Introduction
Cyclin-dependent kinase 5 regulatory subunit
associated protein 1 (CDK5RAP1) is a radical S-adenosyl methionine
(SAM) enzyme (1) with homology to
the bacterial MiaB protein (2),
which post-synthetically converts the RNA modification
N6-isopentenyladenosine (i6A) into
2-methylthio-N6-isopen-tenyladenosine
(ms2i6A) at A37 (3), as shown in Fig. 1A. It was discovered to inhibit the
active CDK5 kinase and function in codon suppression (4) and stabilization of the codon/anticodon
interaction (5). CDK5 aberrant
regulation can lead to a number of diseases (6). The biochemical link established by
CDK5RAP1 between the enzymatic modification of transfer RNA (tRNA)
tanticodon loops and CDK5 kinase activity is highly unusual,
particularly since the modified base ms2i6A
is known to exist in tRNA of prokaryotic origin (7), particularly in mitochondrial tRNA of
mammals (8).
Breast cancer has long been a leading cause of
mortality in women worldwide (9).
Due to the limited efficacy of traditional therapy, it is necessary
to exploit a new treatment strategy for breast cancer.
Mitochondria-initiated responses are thought to be the major
pathway for apoptosis, and, therefore, targeting the mitochondria
is a novel strategy for cancer therapy (10). Hence, in the present study, we
sought to determine if the mistranslation of
ms2i6A in mitochondrial tRNA caused by
CDK5RAP1 deficiency affects the human breast cancer cell line,
MCF-7 cells.
Cell cycle arrest is an important cause of growth
inhibition. Many anticancer agents reduce malignant growth by
arresting the cell cycle at the G1, S or G2/M phases (11). Arresting the cell cycle is an
effective method to regulate cell cycle progression, and to
contribute to malignant cell proliferation (12). Apart from cell cycle arrest,
apoptosis is another cause of growth inhibition. There is
compelling evidence that excessive reactive oxygen species (ROS)
production surmounts cellular antioxidant defenses, triggering
apoptosis (13), and cancer cells
are more sensitive to rapid increases in ROS levels than normal
cells. Oncogenic transformation elevates basal ROS levels
significantly so that any further acute increases can trigger
reactivation of the apoptotic program in cancer cells (14). Various apoptotic stimuli can rapidly
activate MAPKs, which include phospho-c-Jun N-terminal kinase
(p-JNK) (15). The activation of
JNK is associated with ROS elevation (16). The p-JNK activated through
ROS-dependent pathway induces the overexpression of tumor
suppressors, such as p53, then leads to cell apoptosis (17).
In the present study, to the best of our knowledge,
the hypothesis that CDK5RAP1 deficiency inhibits tumor growth in a
human breast cancer cell line was explored for the first time. The
results showed that CDK5RAP1 deficiency induced MCF-7 cell cycle
arrest and apoptosis, which could be prevented by the pretreatment
with N-acetyl-cysteine (NAC; the inhibitor of ROS), or SP600125
(the inhibitor of JNK), suggesting that the ROS/JNK signaling
pathway is an important mechanism in the apoptosis process. Our
study indicated that this may be a novel therapeutic strategy for
cancer.
Materials and methods
Cell culture
The human breast cancer cell line MCF-7 was
purchased from the American Type Culture Collection (ATCC;
Manassas, VA, USA). This study was performed in accordance with the
Experiment Guidelines of Harbin Medical University (Harbin, China)
and ethical approval was obtained from Harbin Medical University.
MCF-7 cells were cultured in RPMI-1640 medium supplemented with 10%
fetal bovine serum (FBS), 100 U/ml penicillin and 100 mg/ml
streptomycin (GIBCO, Grand Island, NY, USA), and were cultured in
an incubator (Sanyo, Tokyo, Japan) with 5% CO2 at
37°C.
