancer by sulforaphane. Cancer Lett.
269:291–304. 2008.
|
|
5
|
Mahéo K, Morel F, Langouet S, Kramer H, Le
Ferrec E, Ketterer B and Guillouzo A: Inhibition of cytochromes
P-450 and induction of glutathione S-transferases by sulforaphane
in primary human and rat hepatocytes. Cancer Res. 57:3649–3652.
1997.
|
|
6
|
Zhou C, Poulton EJ, Grun F, Bammler TK,
Blumberg B, Thummel KE and Eaton DL: The dietary isothiocyanate
sulforaphane is an antagonist of the human steroid and xenobiotic
nuclear receptor. Mol Pharmacol. 71:220–229. 2007.
|
|
7
|
Saracino MR and Lampe JW: Phytochemical
regulation of UDP-glucuronosyltransferases: implications for cancer
prevention. Nutr Cancer. 59:121–141. 2007.
|
|
8
|
Strassburg CP, Kalthoff S and Ehmer U:
Variability and function of family 1 uridine-5′-diphosphate
glucuronosyltransferases (UGT1A). Crit Rev Clin Lab Sci.
45:485–530. 2008.
|
|
9
|
Wang M, Sun DF, Wang S, Qing Y, Chen S, Wu
D, Lin YM, Luo JZ and Li YQ: Polymorphic expression of
UDP-glucuronosyltransferase UGTlA gene in human colorectal cancer.
PLoS One. 8:e570452013.
|
|
10
|
Shintani T and Klionsky DJ: Autophagy in
health and disease: a double-edged sword. Science. 306:990–995.
2004.
|
|
11
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326.
2010.
|
|
12
|
Chen HY and White E: Role of autophagy in
cancer prevention. Cancer Prev Res. 4:973–983. 2011.
|
|
13
|
Herman-Antosiewicz A, Johnson DE and Singh
SV: Sulforaphane causes autophagy to inhibit release of cytochrome
C and apoptosis in human prostate cancer cells. Cancer Res.
66:5828–5835. 2006.
|
|
14
|
Nishikawa T, Tsuno NH, Okaji Y, Shuno Y,
Sasaki K, Hongo K, Sunami E, Kitayama J, Takahashi K and Nagawa H:
Inhibition of autophagy potentiates sulforaphane-induced apoptosis
in human colon cancer cells. Ann Surg Oncol. 17:592–602. 2010.
|
|
15
|
Kanematsu S, Uehara N, Miki H, Yoshizawa
K, Kawanaka A, Yuri T and Tsubura A: Autophagy inhibition enhances
sulforaphane-induced apoptosis in human breast cancer cells.
Anticancer Res. 30:3381–3390. 2010.
|
|
16
|
Svehliková V, Wang S, Jakubíková J,
Williamson G, Mithen R and Bao Y: Interactions between sulforaphane
and apigenin in the induction of UGT1A1 and GSTA1 in CaCo-2 cells.
Carcinogenesis. 25:1629–1637. 2004.
|
|
17
|
Jakubíková J, Sedlák J, Mithen R and Bao
Y: Role of PI3K/Akt and MEK/ERK signaling pathways in
sulforaphane-and erucin-induced phase II enzymes and MRP2
transcription, G2/M arrest and cell death in Caco-2 cells. Biochem
Pharmacol. 9:1543–1552. 2005.
|
|
18
|
Gardner-Stephen D, Heydel JM, Goyal A, Lu
Y, Xie W, Lindblom T, Mackenzie P and Radominska-Pandya A: Human
PXR variants and their differential effects on the regulation of
human UDP-glucuronosyltransferase gene expression. Drug Metab
Dispos. 32:340–347. 2004.
|
|
19
|
Kuma A, Matsui M and Mizushima N: LC3, an
autophagosome marker, can be incorporated into protein aggregates
independent of autophagy: caution in the interpretation of LC3
localization. Autophagy. 3:323–328. 2007.
|
|
20
|
Tong KI, Kobayashi A, Katsuoka F and
Yamamoto M: Two-site substrate recognition model for the Keap1-Nrf2
system: a hinge and latch mechanism. Biol Chem. 387:1311–1320.
