Introduction
Heat shock protein 90 (HSP90) is a highly conserved
molecular chaperone that participates in stabilizing and activating
a wide range of proteins (referred to as HSP90 client proteins),
many of which are involved in tumor progression via interaction
with more than 20 co-chaperones which contribute to its recognition
of client proteins and modulate its biochemical activities
(1–3). More than 100 client proteins have been
explored, among which certain kinases (such as ERBB2, BRAF, EGFR,
CDK6, AKT) and steroid receptors (GR, PR, AR) are well known. In
malignant cells, an increased expression of HSP90 results in the
subversion of its essential chaperoning functions and protects
mutated and overexpressed oncoproteins from degradation and thereby
promotes cancer cell survival (4).
In addition, compared with the latent, uncomplexed conformation of
HSP90 from normal cells, the activated, multichaperone complexes of
tumor HSP90 have a 100-fold higher binding affinity to
17-allylaminogeldanamycin (17-AAG), a first-in-class HSP90
inhibitor currently in phase II/III clinical trials in adults
(5,6). Oncology trials have demonstrated that
blocking HSP90 function with inhibitors induces client protein
degradation and apoptosis, and inhibits cell proliferation and
tumor growth as well as metastasis in various cancer cells and
tumor xenografts (7–9). Clinical evaluation has also confirmed
that HSP90 inhibitors have clinical activity, especially when
combined with other tumor-specific inhibitors (10–14).
Abundant pre-clinical and clinical data demonstrate that inhibiting
HSP90 is a promising strategy for cancer treatment.
Esophageal squamous cell carcinoma (ESCC) is one of
the most common cancers worldwide. More than half of all patients
suffering from esophageal cancer are diagnosed with an advanced
stage of tumor, with either unresectable tumors or radiographically
visible metastases (15). Advances
in surgical resection and neoadjuvant chemoradiatherapy have yet to
overcome the very low overall survival of esophageal cancer
patients (15,16). However, molecular targeted therapy
is a new therapeutic strategy being put into clinical usage and
offers the possibility of an improvement of the survival rate. It
has been shown that HSP90 is abundantly expressed in esophageal
cancer and the specific inhibition of HSP90 by 17-AAG inhibited
cell proliferation and survival by impeding various cellular
components involving proliferative and survival signaling pathways
(17). Therefore, HSP90 may be an
attractive molecular target in ESCC treatment.
NVP-AUY922 is a synthetic small-molecule inhibitor
which antagonizes the function of HSP90 by blocking ATP binding and
exhibits a potent antitumor effect in different types of cancer
(18–20). Since studies regarding HSP90 and its
inhibitors in ESCC, one of the most aggressive malignancies, are
rare, in the present study we assessed the antiproliferative effect
of NVP-AUY922 in ESCC (TE-1, TE-4, TE-8 and TE-10 cell lines). We
further determined if its antiproliferative effect can be affected
by the expression status of a certain growth-related molecule, such
as the phosphatase and tensin homolog deleted on chromosome 10
(PTEN), a suppressor molecule of PI3K-AKT.
Materials and methods
Cell lines and culture conditions
Human esophageal squamous cell carcinoma (ESCC)
lines TE-1, TE-4, TE-8 and TE-10 were cultured in RPMI-1640 medium
supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin
G sodium, 100 μg/ml streptomycin and maintained in a monolayer
culture at 37°C in humidified air with 5% CO2. Cellular
morphology was observed through a microscope during culture and the
experiments.
Reagents
NVP-AUY922 was synthesized and provided by Novartis
Pharma AG (Basel, Switzerland) through a materials transfer
agreement with Okayama University (Okayama, Japan). Stock solutions
of the compound (10 mmol/l) were reconstituted with dimethyl
sulphoxide (DMSO; Sigma-Aldrich, St. Louis, MO, USA) and stored at
−30°C. Working solutions of NVP-AUY922 with a culture medium were
prepared freshly before use. The final concentration of DMSO in all
cultures was 0.0005%.
