Introduction
According to the results of epidemiologic studies,
green tea consumption has a preventive effect on carcinogenesis
(1–6). It is thought that polyphenols, also
known as catechins, play an important role in the chemopreventive
effects mediated by green tea. (−)-Epigallocatechin gallate (EGCG),
a type of polyphenol, which is the most well known, abundant and
active compound found in green tea, exerts its anticancer effects
in a wide range of malignancies (2,7).
Previous studies have suggested that multiple signaling pathways
and mechanisms are involved in the antitumor activity of EGCG
(1,8–10),
including suppression of various protein kinases (11–13);
disruption of the activation of transcription factors such as EGFR,
NF-κB, AP-1 and STATs (14–16); induction of cell cycle arrest or
apoptosis (17,18); and inhibition of cell migration and
metastasis (19–24).
p53, commonly referred to as the ‘cellular
gatekeeper’ or ‘the guardian of the genome’, is a crucial tumor
suppressor gene that is mutated in more than half of all types of
human cancer. As a transcription factor, p53 functions to regulate
cell fate following various types and levels of cellular stress
through its downstream target genes. In addition to its canonical
functions of inducing DNA repair, cell cycle arrest and apoptosis
(25,26), recent studies have also revealed
that p53 is involved in the regulation of various other cellular
functions, such as senescence, metabolism and autophagy. Due to the
importance of p53, its activation is regulated by complicated
post-translational modifications, such as phosphorylation,
acetylation, ubiquitination and sumoylation (8,27–29).
Previous studies have shown that the phosphorylation and
acetylation of p53 promotes the expression of p53 target genes
(28,30,31),
whereas other modifications, such as ubiquitination and
sumoylation, are considered to be associated with the suppression
of p53-mediated transcription and nuclear export of p53 (32–34).
MDM2, a Ring finger domain-containing protein, is the first
identified E3 ligase that can induce p53 ubiquitination and
proteasomal degradation (33).
However, as a transcriptional target of p53, MDM2 expression is
also regulated in a p53-dependent manner. As p53 increases, the
expression of MDM2 is strengthened, which induces degradation of
p53 and achieves balance.
Previous studies have demonstrated that EGCG
treatment increases the expression levels of p53 in various human
cancer cells (35–38). Previous reports found that cells
expressing wild-type p53 are more sensitive than p53-null or p53
knock down cells to EGCG-induced growth inhibition and apoptosis
(36,39,40).
However, the molecular mechanisms underlying p53 regulation by EGCG
and the anticancer effects of green tea via targeting of the p53
tumor suppressor gene are poorly understood. In the present study,
we demonstrated that EGCG treatment can induce p53 accumulation and
enhance the stability of this protein by disturbing the interaction
between p53 and MDM2. Furthermore, our data showed that EGCG
inhibits p53 ubiquitination in a dose-dependent manner. These
results indicate a novel mechanism for the preventive effects of
EGCG on cancer.
Materials and methods
Cell culture and transfection
All cell lines were obtained from the American Type
Culture Collection and were grown in a 37°C incubator with 5%
CO2 according to the American Type Culture Collection
protocols. For transfection experiments, the Lipofectamine™ 2000
transfection reagent (Invitrogen, Carlsbad, CA, USA) was used
according to the manufacturer’s instructions.
Reagents and antibodies
EGCG, cycloheximide (CHX) and MG132 were obtained
from Sigma (St. Louis, MO, USA). The GFP-p53, His-Ub and HA-MDM2
plasmids were gifts from Dr Bo Liu and were previously described
(41). Anti-p53, anti-MDM2,
anti-β-actin, anti-GFP, anti-HA, anti-His, anti-rabbit IgG-HRP,
anti-mouse IgG-HRP, anti-goat IgG-HRP, and normal mouse/rabbit IgG
were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Anti-phospho-p53 (Ser15), anti-phospho-p53 (Ser20), anti-p21,
anti-lamin B, anti-tubulin and anti-ubiquitin antibodies were
purchased from Cell Signaling Technology, Inc. (Danvers, MA,
USA).
Immunoprecipitation
293T and H1299 cells were first co-transfected with
2 μg of GFP-p53 and 2 μg of HA-MDM2. Twenty-four hours after the
transfection, these cells were treated with 40 μM EGCG for 24 h.
Cells were harvested and washed twice with ice-cold PBS and lysed
in NP40 lysis buffer (50 mmol/l Tris-HCl, pH 8.0; 150 mmol/l NaCl;
0.5% NP40) with protease cocktail (Roche Diagnostics GmbH,
Mannheim, Germany). All immunoprecipitation procedures were carried
out at 4°C. The lysates were then incubated with the appropriate
antibodies, followed by incubation with protein A/G agarose beads.
