Introduction
Adrenocortical carcinoma (ACC) is a rare, but highly
aggressive type of tumor with an incidence of one to two per
million annually (1–3). Furthermore, a higher incidence of ACC
has been reported in females. ACC is associated with a bimodal age
distribution that has two peaks; one representing children in the
first decade of life and the other representing adults in the
fourth to fifth decades of life (4–5). A
small number of cases of ACC have been reported to date, each with
a poor prognosis and a five-year overall survival rate of ~35%
(4,6–8). The
poor prognosis may be attributable in part to the advanced stage at
which the majority of ACCs are detected (4).
Adrenocortical carcinosarcoma is an exceptional
variant of ACC, which is characterized by the presence of
histological regions of carcinoma and sarcoma, with the latter
regularly demonstrating heterologous features, including
rhabdomyoblastic (9–12), chondroid or osteogenic
differentiation (9,13). The term sarcomatoid carcinoma has
also been synonymously used to refer to adrenocortical
carcinosarcoma. In the present study, a case of primary,
non-functional adrenocortical carcinosarcoma is described and a
review of the literature is presented to raise awareness of this
particularly rare type of malignant neoplasm with a poor diagnosis
and prognosis. To the best of our knowledge, the present case is
the first to report an incidence of adrenocortical carcinosarcoma
in China. Written informed consent was obtained from the family of
the patient.
Case report
Patient information
In November 2013, a 63-year-old female was admitted
to the Second Xiangya Hospital (Changsha, China) with left flank
pain, fatigue, decreased appetite and weight loss that had
persisted for three months. These clinical symptoms led to the
discovery of a 7-cm heterogeneous hypoechoic left adrenal mass on
an abdominal ultrasound. On admission to the Department of Urology
at the Second Xiangya Hospital, the results of the physical
examination were normal and there was no palpable abdominal mass.
The patient exhibited no clinical features associated with
excessive steroid hormone or catecholamine levels and had no
notable family medical history. Furthermore, laboratory studies of
the adrenal hormone levels of cortisol, aldosterone and
