Introduction
Growing tumors often develop areas of hypoxia due to
an insufficient supply of oxygen as a consequence of irregular and
functionally defective vasculature. Hypoxia then creates selective
pressure resulting in the expansion of adaptable cells with a more
aggressive phenotype and increased metastatic potential. The
primary molecular response to low oxygen is stabilization and
activation of an α subunit of hypoxia-inducible factor (HIF), a key
transactivator of genes involved in the adaptation to hypoxia,
including genes participating in growth arrest, apoptosis,
anaerobic glycolysis, angiogenesis, acidosis, cell adhesion and
cell proliferation (1). Carbonic
anhydrase IX (CA IX) is one of the most prominent hypoxia-inducible
HIF-target molecules (2–4). It belongs to the family of carbonic
anhydrases, which are zinc-binding enzymes that catalyze the
reversible conversion of carbon dioxide to bicarbonate and protons
in a reaction that involves facilitated hydration of CO2
to H2CO3 followed by spontaneous dissociation
of H2CO3 to bicarbonate and protons (2). Via this catalytic activity, carbonic
anhydrases significantly contribute to the modulation of ion
transport and maintenance of an acid-base balance in cells and
tissues of virtually all living organisms. The CA IX isoform is a
highly active transmembrane glycoprotein, which differs from other
carbonic anhydrases by a predominant association with various types
of tumors, including carcinomas of the cervix uteri, kidney, lung,
colon, breast, brain and ovary (5).
A high catalytic performance predisposes CA IX to
function in ion transport and pH control, and tumor cells
efficiently utilize this ability of CA IX to resist a hostile
microenvironment. Indeed, CA IX helps to maintain intracellular pH
of cancer cells within the physiological range, while
simultaneously contributing to the acidification of the
extracellular space (6,7). This has important implications for
cancer progression since a neutral intracellular pH is vital for
cell proliferation and survival, whereas microenvironmental
acidosis contributes to an aggressive tumor phenotype by promoting
invasion and metastasis (8). In
addition, CA IX decreases E-cadherin-mediated cell-cell adhesion
through the competitive binding to β-catenin (9), and increases cell migration via
physical interaction and functional cooperation with bicarbonate
transporters in lamellipodia of moving cells (10). This further supports the
pro-metastatic propensity of tumor cells.
To better understand the role of CA IX in tumor
cells and disclose the underlying molecular mechanisms, we
developed a CA9 mRNA-targeting shRNA inducible system and
studied changes in the gene expression profile connected with CA IX
protein deficiency in the HT-1080 tumor cell line. Using this
approach, we identified a number of potential CA IX downstream
targets belonging to several cancer-related pathways. Among them,
the focal adhesion pathway was one of the most prominent, and thus,
we focused on CA IX involvement in focal adhesion-associated
phenomena.
Materials and methods
Cell culture
HT-1080, HEK 293T and HeLa cells were cultivated in
DMEM with 10% FCS (Lonza BioWhittaker) at 37°C in humidified air
with 5% CO2. Hypoxic experiments were performed in an
anaerobic workstation (Ruskinn Technologies) in a 2% O2,
2% H2, 5% CO2, 91% N2 atmosphere
at 37°C. The conditional shRNA system in HT-1080 cells was
activated with 0.5 μg/ml doxycycline (Clontech, Mountain View, CA,
USA) in cultivation medium.
Antibodies and plasmids
Anti-CA IX mouse monoclonal antibody M75 was
characterized elsewhere (3).
Additional antibodies were as follows: goat polyclonal anti-actin
antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA), mouse
monoclonal anti-HIF-1α antibody (BD Transduction Laboratories),
anti-mouse peroxidase-conjugated antibody (Sigma-Aldrich, St.
Louis, MO, USA) and anti-goat peroxidase-conjugated antibody (Dako,
Carpinteria, CA, USA).
CA9-specific shRNA oligonucleotides were
first cloned into the pSUPER plasmid, which was then digested with
ClaI and EcoRI. The resulting 300-bp fragment was
excised, gel-purified and ligated into the
EcoRI/ClaI-digested pLVTHM lentiviral vector to
replace the pLVTHM H1 promoter with the H1 promoter and shRNA from
pSUPER-shCA9. Recombinant pLVTHM-shCA9 was digested
with FspI and MscI, and the 2.5-kb fragment was
excised, purified and ligated into the inducible pLVET-tTR-KRAB
lentiviral vector digested with FspI and MscI.
Plasmids pSPAX2 and pMD2G encoded packaging and envelope genes,
respectively. A control plasmid bearing nonsense scramble shRNA
(Scr) was constructed using the same method. All plasmids were
obtained from the plasmid repository Addgene.com.
Production and titration of CA9-specific
lentiviral particles
Lentiviral particles were produced in HEK 293T cells
by co-transfection of the packaging plasmid (2.9 μg), the virus
envelope encoding plasmid (1.2 μg), and the vector carrying both
CA9-shRNA (pLVET-shCA9, 4 μg) and the GFP
transduction marker, using the GenePorterII transfection kit
(Genlantis). Supernatant from the transfected cells was collected,
filtered through a 0.22-μm filter and either used directly for
transduction or stored in aliquots at −80°C.
