Introduction
Gastric cancer is one of the most common types of
cancer worldwide and the predominant cause of cancer-related
mortality in Asian countries annually (1,2). The
majority of patients are diagnosed with advanced disease,
particularly in China (3). Advanced
gastric cancer is characterized by a highly invasive and metastatic
malignancy (3). Although
chemotherapy, radiotherapy and targeted therapy have improved the
response rate in advanced gastric cancer patients, metastasis
remains the most common cause of mortality (4). Contributing to this problem is the
unclear metastatic mechanism and the lack of effective biomarkers
for metastasis prediction. Therefore, investigations on the
molecular mechanisms of tumor metastasis will provide potential
molecular targets and biomarkers for the development of effective
therapies for gastric cancer.
Numerous different processes are involved in tumor
metastasis. Growing evidence indicates that
epithelial-to-mesenchymal transition (EMT) is a major contributor
to tumor metastasis in several types of epithelial tumor cells
(5,6). EMT is a developmental process by which
non-motile epithelial cells characterized of cell-cell tight
conjunctions lose their epithelial polarity and become migratory
mesenchymal cells (7). The
insulin-like growth factor-I (IGF-I) axis has been reported to
induce EMT and promote tumor metastasis in prostate and breast
cancer cells (8–10). Meanwhile, basic and clinical studies
have reported that overexpression of IGF-I receptor (IGF-IR) is
associated with enhanced invasiveness in gastrointestinal tumor
cells, and with poor survival in gastric cancer patients (11–13).
However, two recent phase II and III clinical trials reported that
monoclonal antibodies targeting the IGF-IR did not improve overall
survival in breast and pancreatic cancer patients, respectively
(14,15). This failure follows a slew of
setbacks for IGF-IR-targeted therapies. Therefore, exploring the
mechanisms of IGF-I-induced tumor metastasis, finding predictive
biomarkers and selecting suitable patients appear to be important
for further development of IGF-IR-specific targeting therapy.
The phosphoinositide 3-kinase (PI3K)/Akt pathway is
downstream of IGF-I/IGF-IR signaling, and contains important
signaling molecules in the regulation of IGF-IR-mediated EMT
(16–18). Among these effectors, glycogen
synthase kinase 3β (GSK-3β) is a multifunctional kinase capable of
being inactivated by Akt (19,20).
Previous studies have reported that GSK-3β maintains epithelial
phenotypes and inhibits migration in certain types of epithelial
tumor cells (21,22). Consistently, GSK-3β can also repress
IGF-IR-mediated EMT in breast epithelial cells (16). However, the specific effects of
GSK-3β in IGF-I-induced gastric cancer EMT remain unclear.
Consequently, in the present study, the direct role
of IGF-I in promoting EMT in gastric cancer and the role of the
PI3K/Akt signaling pathway, GSK-3β and ZEB2 in this process was
investigated.
Materials and methods
Cell culture
The BGC823 human gastric cell line was obtained from
the Cell Bank of Type Culture Collection of the Chinese Academy of
Sciences (Shanghai, China). The cells were maintained in RPMI-1640
medium (Gibco-BRL, Carlsbad, CA, USA) supplemented with 10% fetal
bovine serum (Gibco-BRL), penicillin (100 U/ml; Invitrogen Life
Technologies, Inc., Carlsbad, CA, USA) and streptomycin (100 mg/ml;
Invitrogen Life Technologies, Inc.) in a humidified atmosphere of
5% CO2 and 95% air, at 37°C. The cells were
serum-starved overnight before human recombinant IGF-I (100 ng/ml;
R&D Systems, Wiesbaden, Germany) treatment. All the cells used
for the experiments were subcultured every 2–3 days and harvested
in the logarithmic phase of growth.
Reagents and antibodies
IGF-I was purchased from R&D Systems, while
specific PI3K/Akt inhibitor, LY294002, and GSK-3β inhibitor,
AR-A01448, were purchased from Sigma-Aldrich (St. Louis, MO, USA).
The dual IGF-IR/IR inhibitor, OSI-906, was purchased from
SelleckBio (Houston, TX, USA). Monoclonal rabbit anti-human
E-cadherin, monoclonal rabbit anti-human vimentin, monoclonal
rabbit anti-human IGF-IR, monoclonal rabbit anti-human ZEB1,
monoclonal rabbit anti-human GSK-3β, polyclonal rabbit anti-human
phospho-IGF-IR (Tyr1131) and monoclonal rabbit anti-human
phospho-GSK-3β (Ser9) antibodies were purchased from Cell Signaling
Technology, Inc. (Beverly, MA), while polyclonal rabbit anti-human
Twist2 antibody was purchased from Abcam (Cambridge, MA, USA).
Monoclonal mouse anti-human ZEB2, monoclonal rabbit anti-human
Twist1, polyclonal rabbit anti-human actin, monoclonal rabbit
anti-human Akt and polyclonal rabbit anti-human phospho-Akt
(Ser473) antibodies were purchased from Santa Cruz Biotechnology,
Inc. (Santa Cruz, CA, USA).