Small interfering RNA (siRNA)
transfection
CDK5RAP1 siRNA and non-targeted negative control
siRNA were purchased from Santa Cruz Biotechnology Inc. (Santa
Cruz, CA, USA). MCF-7 cells were seeded onto 6-well plates at the
recommended density (1×105 cells/well) and grown to
60–80% confluence prior to transfection. siRNAs were transfected
into MCF-7 cells with siRNA Transfection Reagent (Santa Cruz
Biotechnology Inc.) according to the manufacturer’s instructions.
MCF-7 cells were further incubated for another 48 h and then used
for experiments.
Quantitative polymerase chain reaction
(qPCR)
CDK5RAP1 siRNA and negative control siRNA were
transfected into MCF-7 cells, and the cells were further incubated
for 48 h. Total RNA was extracted from MCF-7 cells and relative
mRNA was normalized to 18s. The following primers (Hokkaido System
Science Co. Ltd. Sapporo, Japan) were used: CDK5RAP1 forward,
5′-ATGGCTGCCAGATGAATG TGA-3′ and reverse,
5′-CTCTTGGAGGTTACTGGTCCG-3′; 18s forward,
5′-GTAACCCGTTGAACCCCATT-3′ and reverse, 5′-CCATCCAATCGGTAGTAGCG-3′.
qPCR was performed using the ABI 7300 Fast real-time PCR system
(Applied Biosystems, Foster City, CA, USA).
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay
The viability of normal MCF-7 cells and
CDK5RAP1-deficient MCF-7 cells was determined by a colorimetric MTT
assay according to the method described previously (18). Absorbance at 550 nm was determined
by an MTP-800 microplate reader (Corona Electric, Tokyo, Japan).
Absorbance at 690 nm was also measured to compensate for any
interfering effects of cell debris and the microtiter plate.
Percentage of viable cell number was calculated as: Optical density
(OD) of treated sample/OD of untreated control ×100.
Cell cycle analysis
CDK5RAP1 siRNA and negative control siRNA were
transfected into MCF-7 cells, and the cells were further incubated
for 48 h. Then, MCF-7 cells were trypsinized and fixed in 99%
ethanol at −20°C for 2 h, washed and resuspended in 420 μl PBS.
Subsequently, samples were first incubated with RNase A (Sigma,
Shanghai, China) (50 μl of a 10 mg/ml solution) at 37°C for 30 min,
and then PI (20 μl of a 0.2 mg/ml solution) at room temperature for
10 min. DNA content was analyzed by flow cytometry using a
FACSCalibur and CellQuest software (Becton Dickinson, Franklin
Lake, NJ, USA), as previously described (19).
Apoptosis assay
MCF-7 cell apoptosis staining was performed using an
Annexin V (cell apoptosis signaling component)-Biotin Apoptosis kit
as per the manufacturer’s instructions (Mountain View, CA, USA).
The MCF-7 cells were seeded in a 6-well plate at the density of
1×105 cells/well and were pretreated or non-treated with
NAC (5 mM) or SP600125 (5 μM) (Sigma) for 1 h prior to CDK5RAP1
siRNA transfection. After transfection with CDK5RAP1 siRNA or
control siRNA, MCF-7 cells were further incubated for 48 h. Stained
cells were analyzed using FACSCalibur™ Flow Cytometry (BD
Biosciences, San Jose, CA, USA) with CellQuest software. Ten
thousand events were collected for each sample.
Nuclear staining with Hoechst 33342 for
morphological evaluation
MCF-7 cells were plated in 6-well plates at the
density of 1×105 cells/well. CDK5RAP1 siRNA and negative
control siRNA were transfected into MCF-7 cells, and the cells were
further incubated for 48 h. Then, the cells were washed with PBS,
fixed in 4% paraformaldehyde (Bioss, Beijing, China) for 30 min and
then stained with 20 mg/ml Hoechst 33342 for 15 min at room
temperature in the dark. Cells were then assessed by fluorescence
microscopy for morphological changes.