The virus was titrated by transducing HeLa cells
with aliquots of viral stock in cultivation medium mixed with
Polybrene (5 μg/ml). The transduced cells were incubated for 3 days
in the presence of doxycycline and analyzed on the EasyCyte 6HT
flow cytometer (Millipore) for expression of the GFP transduction
marker. The titer was calculated from the number of plated cells
multiplied by the proportion of transduced cells and divided by the
volume of added viral stock.
Transduction of HT-1080 cells
The HT-1080 cells were mixed with the lentivirus
particles at MOI of 10 and plated into 96-well plates. On the next
day, the medium with doxycycline was replaced. Two days later, the
cells were examined for GFP expression and then seeded at a density
of 500 cells per 100-mm dish to form colonies in the presence of
doxycycline. The colonies expressing GFP were picked up, expanded
and used for further study.
Reverse transcription PCR
Total RNA was extracted using a GeneJET RNA
Purification kit (Fermentas) and transcribed to cDNA using a High
Capacity cDNA Reverse Transcription kit (Applied Biosystems).
Quantitative PCR was performed with Power SYBR® Green
PCR Master Mix on a StepOne Real-Time PCR system (Applied
Biosystems). Real-time PCR was performed using the following
primers: MMP9 S, atcttccaa ggccaatcctact and A,
ctatccagctcaccggtctc; MMP3 S, gtttgttaggagaaaggacagtgg and A,
tatgagcagcaacgagaaataa; Col4A1 S, ggacaaaagggtgatactgga and A,
gccattgcatcctggaatac; Col4A2 S, agggtgaaaagggtgacgta and A,
tgcctcttattcctggttcc; Itg β3 S, acagtctgtgatgaaaagattgg and A,
cagccccaaagagggataat β-actin S, ccaaccgcgagaagatgacc and A,
gatcttcatgaggtagtcagt.
Immunoblotting
The cells were grown for 3 days in medium containing
doxycycline to induce CA9 silencing. On the fourth day, the
cells were moved into hypoxia for 24/48 h to induce expression of
the CA IX protein. After incubation in hypoxia samples were lysed.
Samples consisting of 100 μg total proteins were separated in 10%
SDS-PAGE and immunoblotting was performed as described elsewhere
(11).
Immunofluorescence
Cells grown on glass coverslips with or without
collagen coating were fixed in 4% paraformaldehyde and
permeabilized with 0.1% Triton X-100. Immunofluorescence staining
and confocal microscopy were carried out as previously described
(10).
Microarray analysis
Affymetrix GeneChip Human Gene 1.0 ST (Affymetrix)
was used for the microarray analysis following the standard
protocol (starting with 100 ng of total RNA). The analysis was
performed in three replicates. The raw data were analyzed with
Partek Genomics Suite (Partek Inc.). All hybridizations passed
quality control. The data were background-corrected by the GC
Robust Multi-Array Average (GC-RMA) algorithm, variance-stabilized
by logarithmic transformation and normalized with the probeset
level quantile method. One-way analysis of variance (ANOVA) was
performed to identify genes varying significantly across two
compared groups. Signaling Pathway Impact Analysis (SPIA) was
performed using SPIA package of the Bioconductor within the R
environment (http://www.r-project.org).
Spreading assay, wound healing assay and
Matrigel invasion
Following treatment with doxycycline and exposure to
hypoxia, HT-1080 cells were seeded on collagen-coated coverslips
and left to attach for 15 and 30 min, respectively. The cells were
then fixed with 2% paraformaldehyde in PBS for 10 min at room
temperature (RT), washed with PBS, stained with 0.5% Coomassie blue
for 5 min at RT and washed 3 times with PBS. Images of the fixed
cells were analyzed using ImageJ software (http://rsb.info.nih.gov/ij/). At least 500 cells were
analyzed for each sample, and cell-spreading areas of
CA9-silenced HT-1080 cells and relevant control samples were
calculated and compared using the t-test.
For the wound healing assay, HT-1080 cells were
pre-cultivated in hypoxia for 24 h with or without doxycycline. On
the following day, the confluent monolayer was wounded with a
sterile micropipette tip. The wound healing assay was performed in
DMEM with 1% FCS containing 20 ng/ml HGF in normoxic conditions,
without the presence of doxycycline. The cells were photographed at
20 positions for each sample immediately after wound initiation and
after 8 h using an inverted Zeiss microscope (Axiovert 40 CFL), 10×
objective. Wound healing was quantified using ImageJ software (mean
± standard deviation) and results were compared by the t-test
(**P<0.01).
Matrigel invasion was performed using BD Falcon HTS
FluoroBlok 8.0-μm colored PET membrane inserts for 24-well plates
(BD Biosciences) using HT-1080 cells labeled with a lipophilic
fluorescent dye DiO (Invitrogen) as previously described (10).