Trypan blue exclusion assay and
IC50 calculation
TE-1, TE-4, TE-8 and TE-10 cells were seeded in
12-well plates at a density of 1×105/well for 24 h
before drug treatment. The subconfluent cells were treated with
different concentrations of NVP-AUY922 for 24 h. After treatment,
cells were digested with trypsin, stained with trypan blue and
counted manually with a hemacytometer. Dose-effect plots were
created to calculate the IC50 of NVP-AUY922 for each
cell line using CalcuSyn software (Biosoft).
Western blot analysis
ESCC cells were plated into 6-well plates at a
density of 2.5×105/well for 24 h before drug treatment.
The subconfluent cells were treated with different concentrations
of NVP-AUY922 for 24 h. The culture medium was carefully removed,
washed once in cold PBS, and an appropriate amount of Mammalian
Protein Extraction Reagent (M-PER; Thermo Scientific, Rockford, IL,
USA) was added to the plate. Cell lysate was collected after
shaking gently for 5 min and centrifuged at 15,000 rpm at 4°C for
20 min. The supernatant was transferred to a new tube for protein
determination and western blot analysis. The concentration of
protein lysates was measured with a Bicinchoninic acid (BCA)
protein assay kit (Thermo Scientific). Equal amounts (20 μg) of
protein lysate were electrophoresed under reducing conditions in
5–10% (w/v) SDS-polyacrylamide gels. Proteins were then transferred
to Hybond polyvinylidene difluoride (PVDF) transfer membranes (GE
Healthcare, Buckinghamshire, UK) and incubated with primary
antibodies at 4°C overnight, followed by incubation with
peroxidase-linked secondary antibodies at room temperature for 1 h.
SuperSignal West Pico chemiluminescent substrate (Thermo
Scientific) and chemiluminescence film (GE Healthcare) were used
for signal detection.
The antibodies used for western blot analysis were
the following: PTEN (#9559), AKT (#2938), phospho-AKT (Ser473)
(#4058), ERK1/2 (#9102) and phospho-ERK1/2 (Thr202/Tyr204) (#9101)
were purchased from Cell Signaling Technology, Inc. (Beverly, MA,
USA). Actin (sc-69879) was obtained from Santa Cruz Biotechnology,
Inc. (Santa Cruz, CA, USA). Peroxidase-conjugated secondary
antibodies (goat anti-rabbit IgG and goat anti-mouse IgG) were
obtained from Jackson ImmunoResearch Laboratories, Inc. (West
Grove, PA, USA).
pcDNA3 GFP PTEN transfection and G418
selection
TE-4 cells with an antibiotic-free medium were
seeded in a 60-mm dish for 24 h before transfection. Plasmid pcDNA
GFP PTEN (Addgene, Cambridge, MA, USA) was mixed gently with
Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) and incubated
for 20 min at room temperature. The medium was changed with a fresh
antibiotic-containing medium after adding plasmid-Lipofectamine
2000 complexes for 6 h. To establish a stable GFP-PTEN expressing
cell line, the transfected cells were placed at a 1:10 ratio into a
fresh growth medium 24 h after transfection. The culture medium was
changed with 300 μg/ml of G418-containing medium the following day.
Cell growth was observed every 2–3 days and the medium changed with
the selection drug every 3 days. PTEN transduced TE-4 cells
(TE-4/PTEN cells) were maintained with 300 μg/ml of G418-containing
medium and used for NVP-AUY922 treatment. TE-4 cells were used as a
control group.
Cell growth curve
TE-4 and TE-4/PTEN cells were seeded in a 24-well
plate at a density of 3×103/well. The number of cells
was counted by trypan blue exclusion assay on Days 1, 3, 5 and
7.
siRNA transfection
TE-10 cells with an antibiotic-free medium were
seeded in a 12-well plate at a density of 8×104/well for
24 h before transfection. PTEN siRNA (Cell Signaling Technology,
Inc.) or control siRNA were mixed gently with Lipofectamine 2000
and incubated for 20 min at room temperature. The medium was
changed with a fresh antibiotic containing medium after adding
siRNA-Lipofectamine 2000 complexes for 6 h. The following day, PTEN
siRNA transfected TE-10 cells were applied to drug treatment. TE-10
cells that were transfected with Lipofectamine 2000 only (mock) or
control siRNA were used as a control group.