Detection of intracellular ROS
Intracellular accumulation of ROS was estimated
using the fluorescent dye H2-DCFDA (Life Technologies,
Tokyo, Japan), which is converted to a membrane impermeable and
highly fluorescent compound, dichlorofluorescin diacetate (DCF), in
the cell in the presence of ROS (20). The MCF-7 cells were seeded in a
6-well plate at the density of 1×105 cells/well.
Following transfection with CDK5RAP1 siRNA or control siRNA, MCF-7
cells were further incubated for 48 h. The cells were rinsed with a
serum-free medium and were incubated in 5 μM H2-DCFDA
for 60 min at 37°C. The cells were then examined under a
fluorescence microscope (C1-T-SM; Nikon, Tokyo, Japan), collected
and subjected to a fluorescence spectrophotometer (F-2500; Hitachi,
Tokyo, Japan) to detect the fluorescence of DCF inside cells
(excitation, 488 nm; emission, 521 nm).
Flow cytometry
Intracellular ROS was measured using
2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) (Life
Technologies). The MCF-7 cells were seeded in a 6-well plate at the
density of 1×105 cells/well. CDK5RAP1 siRNA and negative
control siRNA were transfected into MCF-7 cells, and the cells were
further incubated for 48 h. MCF-7 cells were then washed with PBS
and labeled with 10 μM DCFDA for 30 min. Then, excess DCFH-DA was
removed by washing the cells in serum-free RPMI-1640 medium
(Sigma). The fluorescence intensities were measured using a
FACSCalibur flow cytometer (BD Biosciences).
Western blot analysis
Electrophoresis was performed using a vertical slab
gel with 12% polyacrylamide content according to the method
described previously (21). The
transfer of proteins from the SDS polyacrylamide gel to a membrane
was performed electrophoretically according to the method described
previously (22) with certain
modifications using a Semi Dry Electroblotter (Sartorius AG,
Goettingen, Germany) for 90 min with an electric current of 15 V.
The membrane was treated with Block Ace™ (4%) for 30 min at 22°C.
The first reaction was performed using rabbit immunoglobulin (IG) G
antibodies against JNK, p-JNK, p53, caspase-9 and caspase-3 (Sigma)
in PBS containing 0.03% Tween-20 for 1 h at 22°C. Following washing
in the same buffer, the second reaction was performed using
horseradish peroxidase (HRP)-conjugated anti-rabbit goat IgG (20
ng/ml) for 30 min at 22°C. After washing, the enhanced
chemiluminescence (ECL) reaction was performed on the membrane
using the ECL Plus Western Blotting Detection System™ (GE
Healthcare Life Sciences).
Statistical analysis
Data are expressed as the mean ± standard deviation.
Each experiment was repeated at least 3 times. The Student’s t-test
was used and P<0.05 was considered to indicate a statistically
significant difference.
Results
CDK5RAP1 deficiency suppresses tumor
growth in MCF-7 cells
To investigate the effect of CDK5RAP1 deficiency on
the growth of human breast cancer cell line (MCF-7 cells), MCF-7
cells were seeded onto 6-well plates at the recommended density
(1×105 cells/well) and grown to 60–80% confluence prior
to transfection. CDK5RAP1 siRNA and negative control siRNA were
transfected into MCF-7 cells, and then the cells were further
incubated for 48 h. The viability of normal MCF-7 cells and
CDK5RAP1-deficient MCF-7 cells was determined by a colorimetric MTT
assay. The tumor growth was significantly suppressed in the
CDK5RAP1-deficient MCF-7 cells (Fig. 2A
and B; P<0.01).
CDK5RAP1 deficiency arrests MCF-7 cells
at the G2/M phase
CDK5RAP1 siRNA and negative control siRNA were
transfected into MCF-7 cells, and then the cells were further
incubated for 48 h. Cell cycle analysis was performed using a
FACSCalibur and CellQuest software. CDK5RAP1 deficiency arrested
MCF-7 cells at the G2/M phase significantly compared with control
MCF-7 cells (Fig. 2C and D;
P<0.01).
