Introduction
Uterine cancer is the most common gynecological
malignancy in Sweden. It accounts for 5.2% of all female
malignancies, with 1431 new cases reported in 2011 (1). The majority of uterine cancer cases
are endometrial carcinoma, whilst uterine sarcomas are less common,
accounting for <5% of all cases.
The risk of uterine cancer increases with age, and
females are typically affected between 50 and 60 years of age
(2,3). The major risk factors for uterine
cancer include diabetes mellitus, obesity, hypertension, polycystic
ovary syndrome, anovulation, nulliparity and exposure to exogenous
estrogens (2–4). There are two types of endometrial
cancer: Type I and type II. Type I is preceded by endometrial
hyperplasia due to estrogen stimulation. These tumors originate
from endometrioid epithelium and are typically well-differentiated.
Type I is associated with specific genetic alterations, including
microsatellite instability due to methylation of the mismatch
repair gene MLH1, and somatic mutations in KRAS,
CTNNB1, phosphatase and tensin homolog (PTEN) and
PIK3CA genes. Type II diseases are of serous or clear cell
histology, and develop from the atrophic endometrium without
hyperplasia. They are generally poorly differentiated and exhibit
p53 mutations, E-cadherin inactivation and
HER2-amplification (3,5,6). The
majority of cases of uterine cancer are sporadic; however, multiple
instances of endometrial cancer within a family occurs in ~5% of
all cases (7).
Cowden syndrome (CS) is an autosomal dominant
disorder characterized by multiple hamartomas in the breast,
thyroid, colon, kidney and endometrium, and has a worldwide
prevalence of 1 in 250,000 (8).
Germline mutations in PTEN were initially proposed to be
responsible for 80% of CS cases; however, more recent studies
indicate that only 30–35% of all CS cases are caused by PTEN
mutations (9). Recent studies also
indicate that the lifetime risk of developing endometrial carcinoma
in patients with CS is 21–28%, with the highest risk levels
occurring in individuals aged >35 years (10–12).
Diagnosis is determined according to the National Comprehensive
Cancer Network (NCCN) criteria (13).
The gene responsible for CS is the tumor suppressor
gene PTEN, which is located in the 10q23.3 chromosomal
region and consists of nine-exons, encoding the 403 amino acid PTEN
protein. It negatively regulates the phosphoinositide
3-kinase/protein kinase B/mammalian target of rapamycin
(PI3K/AKT/mTOR) pathway by the dephosphorylation of three residues
of phosphatidylinositol (3,4,5)-triphosphate. This decreases the
activity of kinases downstream of PI3K, including phosphoinositide
dependent kinase 1 (PDK-1), AKT, mTOR and ribosomal protein s6
kinase (S6K1). In CS, the loss of activity of PTEN occurs
after inheriting a mutated allele, followed by a second hit
mutation (somatic) of the normal allele, which leads to a loss of
function of the protein product and increased phosphorylation. This
affects various cellular processes and signaling pathways,
including cell cycle progression, metabolism, translation, growth,
migration, invasion, angiogenesis and apoptosis (5,8,14).
Given the low prevalence of CS and the difficulty in
identifying which patients fulfill the NCCN criteria, the present
study aimed to examine whether PTEN mutations are present in
a significant proportion of families with uterine cancer that do
not meet the strict criteria but have a CS-like family hereditary
pattern.
Materials and methods
Uterine cancer patients who underwent surgery
between January 2008 and March 2012 were invited to participate in
the present study. All participants gave their written informed
consent for data collection and genetic analysis. Those who
accepted (index patients) completed a questionnaire regarding the
initial diagnosis and age of onset of coincidental cancers in their
family (the index patient, the first- and second-degree relatives
and first cousins), including colorectal, breast, ovarian and other
cancer types. At the end of the study period in 2012, all index
patients were checked for relapse and/or novel primary tumors via
the Swedish Cancer Registry.
Upon enrollment, all index patients provided a blood
sample for DNA extraction, according to the manufacturer’s
instructions (MagneSil Genomic, Large Volume System, Promega,
Madison, WI, USA; Freedom EVO Tecan robot, serial no. 904004850,
Tecan, Männedorf, Switzerland), at the Department of Clinical
Genetics, Karolinska University Hospital (Stockholm, Sweden), and
their histological results were obtained. Telephone interviews were
conducted to acquire information with regard to cancer diagnosis in
relatives. For all relatives with cancer, the current age or age at
mortality, type of cancer and age at diagnosis was recorded.