Western blot assay
Cells were washed three or four times with 1X
phosphate-buffered saline (PBS), solubilized in 1% Triton lysis
buffer [50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 10 mM EDTA, 100 mM
NaF, 1 mM Na3VO4, 1% Triton X-100
(Sigma-Aldrich), 1 mM PMSF and 2 μg/ml aprotinin] on ice, and then
quantified according to the Lowry method (23). Following this, all the samples were
eluted by boiling water at 100°C for 5 min with 3X sampling buffer.
Total proteins were subjected to sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and electronically
transferred to nitrocellulose membranes. The blots were incubated
with E-cadherin, vimentin, IGF-IR, ZEB1/2, Twist1/2, GSK-3β, Akt,
actin, phospho-IGF-IR (Tyr1131), phospho-GSK-3β(Ser9) or
phospho-Akt (Ser473) antibodies at 4°C overnight, after blocking
with 5% skimmed milk in TBST [10 mM Tris (pH 7.4), 150 mM NaCl and
0.1% Tween-20]. On the following day, the blots were incubated with
monoclonal anti-rabbit or mouse secondary antibodies (Santa Cruz
Biotechnology, Inc.) for 30 min at room temperature. After four
washes with TBST, proteins were detected using an enhanced
chemiluminescence reagent (SuperSignal Western Pico
Chemiluminescent Substrate; Pierce, Rockford, IL, USA) and
visualized with an enhanced chemiliuminescence detection system
(DNR Bio-Imaging Systems, Ltd., Jerusalem, Israel). The images were
then analyzed by National Institutes of Health image software
(http://rsb.info.nih.gov/nih-image/)
for further statistical analysis.
Immunofluorescence
The cells (2×104 cells/well) were seeded
in Lab-Tek chamber slides (Nunc S/A; Polylabo, Strasbourg, France).
After being serum-starved overnight, the cells were treated with or
without IGF-I (100 ng/ml) for 48 h and fixed with 3.3%
paraformaldehyde for 15 min, followed by rinsing with 1X PBS three
times at room temperature. For morphological analysis, cells were
permeabilized with 0.2% Triton X-100 for 5 min, blocked with 5%
bovine serum albumin (Sigma-Aldrich) in 1X PBS for 1 h at room
temperature, and then incubated with anti-E-cadherin or
anti-Vimentin antibody overnight at 4°C. On the next day, alexa
Fluor 546-conjugated goat monoclonal anti-rabbit IgG or Alexa Fluor
488-conjugated goat monoclonal anti-rabbit IgG (Molecular Probes,
Eugene, OR, USA) were added in blocking solution for 1 h at room
temperature in the dark. 4′6-diamidino-2-phenylindole was used to
stain nuclei for 5 min. The cells were visualized by fluorescence
microscopy (BX61; Olympus, Tokyo, Japan) following mounting using
the SlowFade Antifade kit (Molecular Probes).
Statistical analysis
All data presented in the study are expressed as the
mean ± standard deviation. Representative results were from at