The protein-antibody complexes were recovered and subjected to
western blot analysis after separation by SDS-PAGE.
Western blotting
Cells were harvested by trypsinization and pelleted
by centrifugation. Cell pellets were lysed in NP40 lysis buffer
supplemented with protease inhibitors. Protein concentrations were
determined using the Bradford assay (Bio-Rad Laboratories
Philadelphia, PA, USA). Proteins were separated by SDS-PAGE and
electrically transferred to a polyvinylidene difluoride membrane
(Millipore, Billerica, MA, USA). After blocking in 5% non-fat dry
milk in TBS, the membranes were hybridized to specific primary
antibodies overnight at 4°C, washed three times with TBS Tween-20,
and then incubated with secondary antibodies conjugated with
horseradish peroxidase for 1 h at room temperature. Next, the
membranes were washed three times in TBS Tween-20 at room
temperature. The protein bands were visualized using ECL
chemiluminescence reagents (Pierce Chemical Co., Rockford, IL, USA)
according to the manufacturer’s protocol.
Measurement of p53 and MDM2
half-lives
For p53 and MDM2 protein stability experiments,
after the treatment of A549 cells with 40 μmol of EGCG for 24 h, 30
μg/ml of CHX was added to inhibit protein synthesis. Subsequent
time points for incubation in medium containing EGCG and CHX were
0, 20, 40, 60 or 90 min as indicated. Cells were then processed as
previously described for western blotting and quantified by
densitometry.
Luciferase reporter assay
The pGL3-p53 firefly luciferase reporter plasmid and
the internal control pRL-SV40 (Renilla luciferase) plasmid were
purchased from Promega. A549 cells were cultured in 24-well plates
and co-transfected with the indicated plasmids using Lipofectamine
2000 (Invitrogen) according to the manufacturer’s instructions.
Each transfection contained 800 ng of firefly luciferase reporter
plasmid and 80 ng of pRL-SV40 plasmid. Twenty-four hours after the
transfection, the cells were treated with different concentrations
of EGCG for 24 h. Cell lysates were analyzed for firefly and
Renilla luciferase activities according to the Dual Luciferase
Reporter Assay kit (Promega) protocol. The firefly luciferase
activity was normalized to the value of pRL-SV40 activity for
transfection efficiency.
Soft agar colony assay
To examine the anchorage-independent growth, lung
cancer cells were suspended (10,000 cells/ml) in 1 ml of 0.3% agar
with Eagle’s basal medium containing 10% FBS, 1% antibiotics, and
different concentrations of EGCG (0, 1, 5, 10, 20 and 40 μmol/l)
overlaid into 6-well plates containing a 0.6% agar base. The
cultures were maintained in a 37°C, 5% CO2 incubator for
1–2 weeks, and then colonies were counted under a microscope using
the Image-Pro Plus software program (Media Cybernetics, Silver
Spring, MD, USA).
Ubiquitination assay
H1299 and 293T cells were co-transfected with 2 μg
of GFP-p53, 1 μg of HA-MDM2 and 0.5 μg His-Ub plasmids. Twenty-four
hours after the transfection, the cells were treated with different
concentrations of EGCG for a further 24 h. Subsequently, 20 μM of
MG132 was added to the culture medium for 6 h prior to harvesting.
Cells were split into two aliquots, one for immunoblotting and the
other for a ubiquitination assay. For the ubiquitination assay, we
first used the anti-GFP antibody to immunoprecipitate p53 and then
detected the ubiquitination using a His-tag antibody. A549 cells
were pre-treated with EGCG for 24 h and MG132 for 6 h. Endogenous
p53 was immunoprecipitated from 1 mg of protein lysate using the
p53 antibody and then immunoblotted with the anti-ubiquitin
antibody to capture polyubiquitinated p53.
Statistical analysis
All the statistical analyses were performed using
the SPSS software (version 13.0). The experiments were performed in
triplicate. The quantitative data are expressed as mean values ±
standard deviation. The significant differences between two groups
were assessed by a two-tailed Student’s t-test. P<0.05 was
considered to represent a statistically significant difference.