Histological verification of cancer diagnoses in relatives was
obtained from the Swedish Cancer Registry, patients’ medical
records and/or their death certificates. All data was acquired
following the written consent of the affected relative (or, if
deceased, from their closest living relative).
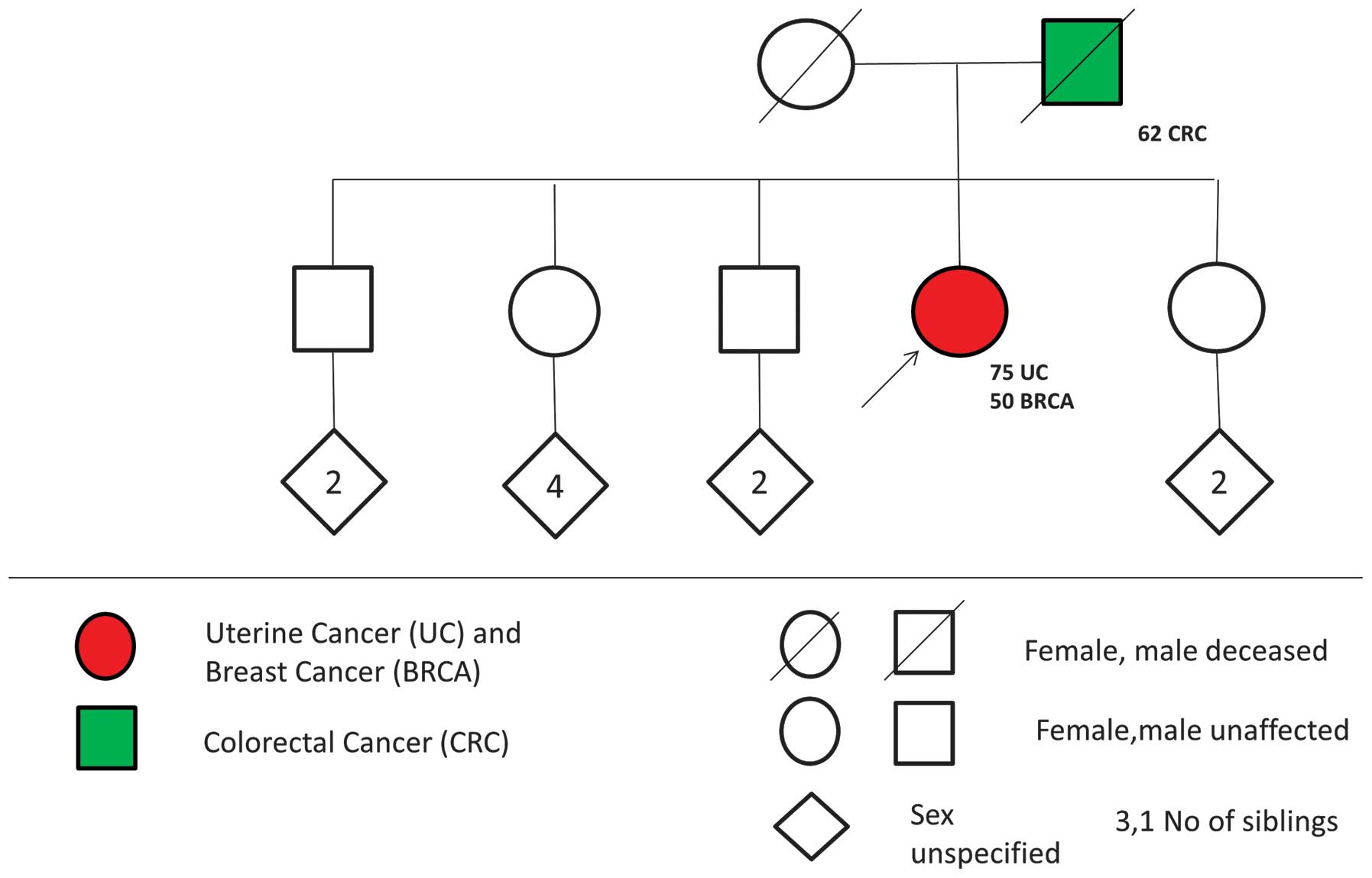
Pedigrees were constructed for each index patient
based on the information provided in the questionnaires and the
telephone interview. All pedigrees were evaluated for the possible
presence of CS using the NCCN guidelines. CS-like families were
defined by the presence of at least one uterine cancer and one
breast cancer, as well as at least one additional Cowden-associated
tumor (uterine, breast, thyroid, colon or kidney cancer), in the
same individual or first-degree relatives (Fig. 1).