Results
Silencing of CA9
The CA IX protein plays an important role in hypoxic
tumor cells as it helps them survive hostile conditions through
efficient buffering of intracellular pH coupled with excretion of
protons to the extracellular space. To elucidate CA IX-regulated
molecular pathways, we investigated the effects of CA IX deficiency
in tumor cells using an HT-1080 human fibrosarcoma model. These
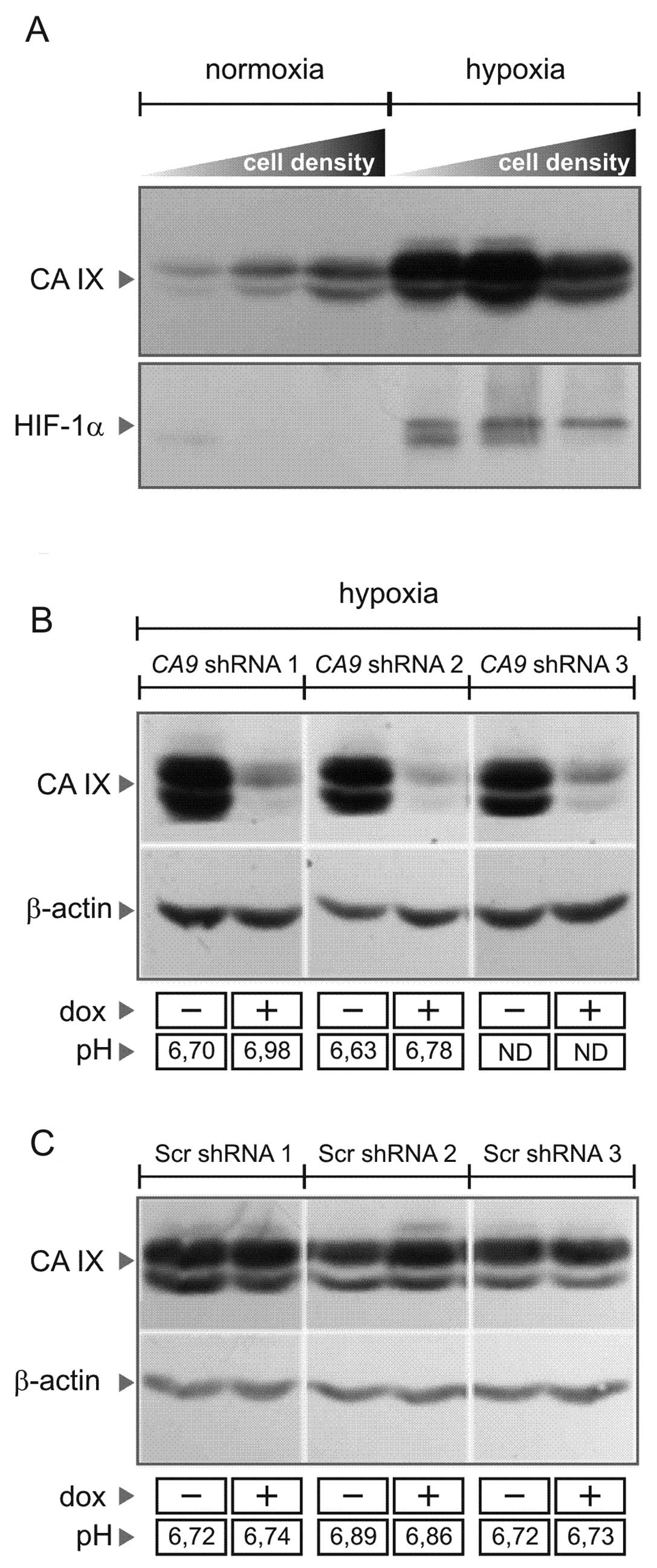
cells display hypoxia-inducible and density-dependent expression of
CA IX (Fig. 1A), similarly as it
was shown for many other tumor cell lines (12,13).
Conditional shRNA-mediated silencing of CA9 was achieved
using a tightly regulated pLVET-tTR-KRAB lentiviral vector, in
which shRNA expression from the wild-type H1 promoter is governed
by tTR-KRAB protein that epigenetically silences the integrated
lentivirus in the absence of doxycycline (14). Several clonal cell lines targeting
CA9 were selected according to immunoblotting analysis and
were further studied in comparison with the scrambled (Scr) shRNA
clones (Fig. 1C). Compared to the
control Scr cells, the CA9-silenced cells exposed to hypoxia
showed a decreased extracellular acidification capacity (Fig. 1B), suggesting that they lost the
pH-regulating function of CA IX and that our shRNA system was
functional.
Gene expression profiling of the control
vs. the CA9-silenced cells
To search for molecular signatures and pathways
affected by CA IX protein deficiency, we performed pairwise gene
expression profiling of the hypoxic cells bearing nonsense Scr
shRNA vs. CA9 shRNA on the HT-1080 cell line background.
Microarray experiments were carried out using three independent
clones established from each of the CA9 shRNA and Scr shRNA
cells. Differential gene expression analysis was performed by
filtering the microarray data for genes that showed at least a
2-fold change in gene expression levels in silenced vs. control
cells (P<0.05).
