Introduction
The majority of cases of acute leukemia (AL)
originate from a specific cellular lineage, either lymphoid or
myeloid, and can therefore be classified as acute lymphoblastic
leukemia (ALL) or acute myeloid leukemia (AML). This classification
is based upon morphological features and the cytochemical and
immunophenotypical profile of the blast cells (1). In a small number of cases, the
leukemic cells express markers belonging to more than one lineage.
This may include two separate blast populations, one myeloid and
one lymphoid, or a single blast population that expresses myeloid
and lymphoid markers simultaneously (2). According to the most recent World
Health Organization (WHO) classification (3), these types are considered as AL of
ambiguous lineage. Biphenotypic AL (BAL) represents ~5% of adult AL
cases, and is defined as a single blast population that
co-expresses two lineage markers (4,5).
Burkitt leukemia (BL) is a highly-aggressive, mature
B-cell neoplasm, which is classified as an L3 ALL by the
French-American-British system (6).
BL exhibits unique morphological characteristics and has the
cytogenetic hallmark of t(8;14)(q24;q32) (7). The present study describes a rare case
of T-cell ALL (T-ALL), with cells co-expressing myeloid markers,
which highly resembled the classic Burkitt leukemia morphology.
Case report
On June 9, 2013, a previously healthy 37-year-old
male was admitted to a local hospital with palpitations and night
sweats that had been apparent for one month. A complete blood count
(CBC) revealed a white blood cell (WBC) count of
2.89×109/l (normal range, 4–10×109/l), a red
blood cell count (RBC) of 1.43×1012/l (normal range,
4.4–5.5×1012/l), a hemoglobin (Hb) level of 46 g/l
(normal range, 120–160 g/l) and a platelet (PLT) count of
91×109/l (normal range, 100–300×109/l). A
bone marrow aspiration identified significantly increased numbers
of raw and immature lymphocytes, medium-sized cells containing a
large number of lipid vacuoles with abundant, basophilic cytoplasm,
and round nuclei with clumped chromatin and multiple nucleoli.
According to the morphological bone marrow features, a diagnosis of
Burkitt leukemia was established. Therefore, an RBC transfusion and
vitamin 12 supplements were administered. After five days, the
patient was referred to the Department of Hematology, Union
Hospital, Tongji Medical University of Science and Technology
(Wuhan, China) for further observation. A CBC revealed a WBC count
of 5.24×109/l, a RBC count of 1.82×1012/l, an
Hb level of 59 g/l and a PLT count of 136×109/l. The
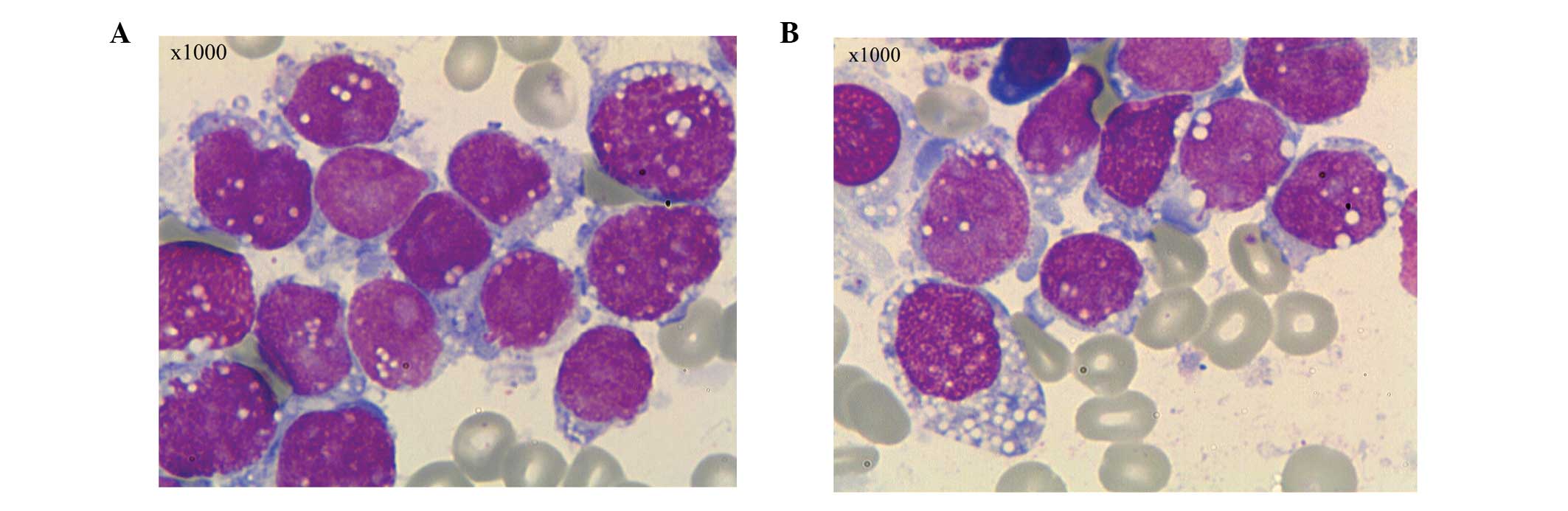
bone marrow and peripheral blood smear revealed 96% and 25% blast
cells, respectively. The blast cells were variable in size,
contained moderate to abundant amounts of cytoplasm and were
stained blue. Certain cells exhibited pseudopodia-like protrusions,