least three independent experiments. Significant differences
between treated and control groups were calculated using the
two-tailed Student’s t-test. P<0.05 was considered to indicate a
statistically significant difference. Statistical analysis was
performed using SPSS version 18.0 software (SPSS, Inc., Chicago,
IL, USA).
Results
IGF-I induces EMT in BGC-823 gastric
cancer cells
To investigate the role of IGF-I in gastric cancer
cells, we treated BGC-823 cells with human recombinant IGF-I (100
ng/ml) for 48 h after overnight serum starvation. As shown in
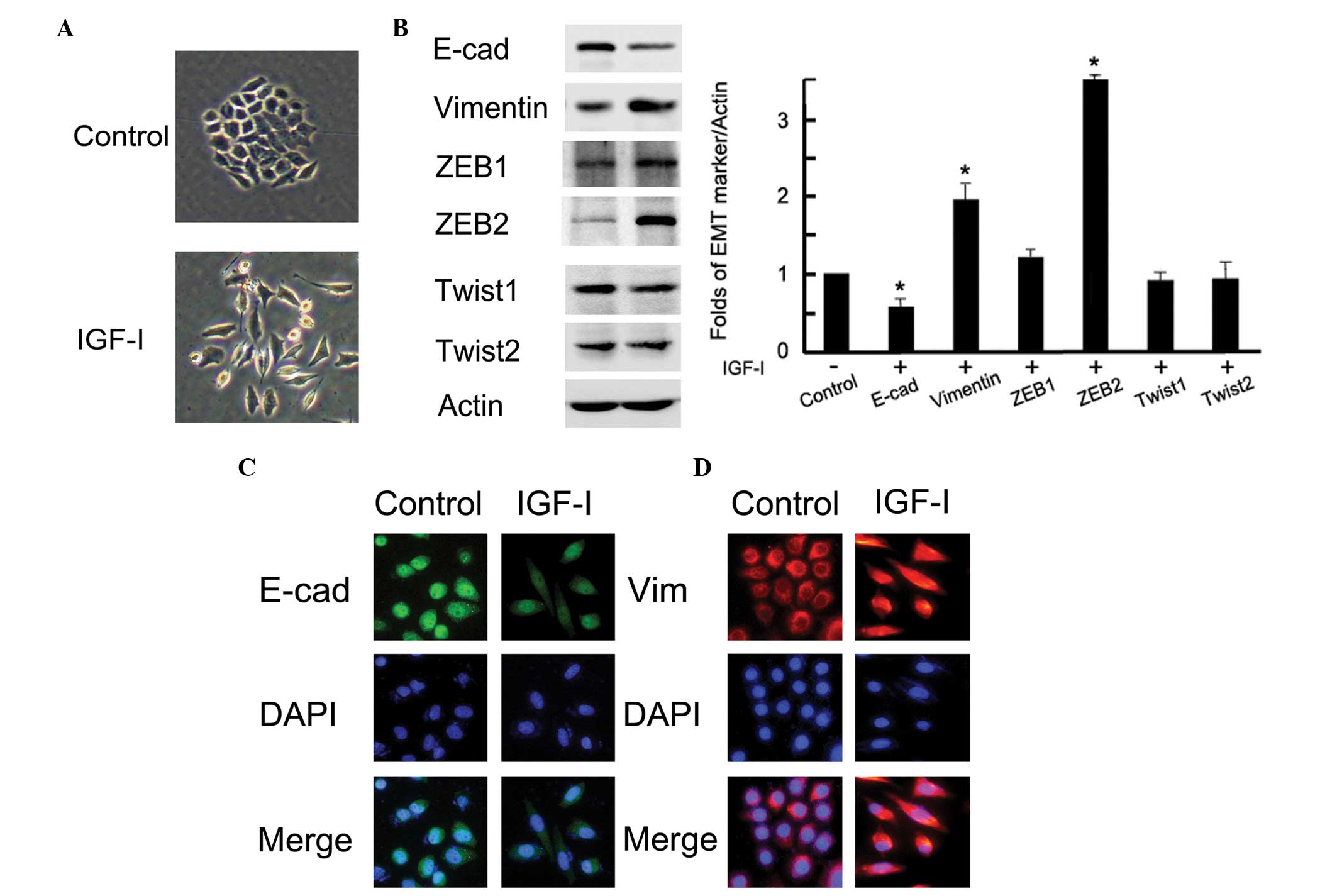
Fig. 1A, the cells with IGF-I
treatment represented a mesenchymal phenotype; a loss of tight
cell-cell junctions, an increase in cell scattering and an
elongation of the cell shape. These morphological changes were
compatible with the characteristics of EMT. Following IGF-I
treatment, both western blotting and immunofluorescence observed
obvious EMT marker switching, as shown by the downregulation of the
epithelial marker E-cadherin and the upregulation of the
mesenchymal marker vimentin in BGC-823 gastric cancer cells
(P<0.05; Fig. 1B and C). In
addition, the expression of the transcription factor ZEB2 was
markedly upregulated after IGF-I treatment (P=0.01); however, the
expression of other measured proteins, ZEB1, Twist1 and Twist2, did
not change (P<0.05; Fig. 1B).
These data demonstrated that IGF-I could upregulate ZEB2 and induce
EMT in BGC-823 gastric cancer cells.
Activation of the PI3K/Akt downstream
pathway is required for IGF-I-induced upregulation of ZEB2
Given that PI3K/Akt is a downstream signaling
pathway of IGF-I/IGF-IR, we further examined the activation level
of this signaling pathway. As shown in Fig. 2A, the phosphorylation levels of
IGF-IR and Akt were time-dependently increased by IGF-I
stimulation. Transient phosphorylation of IGF-IR and Akt was
detected at 3 min to 2 h, and gradually recovered to the baseline
values following exposure to IGF-I for 6 h. Meanwhile, IGF-I
stimulation time-dependently upregulated ZEB2 expression. To
analyze whether ZEB2 activation was dependent on the PI3K/Akt
downstream pathway, BGC-823 cells were pretreated with IGF-IR
inhibitor, OSI-906 (10 μM) and PI3K/Akt inhibitor, LY294002 (100
μM) 2 h to block the signaling pathways prior to IGF-I treatment.
The protein levels of ZEB2 were upregulated after IGF-I treatment
for 2 h. Pretreatment with OSI-906 or LY294002 markedly reversed
the ZEB2 upregulation induced by IGF-I (P<0.05; Fig. 2B and C). These data indicated that
IGF-I-induced ZEB2 upregulation was dependent on the PI3K/Akt
downstream signaling pathway in BGC-823 gastric cancer cells.