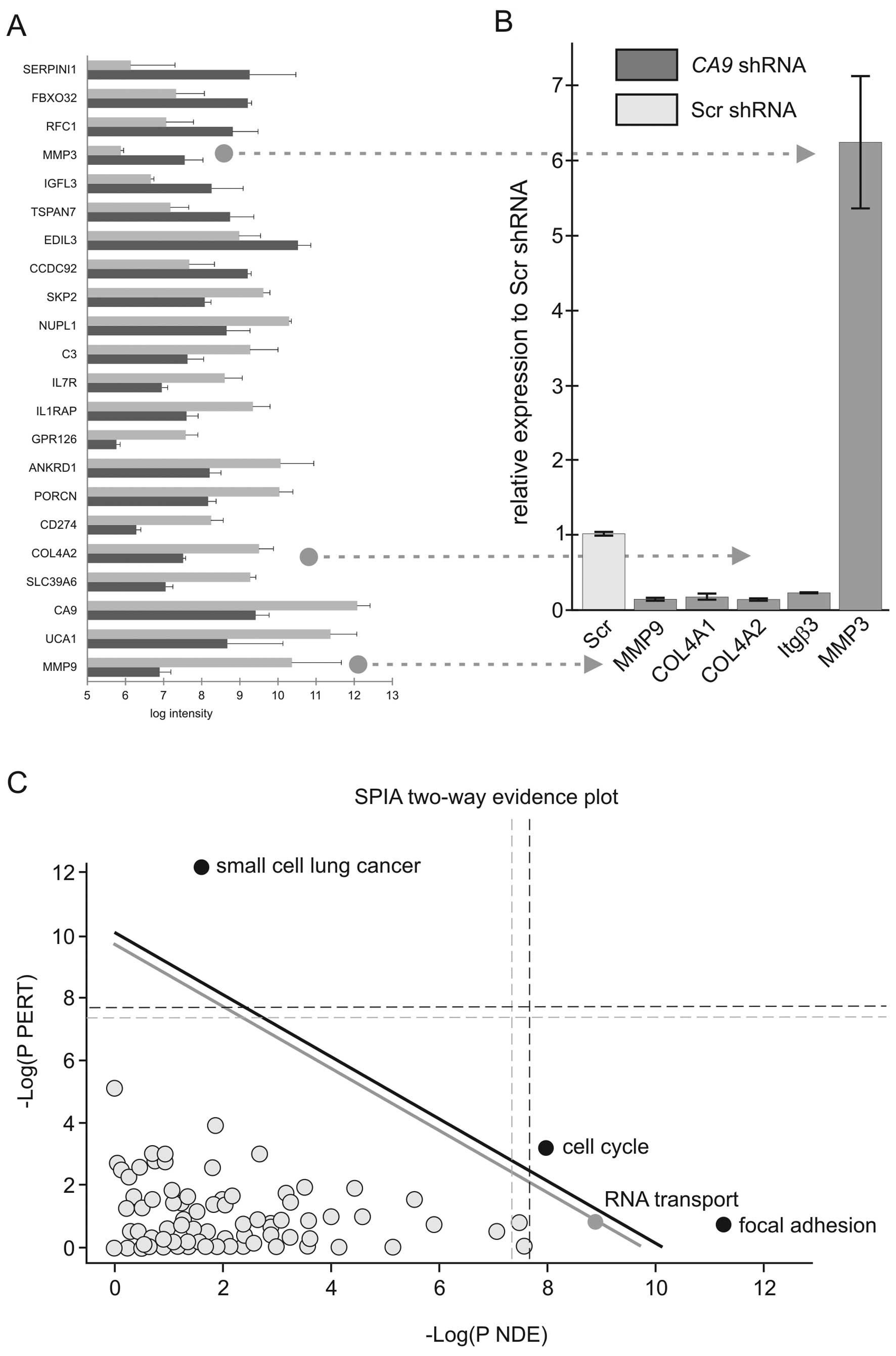
By applying such selection criteria, the expression
levels of 109 genes were found to be significantly altered. Among
them, 62 genes were downregulated and 47 genes were upregulated in
the CA9-silenced cells compared to the Scr controls. The
list of the 22 most highly differentially expressed genes is shown
in Fig. 2A. Presence of CA9
among them confirms efficiency of silencing. Quantitative RT-PCR
performed on two members of the matrix metalloproteinase family
(MMP3 and MMP9) and collagen type IVα2, which are frequently
associated with tumor phenotype, confirmed the microarray results
(Fig. 2B). To identify molecular
pathways deregulated upon CA9 knockdown, we analyzed the
microarray data using the Signaling Pathway Impact Analysis (SPIA)
linked to KEGG (15). The analysis
revealed inhibition of the pathways designated as small-cell lung
cancer, focal adhesion and regulation of actin cytoskeleton in the
CA9-silenced cells. In contrast, pathways involved in cell
cycle, RNA transport and protein processing in endoplasmic
reticulum were found to be activated. SPIA two-way evidence plot
based on combining pPERT and pNDE indicated that the small-cell
lung cancer, cell cycle and focal adhesion pathways were the most
significantly affected (Fig.
2C).
Focal adhesion pathway is inhibited in
the CA9-depleted cells
The ability of cells to form focal adhesions (FAs)
as components of cell motility and migration represents an
important early event in the development of metastasis. CA IX was
previously shown to facilitate cell migration through its
participation in the pH-regulating machinery driving lamellipodial
extensions of moving cells (10).