catecholamines were within the normal limits. 24-h urine volume was
1100 ml (normal range, 1000–1500 ml), 24-h urinary vanillylmandelic
acid level was 33.1 μmol/day (normal range, 0–68.6 μmol/day), the
clinostatic and orthostatic plasma renin activity levels were 270
ng/l/h (normal range, 150–2330 ng/l/h) and 388 ng/l/h (normal
range, 100–6560 ng/l/h), respectively. The clinostatic and
orthostatic plasma aldosterone levels were 56 ng/l (normal range,
30–160 ng/l) and 119 ng/l (normal range, 70–300 ng/l),
respectively. A dynamic, contrast-enhanced abdominal computed
tomography scan showed a 7.6×5.1-cm well-demarcated and
peripherally enhanced left adrenal mass that was impinging on the
pancreas and the spleen, without parenchymal invasion into the
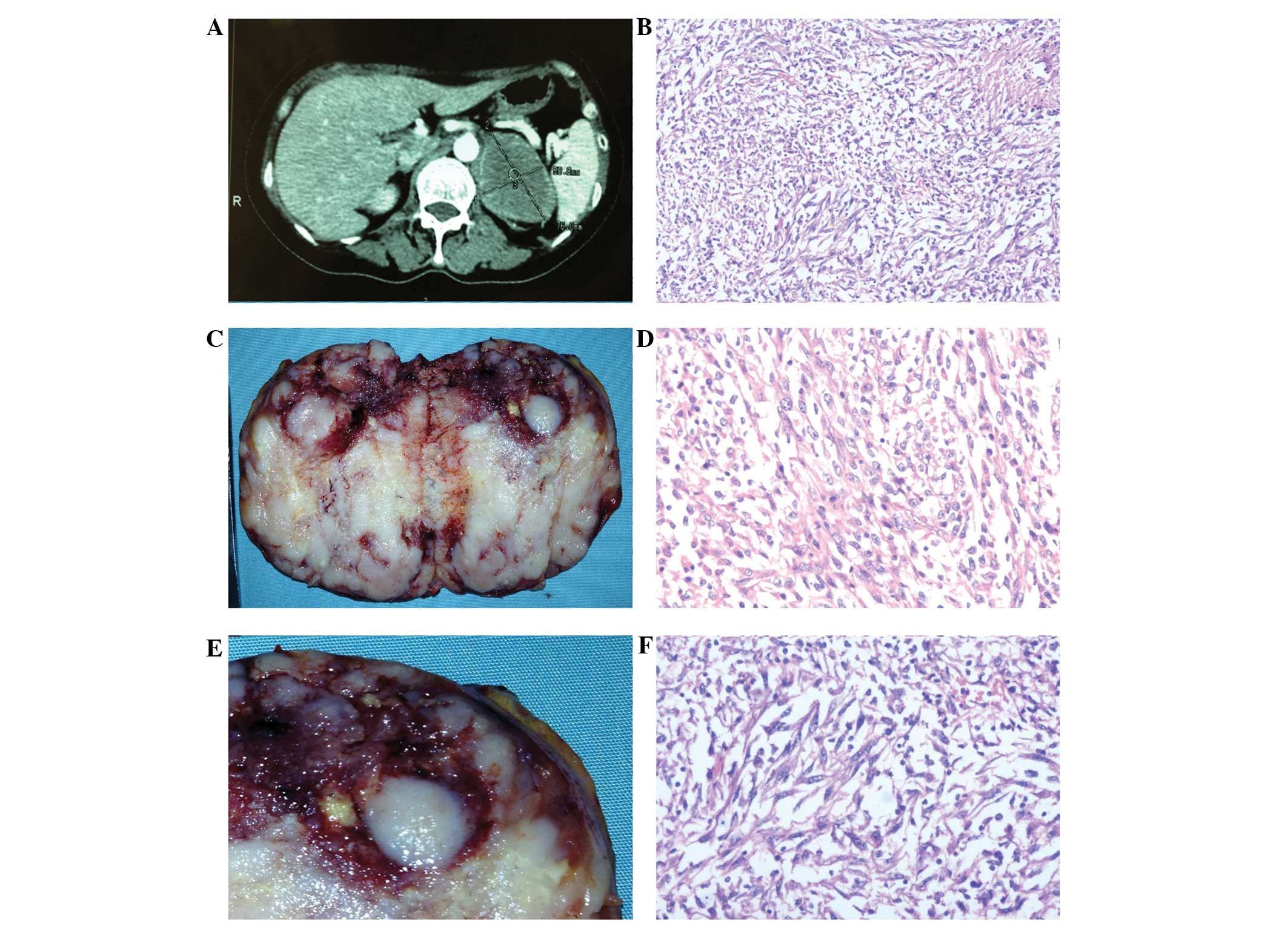
kidney (Fig. 1A). Distant lymph
node, pulmonary or liver metastases were not observed. A
laparoscopic left adrenalectomy was performed and the tumor was
dissected without complication from the left kidney. The patient
did not exhibit signs of tumor recurrence at the one-month
follow-up.
Gross pathology
The gross specimen presented as a well-demarcated,
encapsulated spherical mass, which measured 8×6×4 cm. The cut
surface was firm and varied in color. The majority of the lesion
was pale, however, certain regions (~30% of the tumor area) were
bright yellow (Fig. 1B).
Approximately 20% of the tumor area comprised of numerous foci of
necrosis. There was a small quantity of the residual adrenal gland,
which appeared to be laminated at the periphery of the tumor
(Fig. 1C). The spleen, pancreas and
kidney had not been invaded by the tumor.
Microscopic observations
The tumor comprised of epithelial and spindle cell
components that were multifocally intermingled (Fig. 1D and E). In addition, focal necrosis
and hemorrhaging was observed. The main epithelial component
exhibited poorly differentiated carcinomatous cells, which were
arranged in a diffuse and solid growth pattern. These cells showed
highly atypical nuclei, and an abundant eosinophilic cytoplasm and
usually possessed large, prominent nucleoli (Fig. 1E). Furthermore, spindle-shaped tumor
cells were observed to be arranged in a haphazard fascicular
pattern with highly pleomorphic nuclei (Fig. 1F).