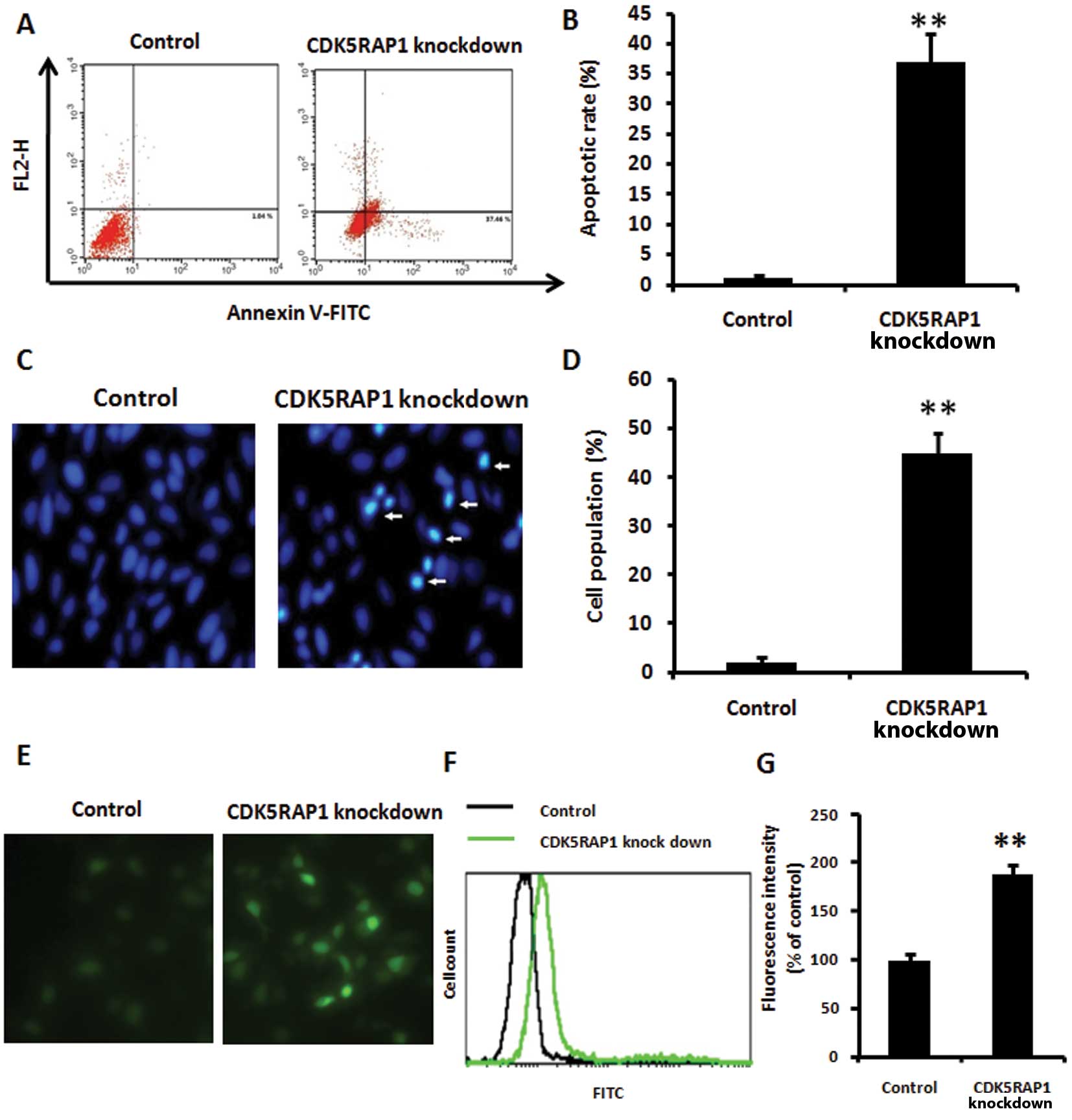
CDK5RAP1 deficiency induces MCF-7 cell
apoptosis
MCF-7 cells were plated in 6-well plates at the
density of 1×105 cells/well. Following transfection with
CDK5RAP1 siRNA or control siRNA, MCF-7 cells were further incubated
for 48 h. The MCF-7 cell apoptosis was performed using an Annexin
V-Biotin Apoptosis kit and nuclear staining with Hoechst 33342 by
fluorescence microscopy. CDK5RAP1 deficiency induced MCF-7 cell
apoptosis significantly compared with control MCF-7 cells (Fig. 3A and B, Annexin V-Biotin; Fig. 3C and D, Hoechst 33342 staining.