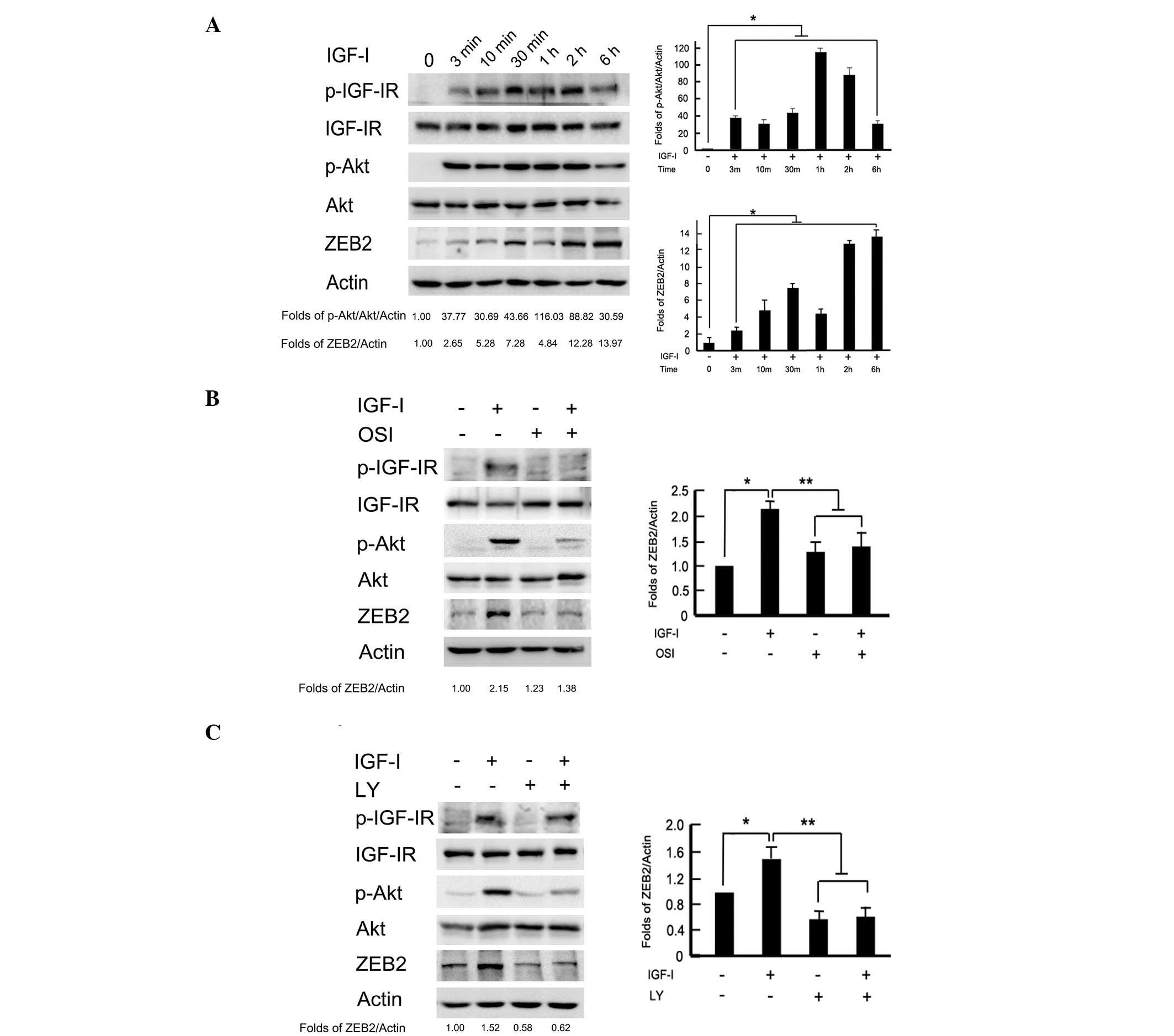 | Figure 2Activation of the PI3K/Akt downstream
pathway was required for IGF-I-induced ZEB2 upregulation. (A)
BGC-823 cells were incubated with IGF-I (100 ng/ml) for the
indicated time periods, and the phosphorylation of IGF-IR and Akt,
as well as ZEB2 expression, were analyzed by western blotting. The
serum-starved cells were pretreated with or without (B) IGF-IR
inhibitor, OSI-906 (10 μM) or (C) PI3K/Akt inhibitor, LY294002 (100
μM) for 2 h, followed by IGF-I (100 ng/ml) stimulation for 1 h.