Variable numbers of cells undergoing mitosis were
identified in different areas. The highest mitotic activity was
observed in the sarcomatous area, however, only occasional
instances of mitosis were observed in the carcinomatous area.
Furthermore, no definite capsular invasion was observed
histologically.
Immunohistochemical findings
An extensive immunohistochemical panel was performed
on formalin-fixed, paraffin-embedded tumor tissue to evaluate the
sarcomatous and carcinomatous components. In addition, negative and
positive controls were performed for each immunohistochemical
stain, including cytokeratin (CK), synapsin (Syn), CD56, CD34,
neuron-specific enolase (NSE), Ki-67, S100, smooth muscle actin,
Bcl-2, human melanoma black 45 (HMB-45) and chromogranin A (CgA).
Negative controls included primary antigens (rabbit anti-human
antibodies), while positive control included the aforementioned
primary antigens and the secondary (goat anti-rabbit) antibodies.
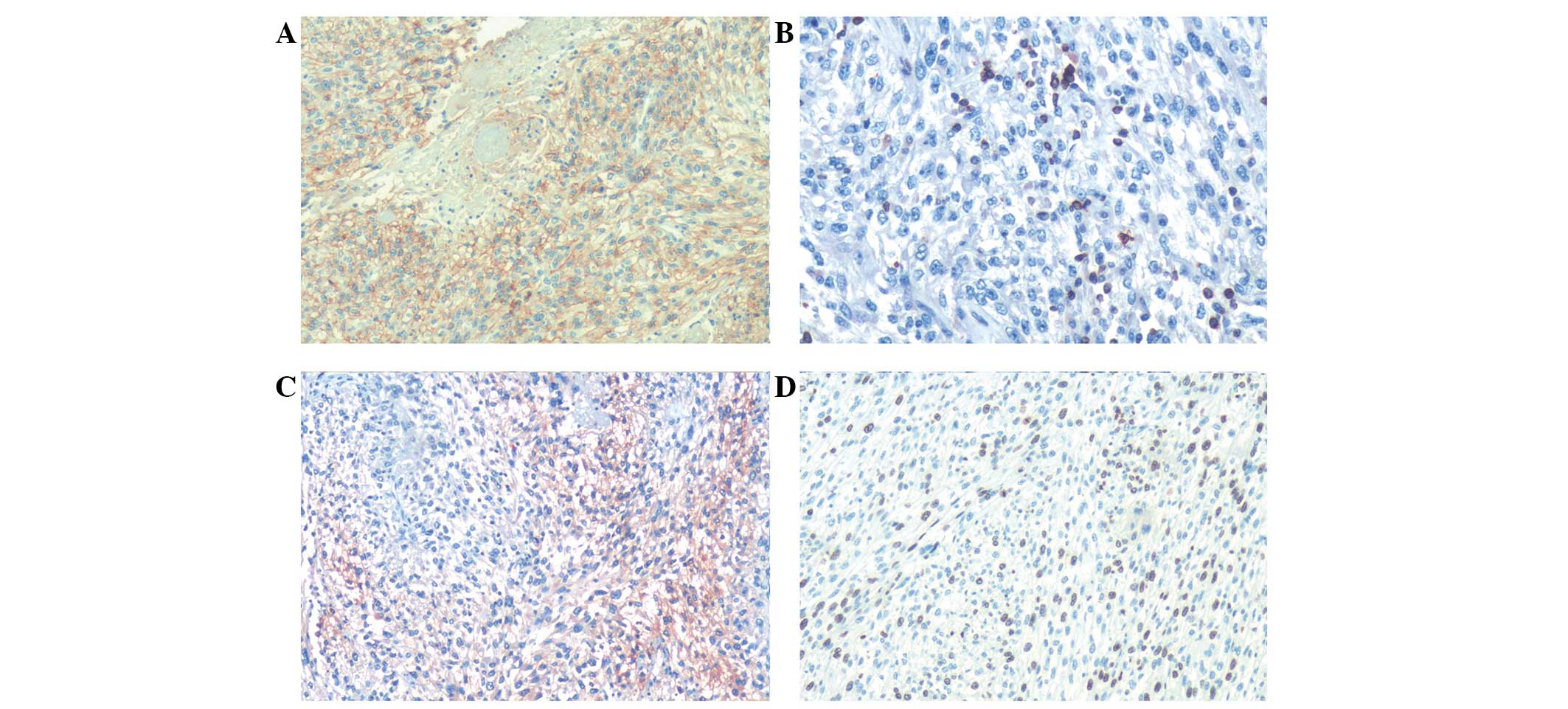
Cluster of differentiation (CD)56 demonstrated diffuse positivity
in the epithelial cells and spindle areas (Fig. 2A). Bcl-2 showed focal positivity in
the sarcomatous areas (Fig. 2B) and
CD34 was weakly positive. NSE was only weakly positive in
sarcomatous components (Fig. 2C)
and Ki-67 demonstrated 70% positivity in the two components
(Fig. 2D). S-100 showed equal
amounts of positivity/negativity, and CK, Syn, smooth muscle actin,
HMB-45 and CgA showed no reactivity in all of the areas.