Genomic DNA was subjected to touchdown polymerase
chain reaction (PCR) for the amplification of all nine exons of the
PTEN gene and their flanking intronic regions. Firstly,
seven cycles of amplification with denaturation at 95°C for 30 sec,
an annealing gradient for 45 sec (Table
I) and extension at 72°C for 45 sec was performed. Next, a
further 30 cycles of amplification were performed with denaturation
at 95°C for 30 sec, annealing at 55–56°C (Table I) for 45 sec and extension at 72°C
for 45 sec. The final extension step was performed as follows: 72°C
for 10 min followed by cooling of the product overnight at 4°C. PCR
amplification was performed in a final volume of 25 μl, using
AmpliTaq Gold DNA Polymerase (Roche Molecular Diagnostics,
Pleasanton, CA, USA), 10 μM final primer concentration (Table 1) and 50 ng template DNA from each
patient, in a 2720 Thermal Cycler (Applied Biosystems, Foster City,
CA, USA) or a DNA Engine Tetrad 2 Peltier Thermal Cycler (Bio-Rad,
Hercules, CA, USA). PCR products were purified with ExoSAP-IT (USB
Affymetrix, Santa Clara, CA, USA) and subsequently sequenced by
Sanger sequencing using a 48-capillary 3730xl DNA Analyzer (Applied
Biosystems). The resulting sequences were analyzed using Seqscape
software version 2.7 (Invitrogen Life Technologies, Carlsbad, CA,
USA), with the reference sequence NM 000314.4.
 | Table IThe primer pairs for every amplicon
(forward and reverse), their corresponding melting and annealing
temperatures, GC% and the polymerase chain reaction product
size. |
Table I
The primer pairs for every amplicon
(forward and reverse), their corresponding melting and annealing
temperatures, GC% and the polymerase chain reaction product
size.
| PTEN exon | Forward primer
5′-3′ | Tm, °C | GC % | Reverse primer
5′-3′ | Tm, °C | GC % | Amplicon size,
bp | Initial temp, °C | Final temp, °C |
|---|
| Exon 1 |
TTCCATCCTGCAGA
AGAAGC | 60.5 | 50 |
CATCCGTCTACTC
CCACGTT | 60.0 | 55 | 230 | 62 | 55 |
| Exon 2 |
TGGGGAAA(A/G)C
TTTCTTTTCA | 57.3 | 35 |
CATCACAAAGTAT
CTTTTTCTGTGG | 59.1 | 36 | 293 | 62 | 55 |
| Exon 3 |
CCATAGAAGGGGT
ATTTGTTGG | 59.6 | 45.5 |
CAATGCTCTTGGA
CTTCTTGA | 58.1 | 42.9 | 364 | 62 | 55 |
| Exon 4 |
AAAGATTCAGGCA
ATGTTTGTT | 57.8 | 31.8 |
TCTCACTCGATAA
TCTGGATGAC | 58.3 | 43.5 | 235 | 62 | 56 |
| Exon 5 |
TGCAACATTTCTAA
AGTTACCTACTTG | 59.3 | 33.3 |
GAAACCCAAAAT
CTGTTTTCCA | 60.2 | 36.4 | 398 | 62 | 56 |
| Exon 6 |
GGCTACGACCCAG
TTACCAT | 58.9 | 55 |
CCTGCATAAATTT
CAAATGTGG | 59.4 | 36.4 | 347 | 61 | 55 |
| Exon 7 |
AAAATCGTTTTTGA
CAGTTTGACA | 60.0 | 29.2 |
CACCAATGCCAG
AGTAAGCA | 59.9 | 50 | 379 | 61 | 55 |
| Exon 8 |
TGTTTAACATAGGT
GACAGATTTTCTT | 58.7 | 29.6 |
CTCCTAGAATTAA
ACACACATCACA | 57.5 | 36 | 396 | 61 | 55 |
| Exon 9 |
TGTTCATCTGCAAA
ATGGAAT | 58.1 | 33.3 |
CTGGTAATCTGAC
ACAATGTCCT | 58.1 | 43.5 | 399 | 61 | 55 |
The study was approved by the Ethics Committee of
Karolinska Institute/Karolinska University Hospital (Stockholm,
Sweden).