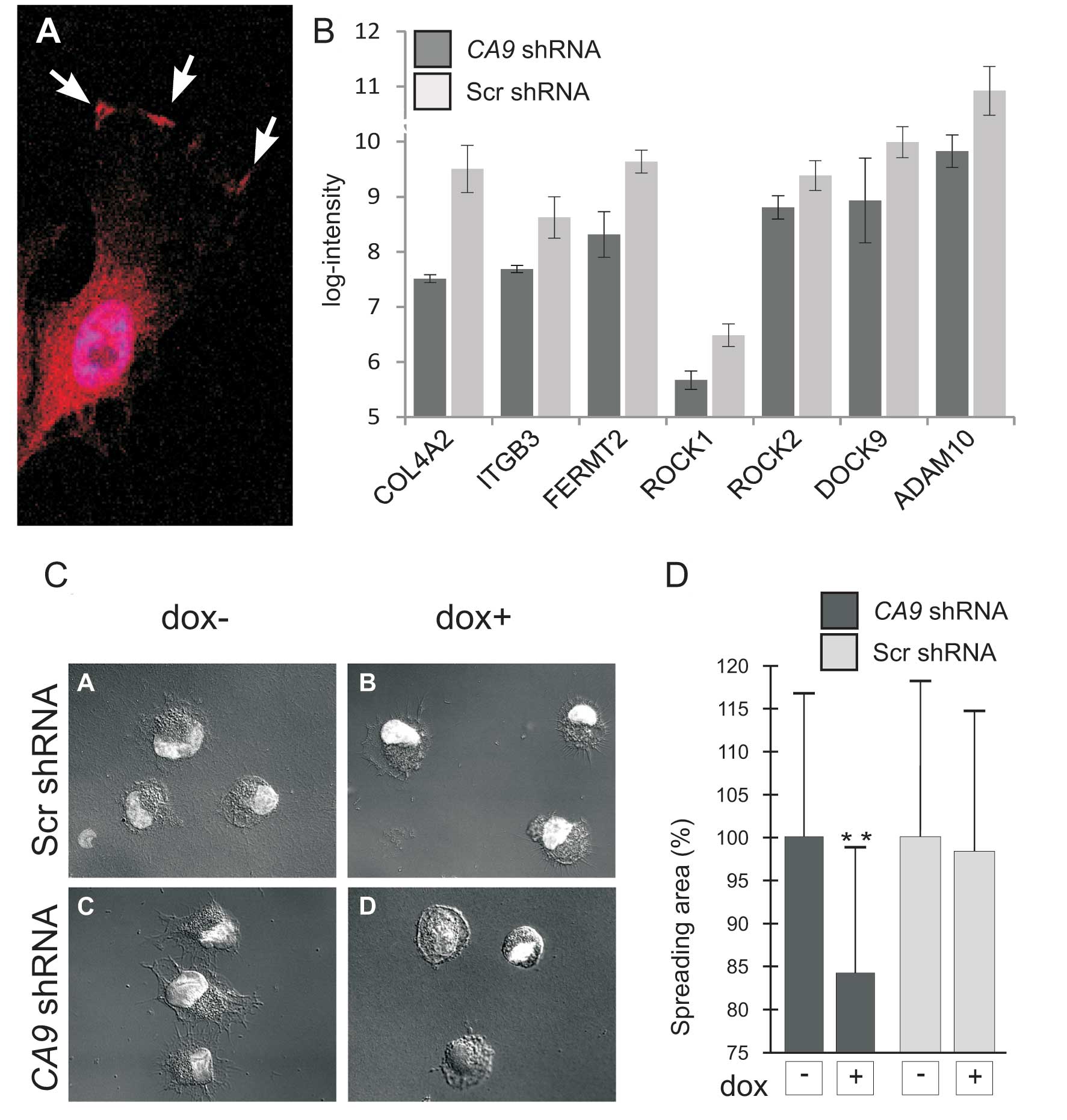
HT-1080 fibrosarcoma cells exhibited similar distribution of CA IX
protein as in epithelial/carcinoma cells indicating that it might
play an analogous role in the migration of tumor cells derived from
fibrosarcoma (Fig. 3A).
Importantly, microarray analysis revealed that the loss of CA IX
resulted in downregulation of several molecules implicated in the
interaction of cells with the extracellular matrix, such as
integrin β3, collagen type IV α2 and α1, dedicator of cytokinesis 9
(DOCK9), fermitin family member 2 (FERMT2), Rho-associated, and
coiled-coil containing protein kinase 1 (ROCK1) (Fig. 3B). This finding suggests that CA IX
affects additional aspects of cell migration, including FA
assembly.