Statistical analysis
The comparison of categorical experimental data was
conducted by Student’s t-test. Data are represented as the mean ±
SD. All P-values are two-sided. A P-value <0.05 was considered
to be statistically significant in all experiments.
Results
NVP-AUY922 potently inhibits the
proliferation of ESCC
To determine the antiproliferative effect of
NVP-AUY922 against HSP90, all four esophageal squamous cancer cell
lines (TE-1, TE-4, TE-8 and TE-10) were treated with different
concentrations (0, 10, 30 and 50 nmol/l) of NVP-AUY922 for 24 h and
their sensitivity to NVP-AUY922 was assessed by trypan blue
exclusion assay. The proliferation of esophageal cancer cells was
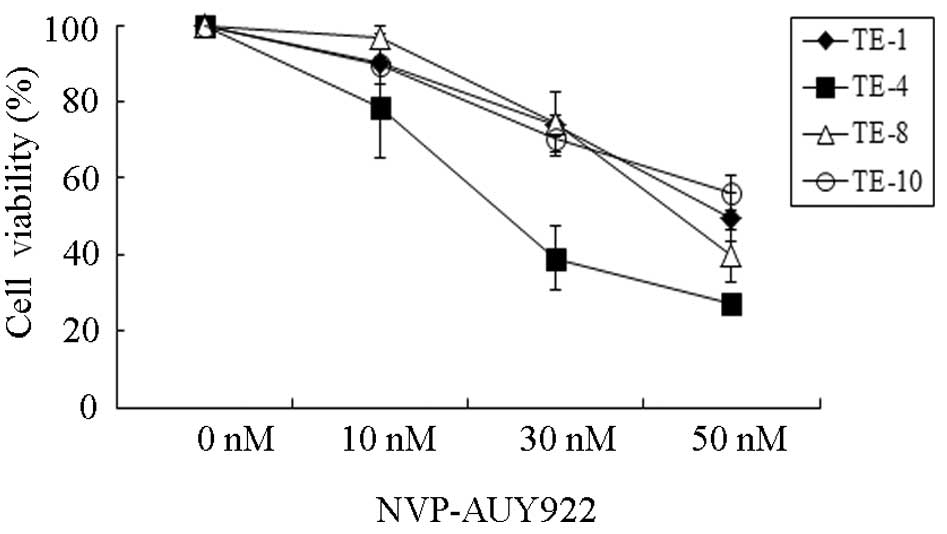
inhibited by NVP-AUY922 in a dose-dependent fashion (Fig. 1). Among the four esophageal cancer
cell lines, TE-4 seemed to be the most sensitive to NVP-AUY922,
especially at a concentration of 30 nmol/l. To quantitatively
examine the effectiveness of NVP-AUY922 in inhibiting cell
proliferation, the half maximal inhibitory concentrations
(IC50) were measured (Table
I). The IC50 of TE-4 was 25.29 nmol/l and those of
TE-1, TE-8 and TE-10 were 72.37, 52.85 and 71.30 nmol/l,
respectively, suggesting that the sensitivity of TE-4 to NVP-AUY922
is 2 to 3-fold higher than that of TE-1, TE-8 or TE-10. These
results demonstrated that NVP-AUY922 exhibits a potent anticancer
effect on esophageal cancers, especially TE-4.
 | Table IIC50 of NVP-AUY922 in
esophageal squamous cell carcinoma. |
Table I
IC50 of NVP-AUY922 in
esophageal squamous cell carcinoma.