Results and Discussion
A cohort of 54 unrelated patients with uterine
cancer and CS-like phenotype was identified. The characteristics of
the patients are shown in Table
II. None of the individuals included in the present study
fulfilled the CS diagnostic criteria. No germline mutations or
polymorphisms were identified in the coding region of PTEN
in the population of CS-like families.
| Table IICharacteristics of the index patients
(n=54). |
Table II
Characteristics of the index patients
(n=54).
|
Characteristics | n/total | % |
|---|
| Age at diagnosis,
years |
| Median
(range) | 75 (45–87) | |
| Histology |
| Endometrioid | 46/54 | 85.2 |
| Serous or
mixed | 4/54 | 7.4 |
| Clear cell | 3/54 | 5.6 |
| Sarcoma | 1/54 | 1.8 |
| Hyperplasia with
atypia | 0/54 | 0 |
| FIGO stage |
| 1A | 35/54 | 64.8 |
| 1B | 10/54 | 18.5 |
| 2 | 5/54 | 9.3 |
| 3A | 1/54 | 1.8 |
| 3B | 0/54 | 0 |
| 3C | 0/54 | 0 |
| 4 | 0/54 | 0 |
| 4B | 3/54 | 5.6 |
| Grade |
| 1 | 21/54 | 38.9 |
| 2 | 22/54 | 40.7 |
| 3 | 11/54 | 20.4 |
| Depth of myometrial
invasion |
| None | 7/54 | 12.9 |
| <50% | 31/54 | 57.4 |
| ≥50% | 14/54 | 26 |
| Through the
serosa | 2/54 | 3.7 |
| Relapse | 0 | |
| Other cancer in the
same individual | 19/54 | 35.2 |
| Breast cancer | 15/19 | 78.9 |
| Colorectal
cancer | 1/19 | 5.3 |
| Combined | 3/19 | 15.8 |
| Other cancer in the
family (FDRs)a | 52 | |
| Breast cancer | 27/52 | 51.9 |
| Colorectal
cancer | 12/52 | 3.8 |
| Endometrial
cancer | 1/52 | 1.9 |
| Combinedb | 12/52 | 3.8 |
As CS is extremely rare, only small-scale studies
have searched for PTEN mutations in families with CS or
CS-like phenotype (5,15–22),
and none of these studies investigated families with uterine
cancer. Patients with uterine cancer and CS-like family history
were referred to our oncogenetic clinic, however, the value of
testing for mutations in PTEN in this patient category is
unclear. Marsh et al (15)
reported one family with a PTEN mutation among 64 CS-like
families with breast and thyroid cancer, suggesting that certain
CS-like families may have CS without fulfilling the strict NCCN
criteria (11). However, the
majority of studies have not identified PTEN mutations in
CS-like families. These studies included families with breast and
thyroid cancer (16,17), hereditary breast cancer (18–20),
breast and central nervous system cancer (21), consecutive cases of thyroid cancer
(22) or consecutive endometrial
cancer (5). The present study
indicates that PTEN mutations are not the cause of cancer in
CS-like families with uterine cancer.
The two major flaws of the current study are the
small number of patients included in the sample, and the lack of
detailed phenotypic evaluation of the patients in order to obtain
information on head circumference, and on non-cancer phenotypes.