Discussion
ACCs containing a component of sarcoma or
sarcoma-like (spindle cells) differentiation are a particularly
rare and aggressive type of neoplasm, and, to the best of our
knowledge, only 12 cases have previously been reported in the
English literature to date (9–20). The
clinical and pathological features of reported patients presenting
with this type of tumor, including the present case, are summarized
in Table I (9–20). The
age of initial presentation ranged from 23 to 79 years, with a mean
of 50.3 years and a median of 45 years, which appears to be similar
to the results of a previous study (mean age, 40–50 years) by
Sasaki et al (11). Eight of
the 13 patients were aged <50 years. Compared with previous
studies of ACC (4), adrenocortical
carcinosarcoma also showed a female preponderance with a
female/male ratio of 1.6:1. Flank/abdominal pain or discomfort was
identified to be a common presentation (10/13 cases) and the
majority of adrenocortical carcinosarcoma presented in the left
adrenal gland (9/12 cases). One tumor was identified during an
investigation of a rectal mass in a pregnant patient (19). In the majority of cases, these
tumors did not exhibit any endocrine dysfunction, although three of
the 13 cases were associated with corticosteroid hypersecretion
(10,13,16).
Generally, the tumors were particularly large (mean size, 14.1 cm;
weight, 1,743 g) and exhibited dramatically aggressive behavior.
All 13 patients succumbed to their cancer within the range of two
days to 14 months following resection, despite aggressive
administration of multimodality therapeutic strategies.
 | Table IClinical and pathological features of
previously reported cases of adrenocortical carcinosarcoma. |
Table I
Clinical and pathological features of
previously reported cases of adrenocortical carcinosarcoma.
| Ref. | Author, year | Age,
years/gender | Clinical
presentation | Endocrine
dysfunction | Location | Size/weight | Sarcomatous
component | Immunostaining
results | Total number of
antibodies | POS |
|---|
|
|---|
| Caricinomatous | Sarcomatous |
|---|
| 14 | Okazumi et al,
1987 | 46/M | Abdominal
distention | No | R | 14 cm, 880 g | Spindle cell | NA | NA | NA | 6 months |
| 15 | Collina et al,
1989 | 68/F | Abdominal
discomfort | No | L | 11 cm
NA | Spindle cell | Low molecular weight
CK, Vim | Low molecular weight
CK, Vim | 7 | 6 months |
| 9 | Decorato et
al, 1990 | 42/F | Abdominal pain | No | L | 19 cm, 1400 g |
Rhabdo-myosarcoma | - | muscle specific
actin | 5 | 7 months |
| 10 | Fischler et
al, 1992 | 29/F | ↓ Weight,
virilization | Yes | L | 12.5 cm, 610 g |
Rhabdo-myosarcoma | Vim, | Vim, HHF and
desmin | 5 | 8 months |
| 13 | Barksdale et
al, 1993 | 79/F | Hypertension | Yes | R | 9 cm, 199 g | Osteosarcoma,
chondrosarcoma | Vim | Vim | 5 | NA |
| 16 | Lee et al,
1997 | 61/M | Flank pain | Yes | R | 12 cm
NA | Spindle cell | CK, focal NSE | CK, Vim | 10 | 2 days |
| 17 | Sturm et al,
2008 | 31/M | Abdominal pain | No | L | 12 cm, 620 g | Spindle cell | CD56, Vim, focal
desmin, CK AE1/AE3, α-inhibin, Syn | CD56, Vim, focal
desmin, HHF35 | 17 | 3 months |
| 18 | Coli et al,
2010 | 75/F | Abdominal pain | No | L | 15 cm