| Cell line | TE-1 | TE-4 | TE-8 | TE-10 |
| IC50
(nM) | 72.37 | 25.29 | 52.85 | 71.30 |
AKT and ERK activity is significantly
suppressed by NVP-AUY922 in PTEN-null cells
To explore the molecular mechanism by which
NVP-AUY922 exerts a higher antiproliferative effect on TE-4, we
first detected the activities of AKT and ERK, two key signaling
molecules in the phosphatidylinositide 3-kinase (PI3K)/AKT axis and
the Ras/Raf/MEK/extracellular signal-regulated kinase (ERK) axis,
respectively, each of which plays an important role in cell
proliferation, migration and metabolism (21,22) of
esophageal cancer cells. TE-4 exhibited higher activity levels of
AKT and ERK than TE-1, TE-8, or TE-10 under regular conditions
(Fig. 2A). Besides regulation by
upstream signaling molecules, AKT activity is also negatively
regulated by PTEN via hydrolyzing the 3-phosphate on PIP3 to
generate PIP2 (23). To clarify
whether PTEN affects AKT activity in esophageal cancer, PTEN
expression was observed by western blot analysis (Fig. 2B). The results showed that TE-1,
TE-8 and TE-10 expressed PTEN at different levels, whereas TE-4
completely lost the expression of PTEN, suggesting that the higher
activity of AKT in TE-4 may be a result of the loss of PTEN.
We next sought to determine the effect of NVP-AUY922
on AKT, a client protein of HSP90, and ERK, a key downstream kinase
of HSP90 client protein Raf. PTEN-null TE-4 and PTEN-proficient
TE-10 were treated with different concentrations of NVP-AUY922 (0,
10, 30 and 50 nmol/l) for 24 h and p-AKT, AKT, p-ERK and ERK were
assessed (Fig. 3). Although TE-4
exhibited a higher activity level of AKT and ERK, 50 nmol/l of
NVP-AUY922 almost completely depleted the phosphorylation of both
AKT and ERK in TE-4, but not in TE-10. The expression of AKT was
also significantly inhibited by NVP-AUY922 in a dose-dependent
manner in TE-4. There was no effect on total ERK in TE-4 or TE-10.
In addition, PTEN was not degraded by NVP-AUY922 in TE-10 (Fig. 3). These findings show that compared
with PTEN-proficient cells, PTEN-null cells lose AKT and ERK
activity more easily under NVP-AUY922 treatment.
The expression of PTEN may be associated
with susceptibility to HSP90 inhibitors
Since PTEN-null TE-4 cells exhibited a higher
sensitivity to NVP-AUY922, we hypothesized that the sensitivity of
ESCC to NVP-AUY922 might be associated with the expression of PTEN.
Two strategies were applied to confirm our hypothesis: one was the
exogenous transduction of PTEN in PTEN-null TE-4 using the PTEN
expression vector pcDNA3 GFP PTEN; the other was the silencing of
PTEN expression in PTEN-proficient TE-10 using PTEN siRNA.
PTEN-null TE-4 was exogenously transduced by pcDNA3 GFP PTEN. After
a certain amount of G418 selection, TE-4/PTEN stably expressed the
fusion protein GFP-PTEN (Fig. 4A).
Although PTEN is a tumor suppression factor, the expression of PTEN
in TE-4 did not affect cell proliferation (Fig. 4B). To determine cell sensitivity
after the transduction of PTEN, TE-4/PTEN cells were treated with
different concentrations of NVP-AUY922 (0, 10, 30 and 50 nmol/l)
for 24 h and cell sensitivity was measured by trypan blue exclusion
assay. As shown in Fig. 4C, the
cell viability of TE-4/PTEN under 10, 30 and 50 nmol/l NVP-AUY922
treatment was increased by 21.5, 16.2 and 17.0%, respectively,
suggesting that PTEN decreases cell sensitivity to NVP-AUY922.