Results
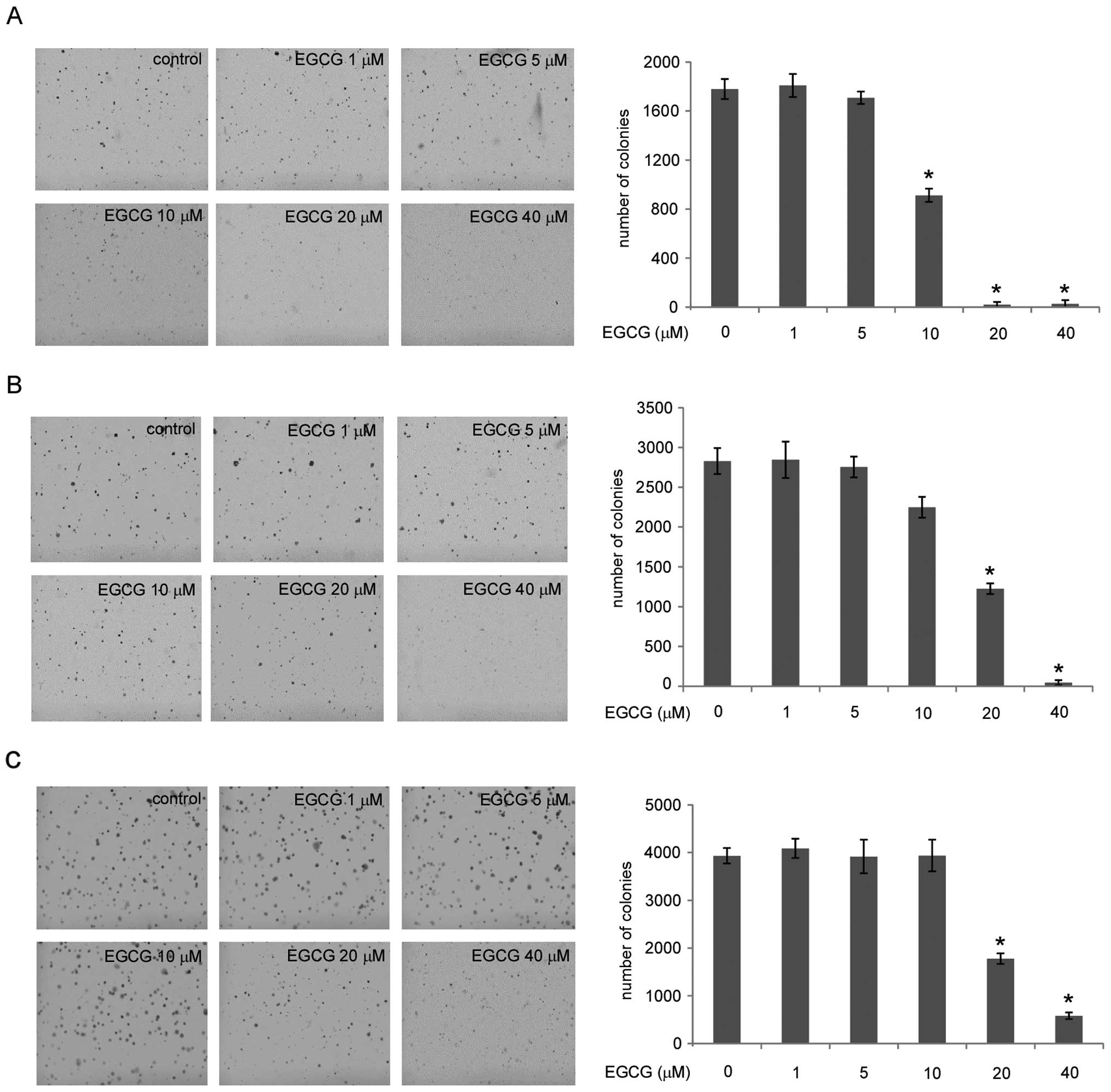
EGCG inhibits the anchorage-independent
growth of human lung cancer cells
Initially, we investigated the effects of EGCG on
the anchorage-independent growth of three different types of human
lung cancer cell lines: A549, H1650 and H460. The results showed
that these three cell lines have different sensitivity to EGCG. At
low concentrations (1–5 μM), EGCG does not substantially inhibit
the anchorage-independent growth of these lung cancer cells. In
H1650 cells, EGCG potently inhibits the anchorage-independent
growth at the concentration of 10 μM, and almost no colonies were
formed at 20 μM (Fig. 1A). In A549
and H460 cells, EGCG showed clear inhibitory effects against
anchorage-independent growth at the concentration of 20 μM
(Fig. 1B and C, respectively).
These results indicate that EGCG inhibits the anchorage-independent
growth of human lung cancer cells in a dose-dependent manner.
EGCG induces p53 accumulation and
upregulates its target genes
Based on the results of the soft agar experiments,
we detected the effects of EGCG on p53 expression in three p53-wild
type human lung cancer cells: A549 (Fig. 2A), H1650 (Fig. 2B) and H460 (Fig. 2C). Our results showed that EGCG
dose-dependently increased the endogenous p53 expression. At the
concentration of 5 μM, the expression of p53 was substantially
increased following the treatment of EGCG for 24 h. In addition, we
investigated the effects of EGCG on its downstream target genes,
p21 and MDM2. Similar to the effect on p53, EGCG enhanced the
expression levels of p21 and MDM2. These data indicate that EGCG
increases the expression of p53 and its target genes.