2006.
|
|
21
|
Wang M, Li YQ, Zhong N, Chen J, Xu XQ and
Yuan MB: Induction of uridine
5′-diphosphate-glucuronosyltransferase gene expression by
sulforaphane and its mechanism: experimental study in human colon
cancel cells. Zhonghua Yi Xue Za Zhi. 85:819–824. 2005.(in
Chinese).
|
|
22
|
Xie W, Yeuh MF, Radominska-Pandya A, Saini
SP, Negishi Y, Bottroff BS, Cabrera GY, Tukey RH and Evans RM:
Control of steroid, heme, and carcinogen metabolism by nuclear
pregnane X receptor and constitutive androstane receptor. Proc Natl
Acad Sci USA. 100:4150–4155. 2003.
|
|
23
|
Kliewer SA, Goodwin B and Willson TM: The
nuclear pregnane X receptor: a key regulator of xenobiotic
metabolism. Endocr Rev. 23:687–702. 2002.
|
|
24
|
Naumann P, Fortunato F, Zentgraf H,
Büchler MW, Herr I and Werner J: Autophagy and cell death signaling
following dietary sulforaphane act independently of each other and
require oxidative stress in pancreatic cancer. Int J Oncol.
39:101–109. 2011.
|
|
25
|
Li J, Hou N, Faried A, Tsutsumi S and
Kuwano H: Inhibition of autophagy augments 5-fluorouracil
chemotherapy in human colon cancer in vitro and in vivo model. Eur
J Cancer. 46:1900–1909. 2010.
|
|
26
|
Liu D, Yang Y, Liu Q and Wang J:
Inhibition of autophagy by 3-MA potentiates cisplatin-induced
apoptosis in esophageal squamous cell carcinoma cells. Med Oncol.
28:105–111. 2011.
|
|
27
|
Wang M, Chen S, Wang S, Sun D, Chen J, Li
Y, Han W, Yang X and Gao HQ: Effects of phytochemicals sulforaphane
on uridine diphosphate-glucuronosyltransferase expression as well
as cell-cycle arrest and apoptosis in human colon cancer Caco-2
cells. Chin J Physiol. 55:134–144. 2012.
|
|
28
|
Buckley DB and Klaassen CD: Induction of
mouse UDP-glucuronosyltransferase mRNA expression in liver and
intestine by activators of aryl-hydrocarbon receptor, constitutive
androstane receptor, pregnane X receptor, peroxisome
proliferator-activated receptor alpha, and nuclear factor erythroid
2-related factor 2. Drug Metab Dispos. 37:847–856. 2009.
|
|
29
|
Sugatani J, Yamakawa K, Tonda E, Nishitani
S, Yoshinari K, Degawa M, Abe I, Noguchi H and Miwa M: The
induction of human UDP-glucuronosyltransferase 1A1 mediated through
a distal enhancer module by flavonoids and xenobiotics. Biochem
Pharmacol. 67:989–1000. 2004.
|
|
30
|
Bozina N, Bradamante V and Lovrić M:
Genetic polymorphism of metabolic enzymes P450 (CYP) as a
susceptibility factor for drug response, toxicity, and cancer risk.
Arh Hig Rada Toksikol. 60:217–242. 2009.
|
|
31
|
Guengerich FP: Cytochrome P-450 3A4:
regulation and role in drug metabolism. Annu Rev Pharmacol Toxicol.
39:1–17. 1999.
|
|
32
|
Kamdem LK, Meineke I, Gödtel-Armbrust U,
Brockmöller J and Wojnowski L: Dominant contribution of P450 3A4 to
the hepatic carcinogenic activation of aflatoxin B1. Chem Res
Toxicol. 19:577–586. 2006.
|
|
33
|
Gross-Steinmeyer K, Stapleton PL, Tracy
JH, Bammler TK, Strom SC and Eaton DL: Sulforaphane- and phenethyl
isothiocyanate-induced inhibition of aflatoxin B1-mediated
genotoxicity in human hepatocytes: role of GSTM1 genotype and
CYP3A4 gene expression. Toxicol Sci. 116:422–432. 2010.
|