P<0.01).
CDK5RAP1 deficiency induces ROS
generation in MCF-7 cells
CDK5RAP1 siRNA and negative control siRNA were
transfected into MCF-7 cells, and then the cells were further
incubated for 48 h. Intracellular accumulation of ROS was estimated
using the fluorescent dye H2-DCFDA (Fig. 3E and G), and flow cytometry using
DCFH-DA (Fig. 3F). CDK5RAP1
deficiency significantly induced ROS generation in MCF-7 cells
(P<0.01).
CDK5RAP1 deficiency upregulates the
expression of p-JNK, p53, caspase-9 and caspase-3 in MCF-7
cells
MCF-7 cells were plated in 6-well plates at the
density of 1×105 cells/well. After transfection with
CDK5RAP1 siRNA or control siRNA, MCF-7 cells were further incubated
for 48 h. The expression levels of p-JNK, p53, caspase-9 and
caspase-3 in MCF-7 cells were measured by western blot analysis.
CDK5RAP1 deficiency upregulated the expression of p-JNK, p53,
caspase-9 and caspase-3 significantly compared with control MCF-7
cells. β-actin was used as the normalization (Fig. 4).
NAC and SP600125 prevent MCF-7 cell
apoptosis induced by CDK5RAP1 deficiency
The MCF-7 cells were seeded in a 6-well plate at the
density of 1×105 cells/well and were pretreated or
non-treated with NAC (5 mM) or SP600125 (5 μM) for 1 h prior to
CDK5RAP1 siRNA transfection. Following transfection with CDK5RAP1
siRNA or control siRNA, MCF-7 cells were further incubated for 48
h. CDK5RAP1 deficiency induced MCF-7 cell apoptosis significantly
compared with control MCF-7 cells, while pretreatment with NAC or
SP600125 prevented the CDK5RAP1 deficiency-induced apoptosis
significantly (Fig. 5A and B;
P<0.01).
NAC and SP600125 prevent the CDK5RAP1
deficiency-induced high expression of p-JNK, p53, caspase-9 and
caspase-3 in MCF-7 cells
The MCF-7 cells were pretreated or non-treated with
NAC (5 mM) or SP600125 (5 μM) for 1 h prior to CDK5RAP1 siRNA
transfection. After transfection with CDK5RAP1 siRNA or control
siRNA, MCF-7 cells were further incubated for 48 h. CDK5RAP1
deficiency upregulated the expression of p-JNK, p53, caspase-9 and
caspase-3 significantly, while pretreatment with NAC or SP600125
prevented the CDK5RAP1 deficiency-induced high expression of p-JNK,
p53, caspase-9 and caspase-3 in MCF-7 cells. β-actin was used as
the normalization (Fig. 5C).
Discussion
The present study demonstrated, to the best of our
knowledge for the first time, that CDK5RAP1 deficiency suppresses
tumor growth and induces cell cycle arrest and apoptosis in a human
breast cancer cell line (MCF-7 cells). CDK5RAP1 is a radical SAM
enzyme (1) with homology to the
bacterial MiaB protein (2), which
post-synthetically converts the RNA modification i6A
into ms2i6A (3–6)
(Fig. 1A). The biochemical link
established by CDK5RAP1 between the enzymatic modification of tRNA
tanticodon loops and CDK5 kinase activity is highly unusual,
particularly since the modified base ms2i6A
is known to exist in tRNA of prokaryotic origin (7), particularly in mitochondrial tRNA of
mammals (8).