EGCG promotes the stability of p53 and
MDM2
As a short-lived protein, p53 is degraded quickly by
ubiquitination under normal physiologic status. Enhancement of
protein stability is the first step for p53 to perform its
functions. To expose the mechanism of p53 upregulation by EGCG, we
first set up a half-life assay to investigate the effects of EGCG
on p53 stability. We adopted CHX treatment to block the protein
synthesis in A549 cells, and western blotting to detect the
expression levels of p53 following treatment with EGCG for 24 h. We
found that EGCG significantly upregulated the half-life of p53 from
~40 min to >90 min. Meanwhile, as its target gene and major
negative regulatory factor, the half-life of MDM2 also clearly
increased after EGCG treatment (Fig.
3). These results demonstrate that EGCG promotes the stability
of p53 and MDM2.
EGCG promotes nuclear localization and
activity of p53
Ubiquitination of p53 results in targeting either
for proteasomal degradation or nuclear export. As an important
transcriptional factor, p53 stability and nuclear localization are
essential for its tumor suppressor function (34), In the nucleus, as a result of
MDM2-mediated ubiquitination, p53 is transported into the cytoplasm
or is degraded by the 26S proteasome. Both in the nucleus and in
the cytoplasm, the phosphorylation of p53 is considered to be a
counteractive post-translational modification that promotes p53
stability and transactivation. Previous studies have reported that
the phosphorylation of p53 at Ser15 or Ser20 increases p53
stability by disrupting the interaction between p53 and MDM2
(42,43). In the present study, we found that
EGCG increased Ser15 and Ser20 phosphorylation of p53 in a
dose-dependent manner (Fig. 4A). We
also confirmed the localization of p53 and MDM2 by cellular
fractionation. As shown in Fig. 4B,
EGCG treatment only slightly promoted the accumulation of p53 in
the cytoplasm. However, in the nucleus, EGCG dose-dependently
increased the accumulation of p53, with a significant increase at
the concentration of 20 μM. By contrast, MDM2 in the nucleus was
mildly decreased as the concentration of EGCG increased.
Furthermore, we tested whether EGCG treatment affects the
transactivation ability of p53 using a Dual Luciferase Reporter
Assay system. As shown in Fig. 4C,
the transactivation ability of p53 was significantly elevated
following EGCG treatment. At the concentration of 20 μM, the
luciferase activity increased nearly 3 times. These results suggest
that EGCG potentiates p53 function in human lung cancer cells.
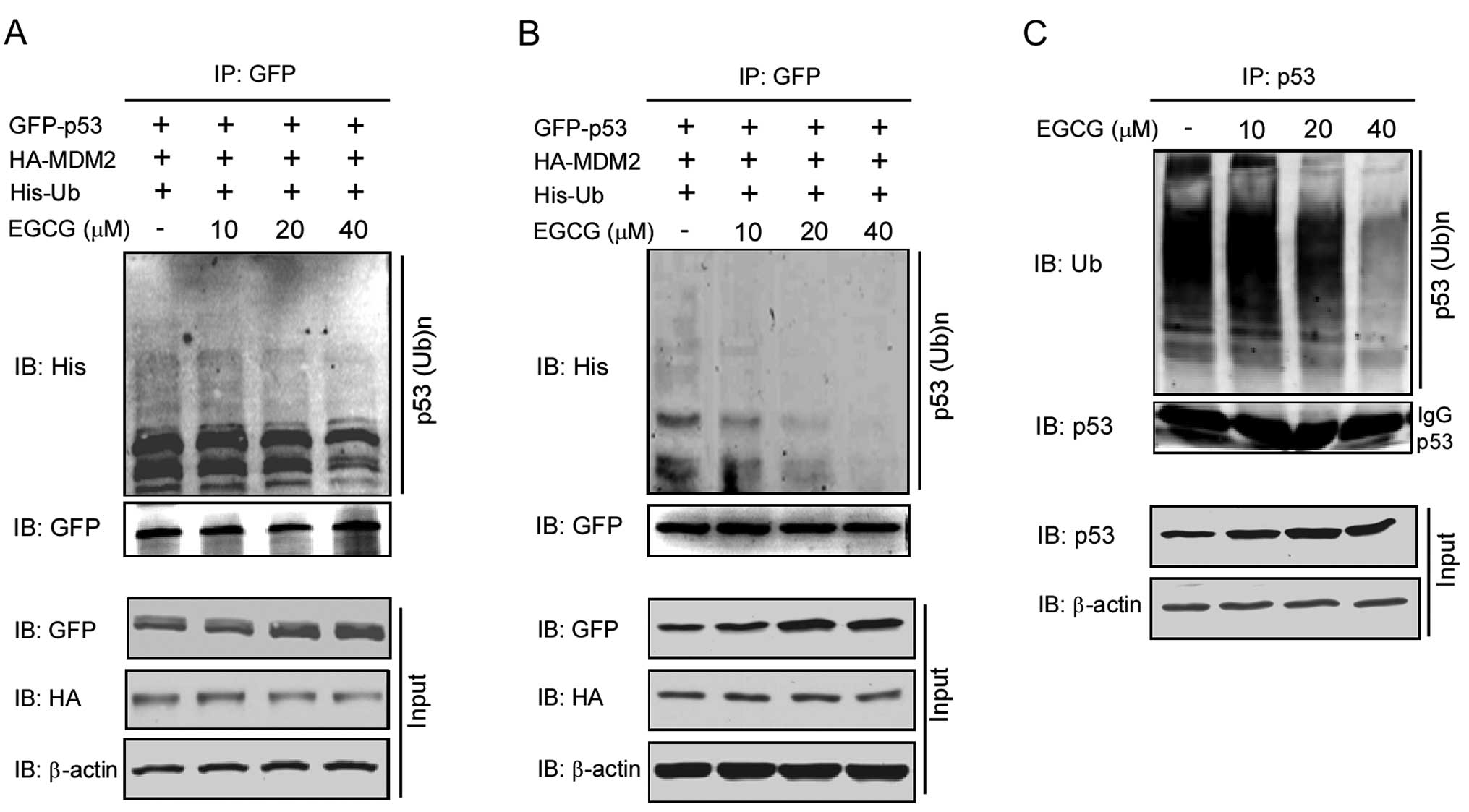
EGCG inhibits proteasomal
degradation-dependent p53 ubiquitination
Based on the observation that EGCG promotes the