but the majority contained immature lacy chromatin, marked
nucleoli, and cytoplasm consisting of abundant lipid vacuoles and
granules. The features of the cells highly resembled those of
Burkitt leukemia cells (Fig. 1).
Cytochemical staining revealed that the cells were negative for
myeloid peroxidase (MPO), acid α-naphthyl acetate esterase (ANAE)
and chloroacetate esterase, but positive for periodic
acid-Schiff.
Immunophenotyping detected a single blast population
with distinct immunoreactivity for T-lymphoid and myeloid antigens.
The blasts expressed cluster of differentiation (CD) 3, 7, 13, 11b,
33, 34, 56 and 38, and human leukocyte antigen-DR. There was no
significant evidence of mature T and B cells in the background
population. G-banding analysis revealed metaphase cells with normal
karyotypes.
The patient received induction chemotherapy with
hyper-Cytoxan (300 mg/m2, i.v., every 12 h, days 1–3),
vincristine (2 mg, i.v., days 4 and 11), Adriamycin (50
mg/m2, i.v., day 4), dexamethasone (40 mg/day, i.v.,
days 1–4 and days 11–14) and Ara-C (70 mg, intrathecal, day 7).
Following once cycle of chemotherapy, the bone marrow aspirate was
reevaluated, which revealed no evidence of leukemia. The patient
attained complete morphological remission, but subsequently
succumbed due to severe complications following a stem cell
transplantation procedure. Written informed consent was obtained
from the patient’s family for the publication of this study.
Discussion
The present study reports a rare case of adult AL,
in which the blast cells co-expressed T-lymphoid and myeloid
antigens, and in which the classical morphological features of
Burkitt leukemia were apparent. BAL is a rare disease, which refers
to a form of AL with a single population of blasts that co-express
markers of two different lineages (4), BAL is often confused with acute
bilineal leukemia, which is composed of a mixed population of
leukemic cells originating from two distinct lineages (8–10). In
1995, a consensus criteria for the diagnosis of BAL was established
by the European Group for the Immunological Characterization of
Leukemias (EGIL) to distinguish between patients with BAL and those
with AL with an aberrant expression of markers from a different
lineage. Within this scoring system, CD markers are assigned a
score of 0.5, 1.0 or 2.0, depending upon whether a particular
antigen originates from a myeloid, B- or T-lymphoid lineage
(Table I) (11). According to the EGIL, a score of
>2 points is sufficient to assign membership in a cell line. If
the score is <2 for the other cell lines, or other markers of
these lines, they should be qualified as aberrant markers. Using
this system, a diagnosis of T-ALL/AML can be established.
 | Table IEuropean Group for the Immunological
Classification of Leukemias scoring system. |
Table I
European Group for the Immunological
Classification of Leukemias scoring system.