Macrocephaly is one of the required major diagnostic criteria for
Cowden syndrome and therefore measurements of head circumference
are important. However, information on head circumference and
discrete mucocutaneous lesions is often lacking on patients with
endometrial cancer and a family history of Cowden-associated
tumors, who are referred for clinical genetic testing of
PTEN in our clinic. The study included the DNA sequencing of
the coding region of PTEN, where 90% of all CS-mutations are
detected. However, deletions/duplications were not investigated, as
larger deletions in PTEN have been demonstrated in only 1%
of all cases (23). The promoter
region of PTEN, where mutations have been observed in 10% of
all patients with classic CS (23),
was also excluded. Despite these limitations, the results indicate
that PTEN mutations are rare in patients with uterine cancer
and other CS-related tumors, and clinical testing of PTEN is
thus not indicated if the NCCN criteria are not met.
Notably, PTEN-negative CS and CS-like
patients have been demonstrated to exhibit hypermethylation of the
bidirectional promoter that regulates PTEN and
KILLIN, which is associated with an increased risk of breast
and kidney cancer (24). Rare cases
also demonstrate mutations in succinate dehydrogenase complex
(SDH) subunit D, SDHB, PI3KCA, AKT1 and
RAS GTPase activating protein genes, suggesting that
PTEN-negative cases of CS are genetically heterogeneous
(25–27). Thus, diagnosis of CS and CS-like
patients may be improved by utilizing a targeted gene panel
including the aforementioned genes, in combination with a
methylation analysis. This was beyond of the scope of the present
study, and may be a perspective for future studies.
In conclusion, germline PTEN mutations are
rare in a population of CS-like families with uterine cancer. The
high cost of routine screening for PTEN mutations among
endometrial cancer patients is not justified at an oncogenetic
clinic, and must be restricted to patients that meet the strict
Cowden criteria. Gynecologists must be aware of the CS criteria in
order to identify potential cases of CS in females where uterine
cancer is the sentinel cancer.
Acknowledgements
Financial support was provided through the regional
agreement on medical training and clinical research (ALF) between
the Stockholm County Council and Karolinska Institute (grant no.
510 222). This study was also supported by grants from the Swedish
Labor Market Insurance (grant no. 100069).
The authors would like to thank Mrs Berith Wejderot
(Department of Women’s and Children’s Health, Karolinska University
Hospital, Stockholm, Sweden), Mrs Margareta Ström (Division of
Obstetrics and Gynecology, Karolinska University Hospital), Mrs
Maria Karlsson (Division of Obstetrics and Gynecology, Karolinska
University Hospital), Dr Olga Romanov (Department of Obstetrics and
Gynecology, South Stockholm General Hospital, Stockholm, Sweden),
Dr Tao Liu (Department of Molecular Medicine and Surgery,
Karolinska University Hospital) and Professor Kristina
Gemzell-Danielsson (Department of Women’s and Children’s Health,
Karolinska University Hospital) for their valuable assistance.
References
|
1
|
Cancer incidence in Sweden in 2011.
National Board of Health and Welfare; Stockholm, Sweden: pp. 23–25.
2012
|
|
2
|
Haidopoulos D, Simou M, Akrivos N,
Rodolakis A, Vlachos G, et al: Risk factors in women 40 years of
age and younger with endometrial carcinoma. Acta Obstet Gynecol
Scand. 89:1326–1330. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sorosky JI: Endometrial cancer. Obstet
Gynecol. 120:383–397. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bansal N, Yendluri V and Wenham RM: The
molecular biology of endometrial cancers and the implications for
pathogenesis, classification, and targeted therapies. Cancer
Control. 16:8–13. 2009.
|
|
5
|
Black D, Bogomolniy F, Robson ME, Offit K,
Barakat RR and Boyd J: Evaluation of germline PTEN mutations in
endometrial cancer patients. Gynecol Oncol. 96:21–24. 2005.
View Article : Google Scholar
|
|
6
|
Peterson LM, Kipp BR, Halling KC, et al:
Molecular characterization of endometrial cancer: a correlative
study assessing microsatellite instability, MLH1 hypermethylation,
DNA mismatch repair protein expression, and PTEN, PIK3CA, KRAS, and
BRAF mutation analysis. Int J Gynecol Pathol. 31:195–205. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Olson SH, Chen C, De Vivo I, Doherty JA,
Hartmuller V, et al: Maximizing resources to study an uncommon
cancer: E2C2 - Epidemiology of Endometrial Cancer Consortium.