Therefore, we decided to further explore this
assumption. We showed that CA IX-deficient cells were less capable
of spreading on the support independently of collagen coating
compared to control cells (Fig. 3C and
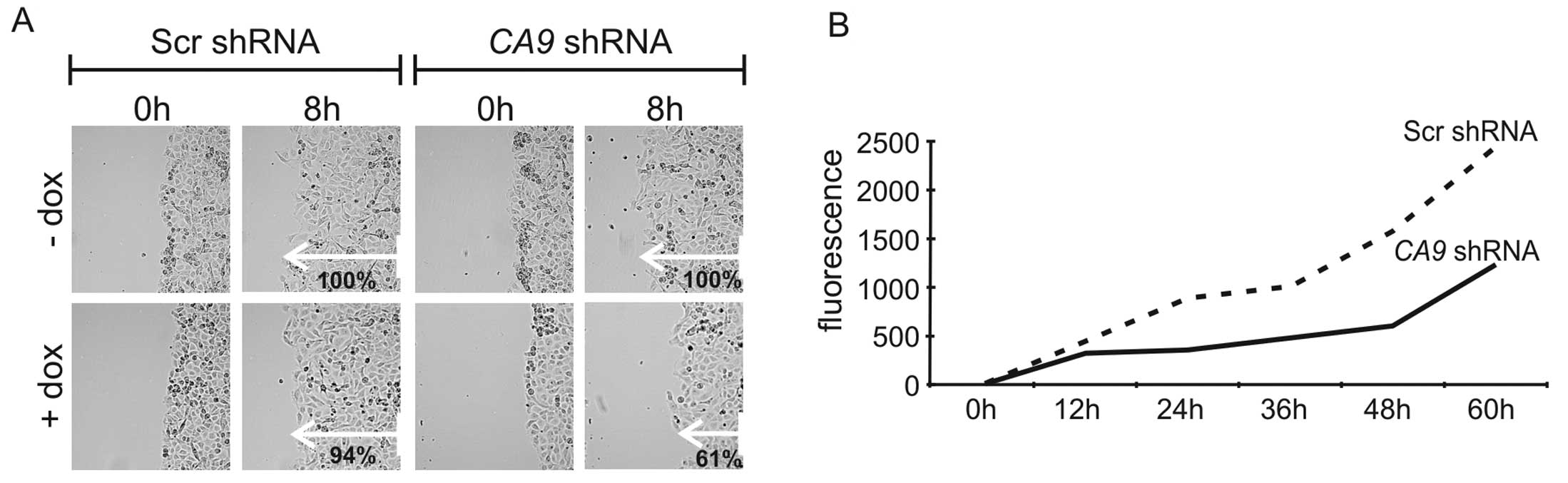
D). In the wound healing assay, decreased expression of CA IX
led to a significant reduction (P<0.01) in the migration rate to
61% in comparison to control cells (Fig. 4A). There was no significant
difference in migration of scrambled cells with or without
doxycycline, and between Scr and CA9 shRNA cells incubated
without doxycycline. To assess the migratory and invasive abilities
of the HT-1080 cells upon knockdown of CA9, we performed a
Matrigel invasion assay that mimics invasion of cells through a
basement membrane. We observed reduced invasion/migration of CA
IX-depleted cells (Fig. 4B). This
could be related to the diminished level of MMP9, which is
essential for invadopodia formation and cleavage of the basement
membrane.
Altogether, these experiments confirmed the
microarray predictions and further underlined the importance of CA
IX in adhesion, migration and invasion of tumor cells under
conditions of limited oxygen tension typical of the tumor
microenvironment.
Discussion
Due to its tight transcriptional control by HIF-1α,
CA IX is frequently used as a favored hypoxia marker in many types
of tumors, where it can serve as a prognostic indicator and
predictive factor (5,16). Moreover, accumulating evidence
suggests that CA IX plays an active role in tumor physiology
principally through its catalytic activity-mediated control of pH
and cell adhesion/migration (6,7). This
evidence is mostly based on functional studies of various cellular
models with CA IX overexpression, mutagenesis or suppression
(6,9,10,17–19).
To date, only one study has reported the gene profiling of
carcinoma cells with forced constitutive overexpression of CA IX.
This study showed the involvement of CA IX in aberrant Rho-GTPase
signal transduction leading to decreased cell adhesion and
augmented cell motility (20).
In the present study, we employed a different
approach, using inducible CA9 mRNA silencing in HT-1080
fibrosarcoma cells. Efficiency of silencing was demonstrated by the
abrogation of the capacity of CA IX to acidify culture media, in
line with the data obtained in a colon adenocarcinoma cell model
(17).
Gene expression profiling of hypoxic HT-1080 cells
revealed that 109 genes were significantly deregulated as a result
of the loss of CA IX. These genes were members of several
cancer-related pathways, with the prominent pathway designated as
‘focal adhesion’ that involves components of cell
adhesion-migration-invasion machinery which was inhibited in CA
IX-depleted cells.
Focal adhesion mediates cell-extracellular matrix
interactions, which are essential for migration and invasion of
tumor cells of both epithelial and fibroblast origin. It is a
complex process that comprises more than 50 proteins from the inner
and outer side of the cell membrane (21). Two members of the type IV collagen
gene family namely COL4A1 and COL4A2 were among the most
downregulated genes within the identified FA pathway. Both are
structural constituents of the extracellular matrix (ECM) important
for proper ECM assembly. Integrin β3, a receptor for fibronectin,
fibrinogen, plasminogen, prothrombin, thrombospondin and
vitronectin, was also downregulated in the CA9-silenced
HT-1080 cells. One of the FA family members downregulated in our
study was FERMT2, which functions in cell shape modulation
(22). Regulation of FA is also
mediated by intracellular signaling from the Rho family of small
GTPases to ROCK-I/ROCK-II kinases (23). Both molecules are responsible for
actomyosin contractility, which is important for cellular movement.
It is notable that both kinases were downregulated in the
CA9-silenced cells. Another signaling molecule negatively
affected by CA IX deficiency was DOCK9 also known as Zizimin1
(24). It is a member of the DOCK
family of guanine nucleotide exchange factors (25). DOCK9 is an activator of Cdc42 which
regulates the actin cytoskeleton, cell migration and several other
cellular activities.
Downregulation of the above-mentioned genes was
clearly mirrored by significantly reduced adhesion and spreading of
CA IX-deficient HT-1080 cells. This resembles formation of aberrant
FA in cells expressing the inactive NHE1 transporter (normally
responsible for export of protons) as well as in cells lacking an
MCT4 lactate transporter (26,27).
All of these three proteins are known to support cell migration by
ensuring a proper pH gradient across the membranes of lamellipodia
(28). It seems plausible that the
pH-regulating function is important also for assembly and/or
disassembly of FAs, which is a rate-limiting process for efficient
cell migration accompanying lamellipodial extension and
retraction.