NA | Spindle cell | CK (MNF-16), Vim,
S-100, melan-A, focal HMB-45 | Vim, SMA, desmin,
caldesmon, myogenin | 20 | 12 months |
| 11 | Sasaki et al,
2010 | 45/M | Abdominal pain | No | L | 17 cm, 2974 g |
Rhabdo-myosarcoma | Vim, Syn, melan-A,
and calretinin | Vim, Syn, melan-A,
and calretinin; desmin, myogenin, myoglobin | 15 | 3 months |
| 19 | Bertolini et
al, 2011 | 23/F | Fatigue, ↓ appetite,
fixed mass in rectum | No | L | 14 cm
NA | Osteosarcomatous | Inhibin,
calretinin | - | NA | 14 months |
| 12 | Thway et al,
2012 | 45/M | Bloating, back
pain | No | L | 24 cm, 6500 g |
Rhabdo-myosarcoma | Melan-A, CD56,
MNF116 | Desmin,
myogenin | 28 | 11 months |
| 20 | Kao et al,
2013 | 45/M | Abdominal pain,
weight loss | No | R | 15 cm, 760 g | Spindle cells,
undifferentiated | melan-A, inhibin,
calretinin, AE1/AE3, Syn, Vim, | Vim, NSE, FLI-1;
Undifferentiated: CD99 | 18 | 7 months ;
alive |
| Current study | Wei et
al | 63/F | Fatigue, flank
pain | No | L | 8 cm
NA | Spindle cells | NSE,
FLI-1
CD56, Ki-67 | CD56, Bcl-2, NSE,
Ki-67 | 11 | 1 month ;
alive |
The present case was extensively sampled, however,
the heterologous elements, such as rhabdomyosarcoma (9) and osteosarcoma (13), that were documented in certain
previous papers were not observed. Additionally, when compared with
previous studies, in this study there were no specific
immunohistochemical staining results. The positive reactivity with
NSE that was only observed in partial sarcomatous areas may provide
evidence for neuroendocrine differentiation in ACCs, which is
consistent with two previous studies; one demonstrated positive
staining for NSE, Syn and neurofilament protein (21), and the other showed positivity for
CK AE1/AE3, Syn and NSE (20).
Notably, the tumor mass observed in the present
case, (size, 8×6×4 cm) was the smallest out of all of the reported
cases. The next smallest, measuring 9×7.5×6.5 cm, was reported in
1993 (13). The tumor sizes
described in the other 11 cases were all >10 cm, which indicates
that although a tumor mass may not be large it may be an
adrenocortical carcinosarcoma. Therefore, the diagnosis of
adrenocortical carcinosarcoma may be complicated.
Furthermore, a compressed rim of normal adrenal
gland was observed adjacent to the tumor capsule in the present
study, which was also described in two previous cases (12,20)
and in a case of large diameter ACC (22). Therefore it may be hypothesized that
the tumor arises from an accessory/ectopic adrenal gland, or
alternatively, from a nodular area of the adrenal gland, which
gradually becomes entirely replaced by the tumor (22).
A challenge during the diagnosis of adrenocortical
carcinosarcoma arises from the difficulty in grossly identifying
the tumor origin. Renal carcinosarcoma, metastatic melanoma and
primary retroperitoneal sarcoma should be considered in the
differential diagnosis, and attention should be given to ACCs
demonstrating negative, or only focally weak, positivity for CKs.
Consequently, it is necessary to adopt immunohistochemical staining
for melan-A, inhibin and calretinin to verify the adrenocortical
origin, particularly for ACC (23–26).