Cell lysates were collected for western blot analysis. PI3K,
phosphoinositide 3-kinase; IGF-I, insulin-like growth factor-I;
OSI, OSI-906; LY, LY294002. |
Activation of the PI3K/Akt downstream
pathway is necessary for IGF-I-induced EMT
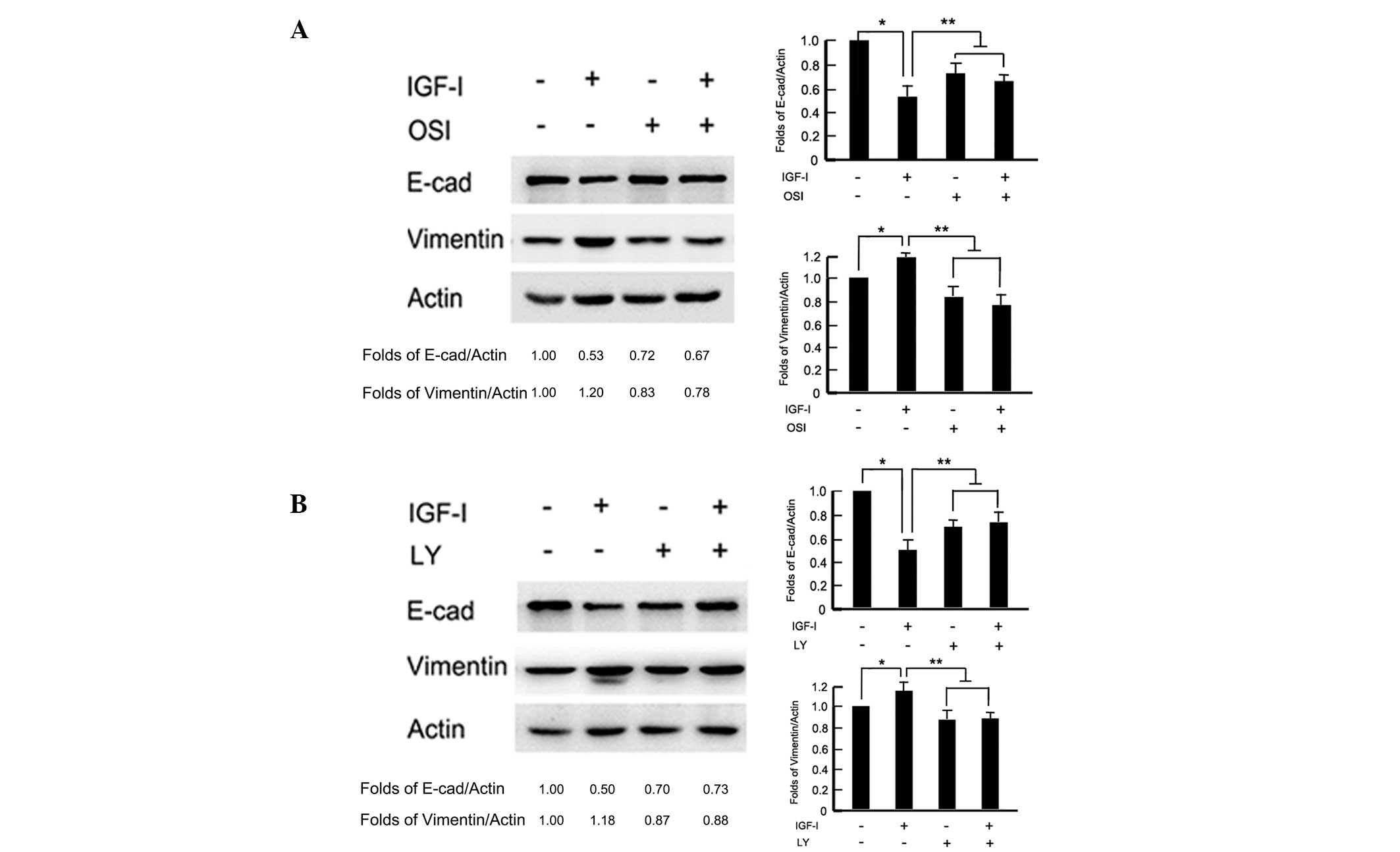
To test whether the PI3K/Akt pathway was involved in
IGF-I-induced EMT, cells were pretreated with the IGF-IR inhibitor,
OSI-906 (10 μM) and PI3K/Akt inhibitor, LY294002 (100 μM) 2 h
before IGF-I stimulation for 48 h, respectively. In the presence of
OSI-906, the BGC-823 cells maintained an epithelial like morphology
with tight cell-cell junctions, following IGF-I treatment for 48 h
(data not shown). Western blot analysis revealed that OSI-906
reversed IGF-I-induced E-cadherin downregulation and vimentin
upregulation (P<0.05; Fig. 3A).
Similarly, blocking the downstream signaling pathway with LY294002
repressed IGF-I-induced cellular morphology changes and attenuated
EMT-associated marker, E-cadherin and vimentin, expression changes
(P<0.05; Fig. 3B). These results
indicated that the PI3K/Akt downstream pathway was necessary for
IGF-I-induced EMT in BGC-823 gastric cancer cells.
A potential PI3K/Akt-GSK-3β-ZEB2
signaling pathway is involved in IGF-I-induced EMT
A previous study has reported that IGF-IR can
modulate GSK-3β activity via Akt in MCF-10A cells (16). To test the effect of PI3K/Akt on
GSK-3β expression, cells were pretreated with the PI3K/Akt
inhibitor, LY294002 (100 μM) for 2 h prior to IGF-I stimulation. As
shown in Fig. 4A western blot
analysis and associated histograms indicated that pretreatment with
LY294002 significantly inhibited the phosphorylation levels of
GSK-3β following IGF-I stimulation (P<0.05). To further examine
the role of GSK-3β in IGF-I-mediated EMT, BGC-823 cells were
pretreated with the GSK-3β inhibitor, AR-A01448 (25 μM) for 2 h
prior to IGF-I treatment for 48 h. Cells were observed to exhibit a
mesenchymal phenotype in the AR-A01448-treated group; a loss of
cell-cell contacts, an elongated cell shape and scattering of cells
were evident (data not shown). Western blotting detected marked
epithelial-mesenchymal phenotype marker switching. In addition,
downregulation of E-cadherin and upregulation of vimentin and ZEB2
were also observed in AR-A01448-treated cells following IGF-I
stimulation (P<0.05; Fig. 4B).