| Points | B-lymphoid
lineage | T-lymphoid
lineage | Myeloid lineage |
|---|
| 2 | CytCD79a | CD3 | MPO |
| CytIgM | anti-TCR | |
| CytCD22 | | |
| 1 | CD19 | CD2 | CD117 |
| CD20 | CD5 | CD13 |
| CD10 | CD8 | CD33 |
| | CD10 | CD65 |
| 0.5 | TdT | TdT | CD14 |
| CD24 | CD7 | CD15 |
| | CD1a | CD64 |
The 2008 WHO classification of hematological tumors
adopted the EGIL criteria for BAL and introduced a novel group of
AL termed ‘AL of ambiguous lineage’. However, the 2008 WHO
classification adopts a more restrictive criteria than the EGIL to
define BAL (12). According to the
2008 WHO criteria, BAL originates from a lineage of cells that
express MPO, CD19 and cytoplasmic CD3 (3). In the present study, MPO expression
was negative, and therefore, the likely diagnosis was T-ALL with
aberrant expression of myeloid markers. Suggs et al
(13) reported that the myeloid
markers CD13 and CD33 are commonly expressed in certain precursor
T-ALLs.
Coche et al (14) described a case of BAL with
Burkitt-like morphology, in which the cells co-expressed the
myeloid markers, IgM, CD79a, 19, 22 and 24, and the B-lymphoid
lineage markers, CD13, 33, 65 and 15. To the best of our knowledge,
the present study was the first to describe a case of T-lymphoid
and myeloid lineage marker co-expression with Burkitt-like cells.
The patient was initially misdiagnosed at the local hospital due to
the atypical cellular morphology, however, the clinical features of
the patient differed from those of the majority of BL cases.
Firstly, BL is a mature B-cell neoplasm. The immunological
phenotype in the present case was markedly different from BL.
Secondly, 80% of BL cases harbor the t(8;14) translocation; in the
remaining 20% of cases, translocations exist between chromosomes 2
and 8, t(2;8)(p12;q24), or between chromosomes 8 and 22,
t(8;22)(q24;q11) (15–17). By contrast, the cytogenetic profile
of the patient in the present study appeared normal.
Of the scoring systems used for the classification
of BAL, EGIL is usually applied during routine clinical practice.
However, certain limitations of this classification system exist.
Firstly, the EGIL does not define lineage-specific markers. Certain
lineage-specific markers, such as CD3, CD22 and MPO, may be only
slightly higher than lineage-associated markers, such as CD7, 13,
19, 20 and 33. This has the potential to lead to an overdiagnosis
of BAL. Secondly, the EGIL is based upon immunological markers and
omitted cytogenetic data, therefore, even well-defined AML may be
misdiagnosed as BAL. Finally, as there are no standard treatment
regimens for BAL, the EGIL is unable to predict optimal therapy
decisions. Therefore, hematologists/oncologists may choose to treat
patients with regimens for either AML or ALL, or both. Improved
lineage definition will provide clinicians with suitable guidelines
for selecting appropriate therapeutic regimens, since the treatment
of AML differs from that of ALL.
At present, there is no uniform agreement regarding
the treatment of patients with BAL. According to the literature
(9,18), ALL-oriented chemotherapy exhibits a
higher complete remission rate compared with AML-oriented
chemotherapy. However, according to the 2008 WHO classification,
the present case was more inclined to be T-ALL with aberrant
myeloid makers, and therefore, the patient was administrated
ALL-based induction chemotherapy. Despite progress in the treatment
of AL (8,19), the prognosis of BAL or T-ALL with
myeloid markers, and the response to drug therapies that target
conventional ALL, remains poor.
To the best of our knowledge, the present study was
the first to report a case of T-ALL with cells co-expressing
myeloid makers and highly resembling the classic morphology of
Burkitt leukemia cells. The diagnosis was determined by the 2008
WHO classification, and not the EGIL. This case demonstrated the
heterogeneity of AL, not only in regard to cellular morphology, but
also immunological performance.
References
|
1
|
Paietta E: Proposals for the immunological
classification of acute leukemias. Leukemia. 9:2147–2148.