PTEN silencing by siRNA in TE-10 cells significantly
suppressed PTEN expression after transfection for 48 h (Fig. 5A). PTEN expression was not changed
in the mock and control siRNA groups. To estimate cell sensitivity
after PTEN silencing, PTEN-silenced TE-10 cells were treated with
different concentrations of NVP-AUY922 (0, 10, 30 and 50 nmol/l)
for 24 h and cell sensitivity was measured by trypan blue exclusion
assay (Fig. 5B). NVP-AUY922
treatment decreased the cell viability by a certain amount. After
treatment with NVP-AUY922 at a concentration of 30 nmol/l, the
viability of cells pretreated with TE-10/mock, TE-10/control and
TE-10/PTEN were 68.5, 70.0 and 48.1% respectively, suggesting that
PTEN silencing decreased cell viability by >20%. These results
support our hypothesis that the expression status of PTEN may
affect the sensitivity to NVP-AUY922 in ESCC cells.
Our results have demonstrated that NVP-AUY922 has a
greater tendency to decrease AKT and ERK activity in PTEN-null
cells (Fig. 3). To confirm the
effect of NVP-AUY922 on AKT activity in PTEN-silencing cells, TE-10
cells that were pretreated with PTEN siRNA were treated with
NVP-AUY922. AKT activity was increased by silencing PTEN in TE-10
cells (Fig. 5C). After drug
treatment, p-AKT was reduced in PTEN-silenced/p-AKT-enhanced TE-10
but not in the control group (Fig.
5C), confirming the possibility that AKT activity affects the
sensitivity of PTEN-silenced cells to NVP-AUY922. Notably, p-ERK
was not decreased by NVP-AUY922, regardless of PTEN silencing.
In summary, NVP-AUY922 has a potent inhibitory
effect on cell proliferation in esophageal cancer cells and PTEN
status can affect its antiproliferative effect most likely via the
regulation of AKT activity.
Discussion
In this study we have demonstrated a promising
antiproliferative effect of HSP90 inhibition by NVP-AUY922 in ESCC
cell lines, particularly in PTEN-loss TE-4 cells. PTEN is a dual
lipid and protein phosphatase and its lipid phosphatase activity
negatively regulates cell survival and proliferation via
inactivating the PI3K/AKT signaling pathway (24,25).
Monoallelic mutations or the complete loss of PTEN at the highest
frequency were estimated in several primary and metastatic cancers
(23). It has been reported that a
decreased expression of nuclear PTEN is associated with disease
advancement and thus may be a significant prognostic marker for
patient suffering from ESCC (26).
Most interestingly, a recent study has demonstrated that
co-expression of HSP90 with phosphatidylinositol-3-kinase
(PI3K)-p110α or the expression of HSP90 along with PTEN loss
predicts significantly worse relapse-free survival (RFS), revealing
strong prognostic significance in patients with invasive breast
cancers (27). However, the
relationship between HSP90 and PTEN expression in ESCC remains to
be examined.
As demonstrated in other tumor types (glioblastoma,
lung cancer and gastric cancer) (9,19,20),
NVP-AUY922 treatment results in a degradation of AKT in TE-4 cells
(Fig. 3). This may be because AKT
as a client protein of HSP90 requires a functional HSP90/CDC37
complex to remain stable and drug treatment induces ubiquitination
of AKT as well as targeting the proteasome, where it is degraded
(28). Downregulation of the PI3
kinase pathway by HSP90 inhibition is partially caused by the
degradation of total AKT expression and partially may be caused by
the degradation of the upstream effectors of AKT (e.g. EGFR and
HER2) (29,30). In addition, ERK, the crucial
signaling transducer of the Ras/Raf/MAPK pathway, was also
inactivated by NVP-AUY922. However, this inactivation is not due to
the decline of protein expression, since HSP90 has no hand in ERK
stability, but possibly due to the degradation of the upstream
regulators of ERK, which require HSP90 for stabilization and
maturation. It was of great interest to us that the effect of
NVP-AUY922 on AKT and ERK was not significant in TE-10 cells. A
good explanation of the lower antiproliferative effect of
NVP-AUY922 in TE-10 compared with TE-4 cells may be the difference
between the expression status of PTEN in TE-4 and TE-10, where TE-4
loses PTEN while TE-10 expresses PTEN proficiently. Our results
therefore suggested that NVP-AUY922 is more effective on AKT and
ERK under PTEN-loss circumstances.