These data indicated that there may be a PI3K/Akt-GSK-3β-ZEB2
signaling pathway involved in IGF-I-induced EMT in BGC-823 gastric
cancer cells.
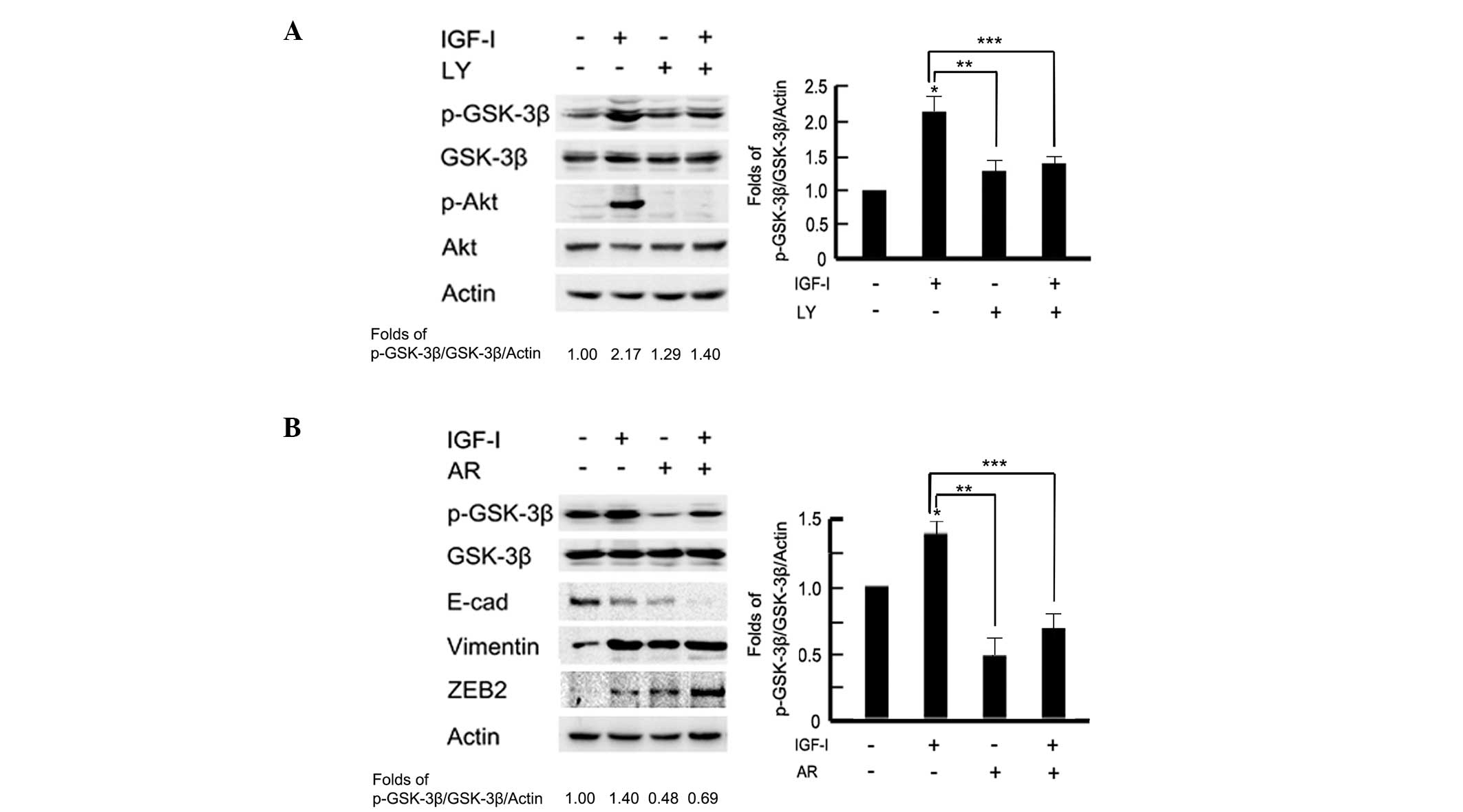 | Figure 4A PI3K/Akt-GSK-3β-ZEB2 signaling
pathway is involved in IGF-I-induced epithelial-to-mesenchymal
transition. The serum-starved BGC-823 cells were pretreated with or
without (A) PI3K/Akt inhibitor, LY294002 (100 μM) or (B) GSK-3β
inhibitor, AR-A01448 (25 μM) for 2 h, followed by IGF-I (100 ng/ml)
treatment for 48 h. Cell lysates were collected for western blot
analysis. The images were analyzed using NIH Image software and
presented as histograms. Data are presented as the mean ± standard
deviation of three independent experiments. *IGF-I
untreated vs. IGF-I treated, P<0.05. **IGF-I treated
vs. LY or AR treated, p < 0.05. *** IGF-I treated vs. IGF-I
combined with LY or AR, P<0.05. The control group was used as
the reference. PI3K, phosphoinositide 3-kinase; IGF-I, insulin-like
growth factor-I; GSK-3β, glycogen synthase kinase 3β; E-cad,
E-cadherin; LY, LY294002; AR, AR-A01448. |
Discussion
As previously reported, transcription factors ZEB1
and Snail are critical transcriptional regulators of
IGF-I/IGF-IR-mediated EMT, and ultimately function as metastasis
promoters through the repression of cell adhesion molecule
E-cadherin in prostate and breast cancer cells (8–10).