1995.PubMed/NCBI
|
|
2
|
Buccheri V, Matutes E, Dyer MJ and
Catovsky D: Lineage commitment in biphenotypic acute leukemia.
Leukemia. 7:919–927. 1993.PubMed/NCBI
|
|
3
|
Swerdlow SH, Campo E, Harris NL, et al:
Acute leukemias of ambiguous lineage. WHO Classification of Tumors
of Haematopoietic and Lymphoid Tissues. IARC; Lyon: pp. 149–155.
2008
|
|
4
|
Matutes E, Morilla R, Farahat N, et al:
Definition of acute biphenotypic leukemia. Haematologica. 82:64–66.
1997.PubMed/NCBI
|
|
5
|
Buccheri V, Matutes E, Dyer MJ and
Catovsky D: Lineage commitment in biphenotypic acute leukemia.
Leukemia. 7:919–927. 1993.PubMed/NCBI
|
|
6
|
Bennett JM, Catovsky D, Daniel MT, et al:
Proposed revised criteria for the classification of acute myeloid
leukemia: A report of the French-American-British Cooperative
Group. Ann Intern Med. 103:620–625. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Harris NL, Jaffe ES, Diebold J, et al:
World Health Organization classification of neoplastic diseases of
the hematopoietic and lymphoid tissues: report of the Clinical
Advisory Committee meeting - Airlie House, Virginia, November 1997.
J Clin Oncol. 17:3835–3849. 1999.PubMed/NCBI
|
|
8
|
Weir EG, Ali Ansari-Lari M, Batista DA, et
al: Acute bilineal leukemia: a rare disease with poor outcome.
Leukemia. 21:2264–2270. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rubnitz JE, Onciu M, Pounds S, et al:
Acute mixed lineage leukemia in children: the experience of St Jude
Children’s Research Hospital. Blood. 113:5083–5089. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Carbonell F, Swansbury J, Min T, et al:
Cytogenetic findings in acute biphenotypic leukaemia. Leukemia.
10:1283–1287. 1996.PubMed/NCBI
|
|
11
|
Bene MC, Castoldi G, Knapp W, et al:
Proposals for the immunological classification of acute leukemias.
European Group for the Immunological Characterization of Leukemias
(EGIL). Leukemia. 9:1783–1786. 1995.PubMed/NCBI
|
|
12
|
Vardiman JW, Thiele J, Arber DA, et al:
The 2008 revision of the World Health Organization (WHO)
classification of myeloid neoplasms and acute leukemia: rationale
and important changes. Blood. 114:937–951. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Suggs JL, Cruse JM and Lewis RE: Aberrant
myeloid marker expression in precursor B-cell and T-cell leukemias.
Exp Mol Pathol. 83:471–473. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Coche D, Bergues B, Harrivel V and
Guillaume N: Biphenotypic acute leukaemia with Burkitt-like
cytology. Ann Biol Clin (Paris). 67:437–440. 2009.(In French).
|
|
15
|
Hecht JL and Aster KC: Molecular biology
of Burkitt’s lymphoma. J Clin Oncol. 18:3707–3721. 2000.PubMed/NCBI
|
|
16
|
Neri A, Barriga M, Knowles DM, et al:
Different regions of the immunoglobulin heavy-chain locus are
involved in chromosomal translocations in distinct pathogenetic
forms of Burkitt lymphoma. Proc Natl Acad Sci USA. 85:2748–2752.
1988. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gerbitz A, Mautner J, Geltinger C,
Hörtnagel K, Christoph B, Asenbauer H, Klobeck G, Polack A and
Bornkamm GW: Deregulation of the proto-oncogene c-myc through
t(8;22) translocation in Burkitt’s lymphoma. Oncogene.
18:1745–1753. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Aribi A, Bueso-Ramos C, Estey E, et al:
Biphenotypic acute leukaemia: a case series. Br J Haematol.
138:213–216. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zucchini A, Fattori PP, Lanza F, et al:
Biphenotypic acute leukemia: a case report. J Biol Regul Homeost
Agents. 18:387–391. 2004.
|