Cell sensitivity to NVP-AUY922 was decreased by the
exogenous transduction of PTEN in PTEN-null TE-4 cells (Fig. 4C), while it was increased by the
endogenous silencing of PTEN in PTEN-proficient TE-10 (Fig. 5B). These results are consistent with
the assessment of sensitivity of TE-10 and TE-4 to NVP-AUY922, and
demonstrated that the expression of PTEN is associated with HSP90
inhibition. This might be confusing because PTEN is a tumor
suppressor protein and negatively regulates the PI3 kinase/AKT
signaling pathway. The inactivation or loss of PTEN should induce
the aberrant activation of AKT and thereby promote cell
proliferation and tumorigenesis. Recent studies have reported that
the loss of PTEN contributes to drug resistance, for example,
gefitinib and erlotinib targeting EGFR in non-small cell lung
cancer (NSCLC) and PLX4720 targeting BRAF in melanomas (31,32).
In contrast to these studies, however, targeting HSP90 by its
specific inhibitors in PTEN-null cancer cells has been demonstrated
as a promising strategy. Eccles et al(18) described that NVP-AUY922 exhibited
potent antitumor efficacy in PTEN-null human glioblastoma
xenografts and in a prostate carcinoma xenograft model. NXD30001, a
new HSP90 inhibitor, also significantly inhibited the growth of
glioblastoma multiforme, in which the loss of PTEN drives the
enhanced EGFR-PI3K-Akt axis (33).
Furthermore, although the anticancer effect of HSP90 inhibitors are
evaluated in PTEN-null cancer cells, the role of PTEN expression in
HSP90 inhibition was not determined previously. This was further
evaluated by our study. We have confirmed that a decline in PTEN
expression confers to HSP90 inhibition causing increased
sensitivity in cancer cells, suggesting that HSP90 treatment with
consideration of PTEN expression may be a novel potential
therapeutic strategy for cancer treatment.
The activity of AKT is elevated by PTEN-loss
(Fig. 3) or PTEN silencing
(Fig. 5C) and is susceptible to
HSP90 inhibition. One possible explanation is that PTEN-deficient
cancer cells might be addicted to AKT and thus hypersensitive to
AKT inhibition. NVP-AUY922, a specific inhibitor of HSP90, can
significantly degrade AKT. Indeed, a recent study by Darido et
al(34) indicated that PTEN is
an important downstream effector of Grhl3 (Grainy headlike 3, a
transcriptional factor essential for epidermal development) tumor
suppression activity, and Grhl3/PTEN-deficient squamous cell
carcinoma exhibited oncogene addiction to the PI3K/AKT signaling
pathway. It will be enlightening to determine whether
PTEN-downregulated or PTEN-loss ESCC cells display real oncogene
addiction to AKT by using direct inhibitors of AKT.
In conclusion, we demonstrated that the novel HSP90
inhibitor NVP-AUY922 exhibits a potent antitumor effect in ESCC.
PTEN expression may affect HSP90 inhibition via the regulation of
PI3K/AKT signaling, providing a novel therapeutic alternative for
cancer treatment.
Acknowledgements
We are grateful to Mr. Toru Tanida (Okayama
University) and Ms. Noriko Miyake (Kawasaki Medical School) for
their technical assistance and to Dr Minoru Haisa (Okayama
Citizens’ Hospital) and Dr Takako Yamada (Kawasaki Medical School)
for useful discussions.