stability of p53, we further investigated the effects of EGCG on
p53 ubiquitination. Plasmids GFP-p53 (3 μg), HA-MDM2 (0.5 μg) and
His-Ub (0.5 μg) were co-transfected into 293T cells, which were
used as an overexpression system. As shown in Fig. 5A, EGCG treatment substantially
decreased the ubiquitination of p53 with significant inhibition
observed at the concentration of 20 μM. We also tested this
phenomenon by transfection of these plasmids in p53-deficient H1299
cells. From the results shown in Fig.
5B, EGCG also dose-dependently inhibited the ubiquitination of
p53. In addition to the overexpression system, we also studied the
effects of EGCG on the regulation of endogenous p53 ubiquitination
(Fig. 5C). In the p53-wild type
A549 cell line treated with EGCG at the concentration of 20 μM for
24 h, the ubiquitination of p53 was markedly decreased. These
results suggest that EGCG inhibits p53 ubiquitination.
EGCG inhibits the interaction of p53 and
MDM2
The results of the above experiments demonstrated
that EGCG inhibits the ubiquitination of p53, promotes its
stability and increases its expression. As a downstream target gene
of p53, the expression levels of MDM2 also increased with the
accumulation of p53 in the nucleus. However, as an important
negative regulator of p53, the increase in MDM2 expression may
cause the ubiquitination and subsequent degradation of p53. The
previous results confirmed that EGCG markedly inhibits the
ubiquitination of p53 even in the case of MDM2 overexpression.
Therefore, studies were performed to further investigate the
effects of EGCG on the interaction between MDM2 and p53. The
plasmids GFP-p53 (2 μg) and HA-MDM2 (2 μg) were co-transfected into
293T and H1299 cells. Following treatment with 40 μM EGCG for 24 h,
co-immunoprecipitation was performed to detect the interaction
between MDM2 and p53. As shown in Fig.
6A and B, in the overexpression systems 293T and H1299, EGCG
treatment resulted in substantial inhibition of the interaction
between p53 and MDM2. In A549, we observed similar results in which
EGCG treatment inhibited MDM2 binding with p53 (Fig. 6C). These results indicate that EGCG
suppresses MDM2-mediated p53 ubiquitination by disrupting their
interaction.
Discussion
The natural compound EGCG is the major polyphenol
component of green tea. It has been extensively studied due to its
relatively high abundance and strong epidemiologic evidence of
cancer-preventive activity (2,3).
Although several reports have shown that EGCG exerts its anticancer
activity by targeting specific cell signaling pathways, the
underlying mechanism is not yet fully understood.
Previous reports revealed that EGCG treatment
induces lung cancer cell apoptosis or cell cycle arrest (18,35).