ZEB2 is another member of the ZEB family, and is a zinc finger
protein with similar repressor effects to ZEB1 in terms of
E-cadherin transcription (24,25).
The ZEB2/E-cadherin ratio has been reported to be positively
associated with tumor invasiveness and poor prognosis in breast and
ovarian cancer (26). Transforming
growth factor-β-mediated ZEB2 expression has been shown to be
involved in the process of EMT and enhancement of invasive ability
in several types of epithelial tumor cells (27,28);
however, whether IGF-I can upregulate the expression of ZEB2 is yet
to be elucidated. In the present study, it was observed that IGF-I
induced EMT and upregulated ZEB2, but not ZEB1, Twist1 or Twist2,
in gastric cancer BGC-823 cells. Furthermore, inhibition of the
PI3K/Akt signaling pathway reversed ZEB2 upregulation and the
subsequent EMT procession mediated by IGF-I. The results indicated
that IGF-I induced EMT by upregulating ZEB2 expression, which was
due to activating the downstream PI3K/Akt signaling pathway in
BGC-823 gastric cancer cells.
To understand the mechanism by which the PI3K/Akt
signaling pathway mediates ZEB2 expression, other intracellular
downstream effectors of Akt were examined. A previous study has
reported that GSK-3β, a major downstream component of the PI3K/Akt
signaling pathway, is involved in IGF-IR-mediated EMT through a
GSK-3β-NF-κB-Snail signaling pathway in immortalized mammary
epithelial MCF10A cells (16). The
present study demonstrated that GSK-3β maintained the epithelial
phenotype of BGC-823 gastric cancer cells, and PI3K/Akt-GSK-3β
signaling is an upstream factor of ZEB2 activation in the
IGF-I-induced EMT process. These data indicated that a
PI3K/Akt-GSK-3β-ZEB2 signaling pathway, which is involved in
IGF-I-induced EMT, may exist in BGC-823 gastric cancer cells.
Overall, to the best of our knowledge, the present
study is the first to report that IGF-I induces EMT, thereby
upregulating ZEB2 expression, and that a potential
PI3K/Akt-GSK-3β-ZEB2 signaling pathway is involved in IGF-I-induced
EMT in BGC-823 gastric cancer cells. These results may be helpful
for elucidating the mechanisms of IGF-I-mediated EMT procession.
ZEB2 may serve as a clinical biomarker to identify patients who can
benefit from IGF-IR-targeted therapy in gastric cancer.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant nos. 81201802, 81172369 and
81172198), the Specialized Research Fund for the Doctoral Program
of Higher Education (grant nos. 20102104120008 and 20112104110005),
the National Science and Technology Major Project (grant no.
2013ZX09303002), the Natural Science Foundation of Liaoning
Province (grant no. 2014021069), the Specialized Research Fund of
Doctoral Program of Higher Education (grant no. 20112104110005) and
the Technology Plan Project of Liaoning Province (grant nos.
2011404013-1 and 2012225001).
References
|
1
|
Jemal A, Siegel R, Ward E, Hao Y, Xu J and
Thun MJ: Cancer statistics. CA Cancer J Clin. 59:225–249. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Crew KD and Neugut AI: Epidemiology of
gastric cancer. World J Gastroenterol. 12:354–362. 2006.PubMed/NCBI
|
|
3
|
Chen W, Zheng R, Zhang S, Zhao P, Li G, Wu
L and He J: The incidences and mortalities of major cancers in
China, 2009. Chin J Cancer. 32:106–112. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wagner AD, Grothe W, Haerting J, Kleber G,
Grothey A and Fleig WE: Chemotherapy in advanced gastric cancer: a
systematic review and meta-analysis based on aggregate data. J Clin
Oncol. 24:2903–2909. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thompson EW, Newgreen DF and Tarin D:
Carcinoma invasion and metastasis: a role for
epithelial-mesenchymal transition? Cancer Res. 65:5991–5995. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang Y, Wen M, Kwon Y, et al: CUL4A
induces epithelial-mesenchymal transition and promotes cancer
metastasis by regulating ZEB1 expression. Cancer Res. 74:520–531.