CA IX-deficient cells also exhibited reduced
capacity for invasion and migration through Matrigel. This could be
due to the fact that several FA-related genes, downregulated in the
absence of CA IX, also participate in cell invasion, such as
integrin subunits β3 and α6 (29),
and matrix metalloproteinase MMP9 (30). In contrast, MMP2 and MMP3 were
upregulated in the CA9-silenced cells. It is well known that
protonation of MMP3 itself and protonation of fibrinogen as a
substrate for MMP2 influence their activity and proteolysis
(31–33). Since CA IX contributes to acidic
pericellular pH by generation of extracellular protons, its loss
may lead to reduced MMP2 and MMP3 activity despite their increased
levels. Furthermore, MMP2-mediated collagenolysis depends on MMP2
interaction with β3 integrin (34,35),
and thus lower β3 integrin subunit expression can contribute to
MMP2 inactivation and reduced invasion of CA IX-deficient cells. An
additional molecule downregulated in the CA9-silenced
HT-1080 cells potentially affecting their adhesion and invasion was
ADAM10, which acts through shedding of many cell surface molecules
including the key cell-cell adhesion molecule E-cadherin (36).
Altogether, our study provided the first molecular
insight into downstream signal transduction pathways driven by CA
IX in tumor cells using an inducible shRNA-based silencing
approach. In accordance with independent functional studies, CA
IX-emitted signaling was found to be important in several key
aspects of cell adhesion, migration and invasion mediated by
molecules that are central players in these phenomena. In addition
to the known CA IX effects on destabilization of cell-cell contacts
and stimulation of epithelial cell motility/migration, we
demonstrated here for the first time its direct role in focal
adhesion and invasion of tumor fibroblasts. This suggests that CA
IX operates at different stages of the metastatic cascade and in
various cell types. Notably, both direct and indirect evidence
supports the view that CA IX acts at each stage via its catalytic
activity-mediated pH regulatory capacity, suggesting that pH
regulation is a critical facet of the cancer phenotype. Thus, our
observation that CA IX has an impact on the pro-metastatic behavior
of tumor cells supports recent efforts to explore its suppression
or pharmacologic inhibition for anticancer therapy.
Acknowledgements
This study was supported by grants no. 2/0194/09 and
2/0130/11 from the Scientific Grant Agency of the Ministry of
Education of the Slovak Republic and the Slovak Academy of
Sciences, from the Research and Development Support Agency
(contracts APVV-0108-10 and DO-7RP-0017-09) and from EU (7FP
Collaborative project METOXIA) and from the Research and
Development Operational Program funded by ERDF (project PV-INF-PAT,
ITMS 26240220032).
Abbreviations:
|
CA IX
|
carbonic anhydrase IX protein
|
|
CA9
|
carbonic anhydrase 9 gene or mRNA
|
|
dox
|
doxycycline
|
|
FA
|
focal adhesion
|
|
GFP
|
green fluorescent protein
|
|
HIF
|
hypoxia-inducible factor
|
|
MMP
|
matrix metalloproteinase
|
|
MOI
|
multiplicity of infection
|
|
Scr
|
scrambled
|
|
shRNA
|
short hairpin RNA
|
References
|
1
|
Harris AL: Hypoxia - a key regulatory
factor in tumour growth. Nat Rev Cancer. 2:38–47. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pastorekova S, Parkkila S, Pastorek J and
Supuran CT: Carbonic anhydrases: current state of the art,
therapeutic applications and future prospects. J Enzyme Inhib Med
Chem. 19:199–229. 2004.PubMed/NCBI
|
|
3
|
Pastorek J, Pastorekova S, Callebaut I, et
al: Cloning and characterization of MN, a human tumor-associated
protein with a domain homologous to carbonic anhydrase and a
putative helix-loop-helix DNA binding segment. Oncogene.
9:2877–2888. 1994.
|
|
4
|
Wykoff CC, Beasley NJ, Watson PH, et al:
Hypoxia-inducible expression of tumor-associated carbonic
anhydrases. Cancer Res. 60:7075–7083. 2000.PubMed/NCBI
|
|
5
|
Potter C and Harris AL: Hypoxia inducible
carbonic anhydrase IX, marker of tumour hypoxia, survival pathway
and therapy target. Cell Cycle. 3:164–167. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Svastova E, Hulikova A, Rafajova M, et al:
Hypoxia activates the capacity of tumor-associated carbonic
anhydrase IX to acidify extracellular pH. FEBS Lett. 577:439–445.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Swietach P, Hulikova A, Vaughan-Jones RD
and Harris AL: New insights into the physiological role of carbonic
anhydrase IX in tumour pH regulation. Oncogene. 29:6509–6521. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Raghunand N, Gatenby RA and Gillies RJ:
Microenvironmental and cellular consequences of altered blood flow
in tumours. Br J Radiol. 76(Spec No 1): S11–S22. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Svastova E, Zilka N, Zat’ovicova M, et al:
Carbonic anhydrase IX reduces E-cadherin-mediated adhesion of MDCK
cells via interaction with beta-catenin. Exp Cell Res. 290:332–345.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Svastova E, Witarski W, Csaderova L, et
al: Carbonic anhydrase IX interacts with bicarbonate transporters
in lamellipodia and increases cell migration via its catalytic
domain. J Biol Chem. 287:3392–3402. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Takacova M, Holotnakova T, Barathova M,
Pastorekova S, Kopacek J and Pastorek J: Src induces expression of
carbonic anhydrase IX via hypoxia-inducible factor 1. Oncol Rep.