Other groups also found that the tumor suppressor gene p53 may be
involved in the antitumor activity of EGCG since cancer cells
expressing wild-type p53 are more sensitive to EGCG treatment
compared with p53-null or p53 knock down cancer cells (36,40).
In the present study, we first examined the antitumor effects of
EGCG in several human lung cancer cells that express wild-type p53
by soft agar assay. Our results demonstrated that EGCG inhibits the
anchorage-independent growth of human lung cancer cells in a
dose-dependent manner. Western blotting data indicated that EGCG
promotes the accumulation of wild-type p53 and its target genes,
p21 and MDM2. Our data demonstrated and corroborated other studies
showing that the effect of EGCG against lung cancer may partly
depend on p53 activity.
To further investigate p53 stability mediated by
EGCG, the effects of EGCG on the half-life of p53 and MDM2 were
examined using CHX treatment. We found that EGCG significantly
upregulated the half-life of p53 from approximately 40 min to 90
min, as well as that of its target gene, MDM2, from approximately
40 min to over 90 min. These results indicate that p53 is a
potential anticancer target mediated by EGCG.
The activity and localization of p53 are mainly
regulated by post-translational modifications, such as
phosphorylation, acetylation and ubiquitination (27,28,34).
Accumulating evidence demonstrates that p53 phosphorylation at
Ser15 and Ser20 attenuates the binding of p53 to MDM2 and disrupts
MDM2-mediated p53 ubiquitination both in vivo and in
vitro(42,43). In our study, we demonstrated that
the increased p53 expression was accompanied by the phosphorylation
of p53 at Ser15 and Ser20. In addition, by extracting the cytosolic
and nuclear fractions, we further determined p53 subcellular
localization upon EGCG treatment. The results clearly showed that
EGCG potently induced nuclear accumulation of p53 in a
dose-dependent manner while, at the same time, the expression
levels of MDM2 in the nucleus appeared slightly decreased.
Therefore, we hypothesized that EGCG-induced stabilization and
nuclear accumulation of p53 might induce an increase in its
transcriptional activity and result in an upregulation of its
target genes, p21 and MDM2. Thus, we tested p53 transcriptional
activity by setting up a reporter gene assay. Our data confirmed
the hypothesis that EGCG treatment increases p53 transcriptional
activity.
MDM2, a downstream target gene of p53, is the major
E3 ligase responsible for regulating p53 polyubiquitination and
targeting p53 for proteasomal degradation. Therefore, enhanced p53
activity causes increased expression of its own negative regulator,
MDM2, thereby forming an auto-regulatory feedback loop. In the
present study, we found that EGCG treatment sustainably increased
p53 expression, accompanied by MDM2 upregulation. To further
investigate the underlying mechanism, we detected p53
polyubiquitination by immunoprecipitation. Although EGCG treatment
increased MDM2 protein levels, MDM2-mediated p53 ubiquitination was
markedly decreased. Furthermore, our co-immunoprecipitation data
showed that EGCG treatment disrupts the interaction between p53 and
MDM2. This observation suggests that upon EGCG treatment, MDM2 acts
only as a target gene and is upregulated by p53, and that its
activity to induce p53 degradation is suppressed.
Collectively, our study identifies p53 as a
potential target of EGCG to execute its cancer-preventive activity,
and that the natural compound EGCG, inhibits the
anchorage-independent growth of human lung cancer cells by
promoting p53 stability/activity and by inhibiting MDM2-mediated
p53 ubiquitination and degradation. Our study provides a new
mechanism to explain the chemopreventive effect of EGCG on cancer;
further research is required to provide evidence for the clinical
use of EGCG in cancer prevention and therapy.
Acknowledgements
This study was supported by the Nature Scientific
Foundation of Hunan Province (grant no. 08JJ6010) and the Research
Program from the Science Technology Department of Hunan Province
(grant no. 2012FJ4076).
Abbreviations:
|
EGCG
|
epigallocatechin gallate
|
|
CHX
|
cycloheximide
|
References
|
1
|
Chen L and Zhang HY: Cancer preventive
mechanisms of the green tea polyphenol
(−)-epigallocatechin-3-gallate. Molecules. 12:946–957. 2007.
|
|
2
|
Johnson R, Bryant S and Huntley AL: Green
tea and green tea catechin extracts: an overview of the clinical
evidence. Maturitas. 73:280–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fujiki H and Suganuma M: Green tea: an
effective synergist with anticancer drugs for tertiary cancer
prevention. Cancer Lett. 324:119–125. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fujiki H, Imai K, Nakachi K, Shimizu M,
Moriwaki H and Suganuma M: Challenging the effectiveness of green
tea in primary and tertiary cancer prevention. J Cancer Res Clin
Oncol. 138:1259–1270. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Surh YJ: Cancer chemoprevention with
dietary phytochemicals. Nat Rev Cancer. 3:768–780. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang CS and Wang X: Green tea and cancer
prevention. Nutr Cancer. 62:931–937. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Singh BN, Shankar S and Srivastava RK:
Green tea catechin, epigallocatechin-3-gallate (EGCG): mechanisms,
perspectives and clinical applications. Biochem Pharmacol.