Routine histological data from Table
II revealed that the carcinomatous and sarcomatous components
expressed vimentin. A notable feature is that desmin was found to
be highly expressed in the sarcomatous component. Furthermore, the
sarcomatous component of the tumor was positive for HHF, myogenin,
and caldesmon, whereas the epithelial component was diffusely
positive for inhibin, melan-A, S-100 protein, NSE and HMB-45. As a
result, a large number of tumor samples and an extensive panel of
immunohistochemical staining are considered to be necessary to
identify the adrenal origin and the biphasic components of
adrenocortical carcinosarcoma.
| Table IIImmunohistochemical results (not
including cytokeratins) of the reported adrenocortical
carcinosarcoma. |
Table II
Immunohistochemical results (not
including cytokeratins) of the reported adrenocortical
carcinosarcoma.
| A, Carcinomatous
component |
|---|
|
|---|
| Antibody | Check sum | Positive | Positive rate |
|---|
| FLI-1 | 1 | 1 | 100 |
| Melan-A | 5 | 4 | 80 |
| Vimentin | 9 | 7 | 78 |
| CD56 | 4 | 3 | 75 |
| Inhibin | 6 | 3 | 50 |
| Ki-67 | 2 | 1 | 50 |
| Syn | 7 | 3 | 43 |
| NSE | 5 | 2 | 40 |
| HMB-45 | 5 | 1 | 20 |
| Desmin | 7 | 1 | 14 |
| S-100 | 8 | 1 | 13 |
|
| B, Sarcomatous
component |
|
| Antibody | Check sum | Positive | Positive rate |
|
| HHF | 3 | 3 | 100 |
| Bcl-2 | 1 | 1 | 100 |
| FLI-1 | 1 | 1 | 100 |
| Vimentin | 9 | 8 | 89 |
| Desmin | 7 | 5 | 71 |
| CD56 | 4 | 2 | 50 |
| Caldesmon | 2 | 1 | 50 |
| Ki-67 | 2 | 1 | 50 |
| Myoglobin | 2 | 1 | 50 |
| Myogenin | 5 | 2 | 40 |
| NSE | 5 | 2 | 40 |
| Calretinin | 5 | 1 | 20 |
| Melan-A | 5 | 1 | 20 |
| SMA | 6 | 1 | 17 |
| Syn | 7 | 1 | 14 |
In conclusion, a potential diagnosis of
adrenocortical carcinosarcoma should be considered when diagnosing
an adrenal malignancy in adult patients. Extensive sampling of the
tumor together with comprehensive immunohistochemical staining are
required to identify a possible sarcomatous pattern and, as a
result of the poor prognosis associated with adrenocortical
carcinosarcoma, the most aggressive therapeutic regimens are
required.
Acknowledgements
The present study was supported by the Fundamental
Research Funds for the Central Universities of Central South
University in 2013 (grant no. 2013zzts095).
References
|
1
|
Lacroix A: Approach to the patient with
adrenocortical carcinoma. J Clin Endocrinol Metab. 95:4812–4822.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zini L, Porpiglia F and Fassnacht M:
Contemporary management of adrenocortical carcinoma. Eur Urol.
60:1055–1065. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fassnacht M, Libé R, Kroiss M and Allolio
B: Adrenocortical carcinoma: a clinician’s update. Nat Rev
Endocrinol. 7:323–335. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ng L and Libertino JM: Adrenocortical
carcinoma: diagnosis, evaluation and treatment. J Urol. 169:5–11.
2003. View Article : Google Scholar
|
|
5
|
Roman S: Adrenocortical carcinoma. Curr
Opin Oncol. 18:36–42. 2006. View Article : Google Scholar
|
|
6
|
Crucitti F, Bellantone R, Ferrante A, et
al: The Italian Registry for Adrenal Cortical Carcinoma: analysis
of a multiinstitutional series of 129 patients. The ACC Italian
Registry Study Group. Surgery. 119:161–170. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Icard P, Goudet P, Charpenay C, et al:
Adrenocortical carcinomas: surgical trends and results of a
253-patient series from the French Association of Endocrine
Surgeons study group. World J Surg. 25:891–897. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tauchmanovà L, Colao A, Marzano LA, et al:
Andrenocortical carcinomas: twelve-year prospective experience.