23:869–874. 2010.PubMed/NCBI
|
|
12
|
Ihnatko R, Kubes M, Takacova M, et al:
Extracellular acidosis elevates carbonic anhydrase IX in human
glioblastoma cells via transcriptional modulation that does not
depend on hypoxia. Int J Oncol. 29:1025–1033. 2006.
|
|
13
|
Kaluz S, Kaluzova M and Stanbridge EJ:
Expression of the hypoxia marker carbonic anhydrase IX is
critically dependent on SP1 activity. Identification of a novel
type of hypoxia-responsive enhancer. Cancer Res. 63:917–922.
2003.PubMed/NCBI
|
|
14
|
Szulc J, Wiznerowicz M, Sauvain MO, Trono
D and Aebischer P: A versatile tool for conditional gene expression
and knockdown. Nat Methods. 3:109–116. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tarca AL, Draghici S, Khatri P, et al: A
novel signaling pathway impact analysis. Bioinformatics. 25:75–82.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pastorekova S, Ratcliffe PJ and Pastorek
J: Molecular mechanisms of carbonic anhydrase IX-mediated pH
regulation under hypoxia. BJU Int. 101(Suppl 4): 8–15. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chiche J, Ilc K, Laferriere J, et al:
Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell
growth by counteracting acidosis through the regulation of the
intracellular pH. Cancer Res. 69:358–368. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hulikova A, Zatovicova M, Svastova E, et
al: Intact intracellular tail is critical for proper functioning of
the tumor-associated, hypoxia-regulated carbonic anhydrase IX. FEBS
Lett. 583:3563–3568. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ditte P, Dequiedt F, Svastova E, et al:
Phosphorylation of carbonic anhydrase IX controls its ability to
mediate extracellular acidification in hypoxic tumors. Cancer Res.
71:7558–7567. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shin HJ, Rho SB, Jung DC, Han IO, Oh ES
and Kim JY: Carbonic anhydrase IX (CA9) modulates tumor-associated
cell migration and invasion. J Cell Sci. 124:1077–1087. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zamir E and Geiger B: Molecular complexity
and dynamics of cell-matrix adhesions. J Cell Sci. 114:3583–3590.
2001.PubMed/NCBI
|
|
22
|
Tu Y, Wu S, Shi X, Chen K and Wu C:
Migfilin and Mig-2 link focal adhesions to filamin and the actin
cytoskeleton and function in cell shape modulation. Cell.
113:37–47. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sanz-Moreno V, Gadea G, Ahn J, et al: Rac
activation and inactivation control plasticity of tumor cell
movement. Cell. 135:510–523. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Meller N, Irani-Tehrani M, Kiosses WB, Del
Pozo MA and Schwartz MA: Zizimin1, a novel Cdc42 activator, reveals
a new GEF domain for Rho proteins. Nat Cell Biol. 4:639–647. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cote JF and Vuori K: Identification of an
evolutionarily conserved superfamily of DOCK180-related proteins
with guanine nucleotide exchange activity. J Cell Sci.
115:4901–4913. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gallagher SM, Castorino JJ and Philp NJ:
Interaction of monocarboxylate transporter 4 with beta1-integrin
and its role in cell migration. Am J Physiol Cell Physiol.
296:C414–C421. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Denker SP and Barber DL: Cell migration
requires both ion translocation and cytoskeletal anchoring by the
Na-H exchanger NHE1. J Cell Biol. 159:1087–1096. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Stock C and Schwab A: Protons make tumor
cells move like clockwork. Pflugers Arch. 458:981–992. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jin H and Varner J: Integrins: roles in
cancer development and as treatment targets. Br J Cancer.
90:561–565. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nagase H, Visse R and Murphy G: Structure
and function of matrix metalloproteinases and TIMPs. Cardiovasc
Res. 69:562–573. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rupp PA, Visconti RP, Czirok A, Cheresh DA
and Little CD: Matrix metalloproteinase 2-integrin alpha(v)beta3
binding is required for mesenchymal cell invasive activity but not
epithelial locomotion: a computational time-lapse study. Mol Biol
Cell. 19:5529–5540. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Holman CM, Kan CC, Gehring MR and Van Wart
HE: Role of His-224 in the anomalous pH dependence of human
stromelysin-1. Biochemistry. 38:677–681. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Monaco S, Gioia M, Rodriguez J, et al:
Modulation of the proteolytic activity of matrix
metalloproteinase-2 (gelatinase A) on fibrinogen. Biochem J.
402:503–513. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Brooks PC, Stromblad S, Sanders LC, et al:
Localization of matrix metalloproteinase MMP-2 to the surface of
invasive cells by interaction with integrin alpha v beta 3. Cell.
85:683–693. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hornebeck W, Emonard H, Monboisse JC and
Bellon G: Matrix-directed regulation of pericellular proteolysis
and tumor progression. Semin Cancer Biol. 12:231–241. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Maretzky T, Reiss K, Ludwig A, et al:
ADAM10 mediates E-cadherin shedding and regulates epithelial
cell-cell adhesion, migration, and beta-catenin translocation. Proc
Natl Acad Sci USA. 102:9182–9187. 2005. View Article : Google Scholar : PubMed/NCBI
|