Breast cancer has long been a leading cause of
mortality in women of developed and developing countries (9,23).
Cell cycle arrest is an important cause of growth inhibition. Many
anticancer agents exhibit anti-proliferation by inhibiting cell
cycle progression at a particular check point such as G0/G1, S, or
G2/M (11). Deregulation of cell
cycle has been linked with cancer initiation and progression
(24). Arresting the cell cycle is
an effective method to regulate cell cycle progression, and
contribute to malignant cell proliferation (12). It has been reported that the
expression of CDK5RAP1 gene is related to the regulation and
progression of the M phase of the cell cycle (25). In accordance with that, our present
study confirmed that CDK5RAP1 deficiency suppressed tumor growth in
MCF-7 cells and arrested the cells at G2/M phase (Fig. 2). Apart from cell cycle arrest,
apoptosis is another cause of growth inhibition (26). Apoptosis, or programmed cell death,
is an essential mechanism through which many types of
chemotherapeutic agents inhibit tumor growth (27). Mitochondria-initiated responses are
thought to be the major pathway for apoptosis, and, therefore,
targeting the mitochondria is a novel strategy for cancer therapy
(10). Our present study also
confirmed that CDK5RAP1 deficiency induced MCF-7 cell apoptosis
(Fig. 3A–D).
ROS, which is the byproduct of normal cellular
oxidative processes, has been suggested to regulate the process
involved in the initiation of apoptotic signaling (28) and has been implicated in several
oncogenic pathways. Although it has been reported to be a tumor
growth promoter (29), there is
compelling evidence that ROS production surmounts cellular
antioxidant defenses, triggering apoptosis (13), and cancer cells are more sensitive
to rapid increases in ROS levels than normal cells. ROS-mediated
cytotoxicity has also been identified as an important mechanism in
some anticancer agents (30).
Accumulating evidence indicates that many anticancer agents destroy
tumor cells by raising the level of ROS above a toxic threshold
(31). Oncogenic transformation
elevates basal ROS levels significantly so that any further acute
increases can trigger reactivation of the apoptotic program in
cancer cells (14). High level of
ROS can destroy the integrity of plasma membrane, affect dynamic of
actin cytoskeleton and cause DNA damage, cumulatively known as
oxidative stress (32). To
investigate whether CDK5RAP1 deficiency-induced MCF-7 cell
apoptosis is promoted through an increase in ROS production, we
measured ROS levels. Our results showed that CDK5RAP1 deficiency
induced cell ROS generation in MCF-7 cells significantly (Fig. 3E–G).
Various apoptotic stimuli can rapidly activate
MAPKs, which include p-JNK (15).
The activation of JNK is associated with ROS elevation (16). A previous study suggested that
activation of JNK through ROS generation is important for apoptosis
(33). To investigate this
hypothesis, we examined the expression of p-JNK in the
CDK5RAP1-deficient MCF-7 cells. The p-JNK activated through
ROS-dependent pathway induces the overexpression of tumor
suppressors, such as p53 (17).
p53, a tumor suppressor protein, triggers cell cycle arrest to
provide time for self-mediated apoptosis through transcriptional
activation of cyclin-dependent kinase inhibitor (34). In addition to cell cycle arrest, p53
can induce the expression of several factors involved in apoptosis,
such as caspase-9 and caspase-3 (35). The activation of caspase-9 and
caspase-3 damage the cell structure and cause functional disorder
by proteolysis, final induction of apoptosis (36). Our data demonstrated that the
expression of p-JNK, p53, caspase-9 and caspase-3 were all
upregulated in CDK5RAP1-deficient MCF-7 cells (Fig. 4). This suggests that p-JNK, p53,
caspase-9 and caspase-3 are all involved in the apoptosis process.