World J Surg. 28:896–903. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Decorato JW, Gruber H, Petti M and
Levowitz BS: Adrenal carcinosarcoma. J Surg Oncol. 45:134–136.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fischler DF, Nunez C, Levin HS, et al:
Adrenal carcinosarcoma presenting in a woman with clinical signs of
virilization. A case report with immunohistochemical and
ultrastructural findings. Am J Surg Pathol. 16:626–631. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sasaki K, Desimone M, Rao HR, et al:
Adrenocortical carcinosarcoma: a case report and review of the
literature. Diagn Pathol. 5:512010.PubMed/NCBI
|
|
12
|
Thway K, Olmos D, Shah C, et al: Oncocytic
adrenal cortical carcinosarcoma with pleomorphic
rhabdomyosarcomatous metastases. Am J Surg Pathol. 36:470–477.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Barksdale SK, Marincola FM and Jaffe G:
Carcinosarcoma of the adrenal cortex presenting with
mineralocorticoid excess. Am J Surg Pathol. 17:941–945. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Okazumi S, Asano T, Ryu M, et al: Surgical
resection of adrenal carcinoma extending into the vena cava, right
atrium and ventricle: case report and review of the literature.
Nihon Geka Gakkai Zasshi. 88:231–238. 1987.(In Japanese).
PubMed/NCBI
|
|
15
|
Collina G, Maldarizzi F, Betts CM and
Eusebi V: Primary sarcomatoid carcinoma of the adrenal gland. First
case report. Virchows Arch A Pathol Anat Histopathol. 415:161–167.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee MS, Park IA, Chi JG, et al: Adrenal
carcinosarcoma - a case report. J Korean Med Sci. 12:374–377. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sturm N, Moulai N, Laverrière MH, et al:
Primary adrenocortical sarcomatoid carcinoma: case report and
review of literature. Virchows Arch. 452:215–219. 2008. View Article : Google Scholar
|
|
18
|
Coli A, Di Giorgio A, Castri F, et al:
Sarcomatoid carcinoma of the adrenal gland: a case report and
review of literature. Pathol Res Pract. 206:59–65. 2010. View Article : Google Scholar
|
|
19
|
Bertolini F, Rossi G, Fiocchi F, et al:
Primary adrenal gland carcinosarcoma associated with metastatic
rectal cancer: a hitherto unreported collision tumor. Tumori.
97:27e–30e. 2011.PubMed/NCBI
|
|
20
|
Kao CS, Grignon DJ, Ulbright TM and Idrees
MT: A case report of adrenocortical carcinosarcoma with oncocytic
and primitive neuroectodermal-like features. Hum pathol.
44:1947–1955. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Miettinen M: Neuroendocrine
differentiation in adrenocortical carcinoma. New
immunohistochemical findings supported by electron microscopy. Lab
Invest. 66:169–174. 1992.PubMed/NCBI
|
|
22
|
Hoang MP, Ayala AG and Albores-Saavedra J:
Oncocytic adrenocortical carcinoma: a morphologic,
immunohistochemical and ultrastructural study of four cases. Mod
Pathol. 15:973–978. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jorda M, De MB and Nadji M: Calretinin and
inhibin are useful in separating adrenocortical neoplasms from
pheochromocytomas. Appl Immunohistochem Mol Morphol. 10:67–70.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ghorab Z, Jorda M, Ganjei P and Nadji M:
Melan A (A103) is expressed in adrenocortical neoplasms but not in
renal cell and hepatocellular carcinomas. Appl Immunohistochem Mol
Morphol. 11:330–333. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang PJ, Genega EM, Tomaszewski JE, et
al: The role of calretinin, inhibin, melan-A, BCL-2, and C-kit in
differentiating adrenal cortical and medullary tumors: an
immunohistochemical study. Mod Pathol. 16:591–597. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pan CC, Chen PC, Tsay SH and Ho DM:
Differential immunoprofiles of hepatocellular carcinoma, renal cell
carcinoma, and adrenocortical carcinoma: a systemic
immunohistochemical survey using tissue array technique. Appl
Immunohistochem Mol Morphol. 13:347–352. 2005. View Article : Google Scholar : PubMed/NCBI
|