82:1807–1821. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Khan N, Afaq F, Saleem M, Ahmad N and
Mukhtar H: Targeting multiple signaling pathways by green tea
polyphenol (−)-epigallocatechin-3-gallate. Cancer Res.
66:2500–2505. 2006.
|
|
9
|
Shimizu M, Deguchi A, Lim JT, Moriwaki H,
Kopelovich L and Weinstein IB: (−)-Epigallocatechin gallate and
polyphenon E inhibit growth and activation of the epidermal growth
factor receptor and human epidermal growth factor receptor-2
signaling pathways in human colon cancer cells. Clin Cancer Res.
11:2735–2746. 2005.
|
|
10
|
Yang CS, Wang H, Li GX, Yang Z, Guan F and
Jin H: Cancer prevention by tea: evidence from laboratory studies.
Pharmacol Res. 64:113–122. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Levites Y, Amit T, Youdim MB and Mandel S:
Involvement of protein kinase C activation and cell survival/cell
cycle genes in green tea polyphenol (−)-epigallocatechin 3-gallate
neuroprotective action. J Biol Chem. 277:30574–30580.
2002.PubMed/NCBI
|
|
12
|
Relat J, Blancafort A, Oliveras G, Cufi S,
Haro D, Marrero PF and Puig T: Different fatty acid metabolism
effects of (−)-epigallocatechin-3-gallate and C75 in adenocarcinoma
lung cancer. BMC Cancer. 12:2802012.
|
|
13
|
Sah JF, Balasubramanian S, Eckert RL and
Rorke EA: Epigallocatechin-3-gallate inhibits epidermal growth
factor receptor signaling pathway. Evidence for direct inhibition
of ERK1/2 and AKT kinases. J Biol Chem. 279:12755–12762. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kanwar J, Taskeen M, Mohammad I, Huo C,
Chan TH and Dou QP: Recent advances on tea polyphenols. Front
Biosci (Elite Ed). 4:111–131. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pianetti S, Guo S, Kavanagh KT and
Sonenshein GE: Green tea polyphenol epigallocatechin-3 gallate
inhibits Her-2/neu signaling, proliferation, and transformed
phenotype of breast cancer cells. Cancer Res. 62:652–655.
2002.PubMed/NCBI
|
|
16
|
Yang F, Oz HS, Barve S, de Villiers WJ,
McClain CJ and Varilek GW: The green tea polyphenol
(−)-epigallocatechin-3-gallate blocks nuclear factor-kappa B
activation by inhibiting I kappa B kinase activity in the
intestinal epithelial cell line IEC-6. Mol Pharmacol. 60:528–533.
2001.
|
|
17
|
Ahmad N, Feyes DK, Nieminen AL, Agarwal R
and Mukhtar H: Green tea constituent epigallocatechin-3-gallate and
induction of apoptosis and cell cycle arrest in human carcinoma
cells. J Natl Cancer Inst. 89:1881–1886. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Saha A, Kuzuhara T, Echigo N, Suganuma M
and Fujiki H: New role of (−)-epicatechin in enhancing the
induction of growth inhibition and apoptosis in human lung cancer
cells by curcumin. Cancer Prev Res. 3:953–962. 2010.
|
|
19
|
Chang CM, Chang PY, Tu MG, et al:
Epigallocatechin gallate sensitizes CAL-27 human oral squamous cell
carcinoma cells to the anti-metastatic effects of gefitinib
(Iressa) via synergistic suppression of epidermal growth factor
receptor and matrix metalloproteinase-2. Oncol Rep. 28:1799–1807.
2012.
|
|
20
|
Deng YT and Lin JK: EGCG inhibits the
invasion of highly invasive CL1-5 lung cancer cells through
suppressing MMP-2 expression via JNK signaling and induces G2/M
arrest. J Agric Food Chem. 59:13318–13327. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jung YD and Ellis LM: Inhibition of tumour
invasion and angiogenesis by epigallocatechin gallate (EGCG), a
major component of green tea. Int J Exp Pathol. 82:309–316. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu LC, Tsao TC, Hsu SR, Wang HC, Tsai TC,
Kao J and Way TD: EGCG inhibits transforming growth
factor-β-mediated epithelial-to-mesenchymal transition via the
inhibition of Smad2 and Erk1/2 signaling pathways in nonsmall cell
lung cancer cells. J Agric Food Chem. 60:9863–9873. 2012.