References
|
1
|
Zhao R, Davey M, Hsu YC, et al: Navigating
the chaperone network: an integrative map of physical and genetic
interactions mediated by the hsp90 chaperone. Cell. 120:715–727.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Trepel J, Mollapour M, Giaccone G and
Neckers L: Targeting the dynamic HSP90 complex in cancer. Nat Rev
Cancer. 10:537–549. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Taipale M, Jarosz DF and Lindquist S:
HSP90 at the hub of protein homeostasis: emerging mechanistic
insights. Nat Rev Mol Cell Biol. 11:515–528. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Whitesell L and Lindquist SL: HSP90 and
the chaperoning of cancer. Nature Rev Cancer. 5:761–772. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kamal A, Thao L, Sensintaffar J, Zhang L,
Boehm MF, Fritz LC and Burrows FJ: A high-affinity conformation of
Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature.
425:407–410. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kim YS, Alarcon SV, Lee S, Lee MJ,
Giaccone G, Neckers L and Trepel JB: Update on Hsp90 inhibitors in
clinical trial. Curr Top Med Chem. 9:1479–1492. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ge J, Normant E, Porter JR, et al: Design,
synthesis, and biological evaluation of hydroquinone derivatives of
17-amino-17-demethoxygeldanamycin as potent, water-soluble
inhibitors of Hsp90. J Med Chem. 49:4606–4615. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Floris G, Debiec-Rychter M, Wozniak A,
Stefan C, Normant E, Faa G, Machiels K, Vanleeuw U, Sciot R and
Schöffski P: The heat shock Protein 90 inhibitor IPI-504 induces
KIT degradation, tumor shrinkage, and cell proliferation arrest in
xenograft models of gastrointestinal stromal tumors. Mol Cancer
Ther. 10:1897–1908. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gaspar N, Sharp SY, Eccles SA, Gowan S,
Popov S, Jones C, Pearson A, Vassal G and Workman P: Mechanistic
evaluation of the novel HSP90 inhibitor NVP-AUY922 in adult and
pediatric glioblastoma. Mol Cancer Ther. 9:1219–1233. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sausville EA, Tomaszewski JE and Ivy P:
Clinical development of 17-allylamino, 17-demethoxygeldanamycin.
Curr Cancer Drug Targets. 3:377–383. 2003. View Article : Google Scholar
|
|
11
|
Banerji U, O’Donnell A, Scurr M, et al:
Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino,
17-demethoxygeldanamycin in patients with advanced malignancies. J
Clin Oncol. 23:4152–4161. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Modi S, Stopeck AT, Gordon MS, et al:
Combination of trastuzumab and tanespimycin (17-AAG, KOS-953) is
safe and active in trastuzumab-refractory HER-2 overexpressing
breast cancer: a phase I dose-escalation study. J Clin Oncol.
25:5410–5417. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Modi S, Stopeck A, Linden H, et al: HSP90
inhibition is effective in breast cancer: a phase II trial of
tanespimycin (17-AAG) plus trastuzumab in patients with
HER2-positive metastatic breast cancer progressing on trastuzumab.
Clin Cancer Res. 17:5132–5139. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vaishampayan UN, Burger AM, Sausville EA,
Heilbrun LK, Li J, Horiba MN, Egorin MJ, Ivy P, Pacey S and Lorusso
PM: Safety, efficacy, pharmacokinetics, and pharmacodynamics of the
combination of sorafenib and tanespimycin. Clin Cancer Res.
16:3795–3804. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Enzinger PC and Mayer RJ: Esophageal
cancer. N Engl J Med. 349:2241–2252. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jemal A, Siegel R, Ward E, Hao Y, Xu J,
Murray T and Thun MJ: Cancer statistics, 2008. CA Cancer J Clin.
58:71–96. 2008. View Article : Google Scholar
|
|
17
|
Wu X, Wanders A, Wardega P, et al: Hsp90
is expressed and represents a therapeutic target in human
oesophageal cancer using the inhibitor
17-allylamino-17-demethoxygeldanamycin. Br J Cancer. 100:334–343.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Eccles SA, Massey A, Raynaud FI, et al:
NVP-AUY922: a novel heat shock protein 90 inhibitor active against
xenograft tumor growth, angiogenesis, and metastasis. Cancer Res.