Cancer Causes Control. 20:491–496. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Farooq A, Walker LJ, Bowling J and Audisio
RA: Cowden syndrome. Cancer Treat Rev. 36:577–583. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pilarski R, Burt R, Kohlman W, Pho L,
Shannon KM and Swisher E: Cowden syndrome and the PTEN hamartoma
tumor syndrome: systematic review and revised diagnostic criteria.
J Natl Cancer Inst. 105:1607–1616. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nieuwenhuis MH, Kets CM, Murphy-Ryan M, et
al: Cancer risk and genotype-phenotype correlations in PTEN
hamartoma tumor syndrome. Fam Cancer. 13:57–63. 2014. View Article : Google Scholar
|
|
11
|
Bubien V, Bonnet F, Brouste V, et al;
French Cowden Disease Network. High cumulative risks of cancer in
patients with PTEN hamartoma tumour syndrome. J Med Genet.
50:255–263. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tan MH, Mester JL, Ngeow J, Rybicki LA,
Orloff MS and Eng C: Lifetime cancer risks in individuals with
germline PTEN mutations. Clin Cancer Res. 18:400–407. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
National Comprehensive Cancer Network.
NCCN guidelines, version 1.2012. http://www.nccn.org/professionals/physician_gls/f_guidelines.asp.
Accessed September 25, 2012
|
|
14
|
Orloff MS and Eng C: Genetic and
phenotypic heterogeneity in the PTEN hamartoma tumour syndrome.
Oncogene. 27:5387–5397. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Marsh DJ, Dahia PL, Caron S, et al:
Germline PTEN mutations in Cowden syndrome-like families. J Med
Genet. 35:881–885. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rustad CF, Bjørnslett M, Heimdal KR, Mæhle
L, Apold J and Møller P: Germline PTEN mutations are rare and
highly penetrant. Hered Cancer Clin Pract. 4:177–185. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lynch ED, Ostermeyer EA, Lee MK, et al:
Inherited mutations in PTEN that are associated with breast cancer,
cowden disease, and juvenile polyposis. Am J Hum Genet.
61:1254–1260. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Freihoff D, Kempe A, Beste B, et al:
Exclusion of a major role for the PTEN tumour-suppressor gene in
breast carcinomas. Br J Cancer. 79:754–758. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kurian AW, Hare EE, Mills MA, et al:
Clinical evaluation of a multiple-gene sequencing panel for
hereditary cancer risk assessment. J Clin Oncol. 32:2001–2009.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Castéra L, Krieger S, Rousselin A, et al:
Next-generation sequencing for the diagnosis of hereditary breast
and ovarian cancer using genomic capture targeting multiple
candidate genes. Eur J Hum Genet. 22:1305–1313. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Laugé A, Lefebvre C, Laurent-Puig P, et
al: No evidence for germline PTEN mutations in families with breast
and brain tumours. Int J Cancer. 84:216–219. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nagy R, Ganapathi S, Comeras I, et al:
Frequency of germline PTEN mutations in differentiated thyroid
cancer. Thyroid. 21:505–510. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhou XP, Waite KA, Pilarski R, et al:
Germline PTEN promoter mutations and deletions in
Cowden/Bannayan-Riley-Ruvalcaba syndrome result in aberrant PTEN
protein and dysregulation of the phosphoinositol-3-kinase/Akt
pathway. Am J Hum Genet. 73:404–411. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bennett KL, Mester J and Eng C: Germline
epigenetic regulation of KILLIN in Cowden and Cowden-like syndrome.
JAMA. 304:2724–2731. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ni Y, Zbuk KM, Sadler T, et al: Germline
mutations and variants in the succinate dehydrogenase genes in
Cowden and Cowden-like syndromes. Am J Hum Genet. 83:261–268. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Orloff MS, He X, Peterson C, et al:
Germline PIK3CA and AKT1 mutations in Cowden and Cowden-like
syndromes. Am J Hum Genet. 92:76–80. 2013. View Article : Google Scholar :
|
|
27
|
Ngeow J, Ni Y, Tohme R, Song Chen F, Bebek
G and Eng C: Germline alterations in RASAL1 in Cowden syndrome
patients presenting with follicular thyroid cancer and in
individuals with apparently sporadic epithelial thyroid cancer. J
Clin Endocrinol Metab. 99:E1316–E1321. 2014. View Article : Google Scholar : PubMed/NCBI
|