2014. View Article : Google Scholar
|
|
7
|
Lee JM, Dedhar S, Kalluri R and Thompson
EW: The epithelial-mesenchymal transition: new insights in
signaling, development, and disease. J Cell Biol. 172:973–981.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Graham TR, Zhau HE, Odero-Marah VA, et al:
Insulin-like growth factor-I-dependent up-regulation of ZEB1 drives
epithelial-to-mesenchymal transition in human prostate cancer
cells. Cancer Res. 68:2479–2488. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Walsh LA and Damjanovski S: IGF-1
increases invasive potential of MCF 7 breast cancer cells and
induces activation of latent TGF-β1 resulting in epithelial to
mesenchymal transition. Cell Commun Signal. 9:102011. View Article : Google Scholar
|
|
10
|
Lorenzatti G, Huang W, Pal A, Cabanillas
AM and Kleer CG: CCN6 (WISP3) decreases ZEB1-mediated EMT and
invasion by attenuation of IGF-1 receptor signaling in breast
cancer. J Cell Sci. 124:1752–1758. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Adachi Y, Li R, Yamamoto H, et al:
Insulin-like growth factor-I receptor blockade reduces the
invasiveness of gastrointestinal cancers via blocking production of
matrilysin. Carcinogenesis. 30:1305–1313. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ge J, Chen Z, Wu S, et al: Expression
levels of insulin-like growth factor-1 and multidrug
resistance-associated protein-1 indicate poor prognosis in patients
with gastric cancer. Digestion. 80:148–158. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Franciosi CM, Piacentini MG, Conti M, et
al: IGF-1 and IGF-1BP3 in gastric adenocarcinoma. Preliminary
study. Hepatogastroenterology. 50:297–300. 2003.PubMed/NCBI
|
|
14
|
Robertson JF, Ferrero JM, Bourgeois H, et
al: Ganitumab with either exemestane or fulvestrant for
postmenopausal women with advanced, hormone-receptor-positive
breast cancer: a randomised, controlled, double-blind, phase 2
trial. Lancet Oncol. 14:228–235. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Trajkovic-Arsic M, Kalideris E and Siveke
JT: The role of insulin and IGF system in pancreatic cancer. J Mol
Endocrinol. 50:R67–R74. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim HJ, Litzenburger BC, Cui X, et al:
Constitutively active type I insulin-like growth factor receptor
causes transformation and xenograft growth of immortalized mammary
epithelial cells and is accompanied by an epithelial-to-mesenchymal
transition mediated by NF-kappaB and snail. Mol Cell Biol.
27:3165–3175. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sivakumar R, Koga H, Selvendiran K,
Maeyama M, Ueno T and Sata M: Autocrine loop for IGF-I receptor
signaling in SLUG-mediated epithelial-mesenchymal transition. Int J
Oncol. 34:329–338. 2009.PubMed/NCBI
|
|
18
|
Irie HY, Pearline RV, Grueneberg D, et al:
Distinct roles of Akt1 and Akt2 in regulating cell migration and
epithelial-mesenchymal transition. J Cell Biol. 171:1023–1034.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Papkoff J and Aikawa M: WNT-1 and HGF
regulate GSK3 beta activity and beta-catenin signaling in mammary
epithelial cells. Biochem Biophys Res Commun. 247:851–858. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sineva GS and Pospelov VA: Inhibition of
GSK3beta enhances both adhesive and signalling activities of
beta-catenin in mouse embryonic stem cells. Biol Cell. 102:549–560.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ho MY, Tang SJ, Chuang MJ, Cha TL, Li JY,
Sun GH and Sun KH: TNF-α induces epithelial-mesenchymal transition
of renal cell carcinoma cells via a GSK3β-dependent mechanism. Mol
Cancer Res. 10:1109–1119. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zheng H, Li W and Wang Y: Glycogen
synthase kinase-3 beta regulates Snail and β-catenin expression
during Fas-induced epithelial-mesenchymal transition in
gastrointestinal cancer. Eur J Cancer. 49:2734–2746. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lowry OH, Rosebrough NJ, Farr AL and
Randall RJ: Protein measurement with the Folin phenol reagent. J
Biol Chem. 193:265–275. 1951.PubMed/NCBI
|
|
24
|
Miyoshi A, Kitajima Y, Sumi K, Sato K,
Hagiwara A, Koga Y and Miyazaki K: Snail and SIP1 increase cancer
invasion by upregulating MMP family in hepatocellular carcinoma
cells. Br J Cancer. 90:1265–1273. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Remacle JE, Kraft H, Lerchner W, et al:
New mode of DNA binding of multi-zinc finger transcription factors:
deltaEF1 family members bind with two hands to two target sites.
EMBO J. 18:5073–5084. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Elloul S, Elstrand MB, Nesland JM, et al:
Snail, Slug, and Smad-interacting protein 1 as novel parameters of
disease aggressiveness in metastatic ovarian and breast carcinoma.
Cancer. 103:1631–1643. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mikaelian I, Malek M, Gadet R, et al:
Genetic and pharmacologic inhibition of mTORC1 promotes EMT by a
TGF-β-independent mechanism. Cancer Res. 73:6621–6631. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gregory PA, Bracken CP, Smith E, et al: An
autocrine TGF- beta/ZEB/miR-200 signaling network regulates
establishment and maintenance of epithelial-mesenchymal transition.
Mol Biol Cell. 22:1686–1698. 2011. View Article : Google Scholar : PubMed/NCBI
|