|
|
23
|
Singh T and Katiyar SK: Green tea
catechins reduce invasive potential of human melanoma cells by
targeting COX-2, PGE2 receptors and epithelial-to-mesenchymal
transition. PLoS One. 6:e252242011. View Article : Google Scholar
|
|
24
|
Watanabe T, Kuramochi H, Takahashi A, et
al: Higher cell stiffness indicating lower metastatic potential in
B16 melanoma cell variants and in (−)-epigallocatechin
gallate-treated cells. J Cancer Res Clin Oncol. Feb 2–2012.(Epub
ahead of print).
|
|
25
|
Charvet C, Wissler M, Brauns-Schubert P,
et al: Phosphorylation of Tip60 by GSK-3 determines the induction
of PUMA and apoptosis by p53. Mol Cell. 42:584–596. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dai C, Tang Y, Jung SY, Qin J, Aaronson SA
and Gu W: Differential effects on p53-mediated cell cycle arrest
vs. apoptosis by p90. Proc Natl Acad Sci USA. 108:18937–18942.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dai C and Gu W: p53 post-translational
modification: deregulated in tumorigenesis. Trends Mol Med.
16:528–536. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kruse JP and Gu W: SnapShot: p53
post-translational modifications. Cell. 133:930–930.e1. 2008.
View Article : Google Scholar
|
|
29
|
Kruse JP and Gu W: Modes of p53
regulation. Cell. 137:609–622. 2009. View Article : Google Scholar
|
|
30
|
Munoz-Fontela C, Gonzalez D, Marcos-Villar
L, et al: Acetylation is indispensable for p53 antiviral activity.
Cell Cycle. 10:3701–3705. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sakaguchi K, Herrera JE, Saito S, et al:
DNA damage activates p53 through a phosphorylation-acetylation
cascade. Genes Dev. 12:2831–2841. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jimenez GS, Khan SH, Stommel JM and Wahl
GM: p53 regulation by post-translational modification and nuclear
retention in response to diverse stresses. Oncogene. 18:7656–7665.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lee JT and Gu W: The multiple levels of
regulation by p53 ubiquitination. Cell Death Differ. 17:86–92.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yuan J, Luo K, Zhang L, Cheville JC and
Lou Z: USP10 regulates p53 localization and stability by
deubiquitinating p53. Cell. 140:384–396. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Amin AR, Wang D, Zhang H, et al: Enhanced
anti-tumor activity by the combination of the natural compounds
(−)-epigallocatechin-3-gallate and luteolin: potential role of p53.
J Biol Chem. 285:34557–34565. 2010.
|
|
36
|
Hastak K, Gupta S, Ahmad N, Agarwal MK,
Agarwal ML and Mukhtar H: Role of p53 and NF-kappaB in
epigallocatechin-3-gallate-induced apoptosis of LNCaP cells.
Oncogene. 22:4851–4859. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lee MH, Han DW, Hyon SH and Park JC:
Apoptosis of human fibrosarcoma HT-1080 cells by
epigallocatechin-3-O-gallate via induction of p53 and caspases as
well as suppression of Bcl-2 and phosphorylated nuclear
factor-kappaB. Apoptosis. 16:75–85. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Qin J, Chen HG, Yan Q, et al: Protein
phosphatase-2A is a target of epigallocatechin-3-gallate and
modulates p53-Bak apoptotic pathway. Cancer Res. 68:4150–4162.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hastak K, Agarwal MK, Mukhtar H and
Agarwal ML: Ablation of either p21 or Bax prevents p53-dependent
apoptosis induced by green tea polyphenol
epigallocatechin-3-gallate. FASEB J. 19:789–791. 2005.PubMed/NCBI
|
|
40
|
Thakur VS, Ruhul Amin AR, Paul RK, et al:
p53-Dependent p21-mediated growth arrest pre-empts and protects
HCT116 cells from PUMA-mediated apoptosis induced by EGCG. Cancer
Lett. 296:225–232. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhao BX, Chen HZ, Lei NZ, et al: p53
mediates the negative regulation of MDM2 by orphan receptor TR3.
EMBO J. 25:5703–5715. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chehab NH, Malikzay A, Stavridi ES and
Halazonetis TD: Phosphorylation of Ser-20 mediates stabilization of
human p53 in response to DNA damage. Proc Natl Acad Sci USA.
96:13777–13782. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Unger T, Juven-Gershon T, Moallem E, et
al: Critical role for Ser20 of human p53 in the negative regulation
of p53 by Mdm2. EMBO J. 18:1805–1814. 1999. View Article : Google Scholar : PubMed/NCBI
|