68:2850–2860. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ueno T, Tsukuda K, Toyooka S, et al:
Strong anti-tumor effect of NVP-AUY922, a novel Hsp90 inhibitor, on
non-small cell lung cancer. Lung Cancer. 76:26–31. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee KH, Lee JH, Han SW, Im SA, Kim TY, Oh
DY and Bang YJ: Antitumor activity of NVP-AUY922, a novel heat
shock protein 90 inhibitor, in human gastric cancer cells is
mediated through proteasomal degradation of client proteins. Cancer
Sci. 102:1388–1395. 2011. View Article : Google Scholar
|
|
21
|
Manning BD and Cantley LC: AKT/PKB
signaling: navigating downstream. Cell. 129:1261–1274. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Johnson GL and Lapadat R:
Mitogen-activated protein kinase pathways mediated by ERK, JNK, and
p38 protein kinases. Science. 298:1911–1912. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Salmena L, Carracedo A and Pandolfi PP:
Tenets of PTEN Tumor Suppression. Cell. 133:403–414. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Stambolic V, Suzuki A, de la Pompa JL,
Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM,
Siderovski DP and Mak TW: Negative regulation of PKB/Akt-dependent
cell survival by the tumor suppressor PTEN. Cell. 95:29–39. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wu X, Senechal K, Neshat MS, Whang YE and
Sawyers CL: The PTEN/MMAC1 tumor suppressor phosphatase functions
as a negative regulator of the phosphoinositide 3-kinase/Akt
pathway. Proc Natl Acad Sci USA. 95:15587–15591. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tachibana M, Shibakita M, Ohno S, Kinugasa
S, Yoshimura H, Ueda S, Fujii T, Rahman MA, Dhar DK and Nagasue N:
Expression and prognostic significance of PTEN product protein in
patients with esophageal squamous cell carcinoma. Cancer.
94:1955–1960. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Song CH, Park SY, Eom KY, Kim JH, Kim SW,
Kim JS and Kim IA: Potential prognostic value of heat-shock protein
90 in the presence of phosphatidylinositol-3-kinase overexpression
or loss of PTEN, in invasive breast cancers. Breast Cancer Res.
12:R202010. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Basso AD, Solit DB, Chiosis G, Giri B,
Tsichlis P and Rosen N: Akt forms an intracellular complex with
heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by
inhibitors of Hsp90 function. J Biol Chem. 277:39858–39866. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jiao Y, Ou W, Meng F, Zhou H and Wang A:
Targeting HSP90 in ovarian cancers with multiple receptor tyrosine
kinase coactivation. Mol Cancer. 10:1252011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Scaltriti M, Serra V, Normant E, et al:
Antitumor activity of the Hsp90 inhibitor IPI-504 in HER2-positive
trastuzumab-resistant breast cancer. Mol Cancer Ther. 10:817–824.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yamamoto C, Basaki Y, Kawahara A, et al:
Loss of PTEN expression by blocking nuclear translocation of EGR1
in gefitinib-resistant lung cancer cells harboring epidermal growth
factor receptor-activating mutations. Cancer Res. 70:8715–8725.
2010. View Article : Google Scholar
|
|
32
|
Paraiso KH, Xiang Y, Rebecca VW, et al:
PTEN loss confers BRAF inhibitor resistance to melanoma cells
through the suppression of BIM expression. Cancer Res.
71:2750–2760. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhu H, Woolfenden S, Bronson RT, Jaffer
ZM, Barluenga S, Winssinger N, Rubenstein AE, Chen R and Charest A:
The novel Hsp90 inhibitor NXD30001 induces tumor regression in a
genetically engineered mouse model of glioblastoma multiforme. Mol
Cancer Ther. 9:2618–2626. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Darido C, Georgy SR, Wilanowski T, et al:
Targeting of the tumor suppressor GRHL3 by a miR-21-dependent
proto-oncogenic network results in PTEN loss and tumorigenesis.
Cancer Cell. 20:635–648. 2011. View Article : Google Scholar : PubMed/NCBI
|