As shown in Fig. 5, pretreatment
with NAC (the inhibitor of ROS) or SP600125 (the inhibitor of JNK),
prevented the apoptosis and the high expression of p-JNK, p53,
caspase-9 and caspase-3 in CDK5RAP1-deficient MCF-7 cells. These
results clearly indicate that CDK5RAP1 deficiency induces the
mitochondrial apoptosis by the ROS/JNK signaling pathway.
CDK5RAP1 deficiency induces cell cycle arrest and
apoptosis in MCF-7 cells via ROS generation, resulting p-JNK and
p53 activation, increase in cleavage of caspase-9 and caspase-3,
according to the mechanism described in Fig. 6. Although our data provided evidence
that tumor growth was markedly inhibited in the CDK5RAP1-deficient
MCF-7 cells, the complex process and mechanism require further
investigation in the future.
In the present study, to the best of our knowledge,
we demonstrated for the first time that CDK5RAP1 deficiency induces
cell cycle arrest and apoptosis in human breast cancer MCF-7 cells
by the ROS/JNK signaling pathway. The potential of CDK5RAP1
deficiency in cancer cells is expected to provide key insight into
the development of novel clinical treatments for cancer.
References
|
1
|
Atta M, Mulliez E, Arragain S, Forouhar F,
Hunt JF and Fontecave M: S-Adenosylmethionine-dependent
radical-based modification of biological macromolecules. Curr Opin
Struct Biol. 20:684–692. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kaminska KH, Baraniak U, Boniecki M,
Nowaczyk K, Czerwoniec A and Bujnicki JM: Structural bioinformatics
analysis of enzymes involved in the biosynthesis pathway of the
hypermodified nucleoside ms(2)io(6)A37 in
tRNA. Proteins. 70:1–18. 2008. View Article : Google Scholar
|
|
3
|
Pierrel F, Douki T, Fontecave M and Atta
M: MiaB protein is a bifunctional radical-S-adenosylmethionine
enzyme involved in thiolation and methylation of tRNA. J Biol Chem.
279:47555–47563. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bouadloun F, Srichaiyo T, Isaksson LA and
Bjork GR: Influence of modification next to the anticodon in tRNA
on codon context sensitivity of translational suppression and
accuracy. J Bacteriol. 166:1022–1027. 1986.PubMed/NCBI
|
|
5
|
Jenner LB, Demeshkina N, Yusupova G and
Yusupov M: Structural aspects of messenger RNA reading frame
maintenance by the ribosome. Nat Struct Mol Biol. 17:555–560. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gong CX and Iqbal K: Hyperphosphorylation
of microtubule-associated protein tau: a promising therapeutic
target for Alzheimer disease. Curr Med Chem. 15:2321–2328. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Esberg B, Leung HC, Tsui HC, Bjork GR and
Winkler ME: Identification of the miaB gene, involved in
methylthiolation of isopentenylated A37 derivatives in the tRNA of
Salmonella typhimurium and Escherichia coli. J Bacteriol.
181:7256–7265. 1999.PubMed/NCBI
|
|
8
|
Globisch D, Pearson D, Hienzsch A, Brückl
T, Wagner M, Thoma I, Thumbs P, Reiter V, Kneuttinger AC, Müller M,
Sieber SA and Carell T: Systems-based analysis of modified tRNA
bases. Angew Chem Int Ed Engl. 50:9739–9742. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Brody JG, Rudel RA, Michels KB, Moysich
KB, Bernstein L, Attfield KR and Gray S: Environmental pollutants,
diet, physical activity, body size, and breast cancer: where do we
stand in research to identify opportunities for prevention? Cancer.
109:2627–2634. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fulda S and Debatin KM: Targeting
apoptosis pathways in cancer therapy. Curr Cancer Drug Targets.
4:569–576. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gamet-Payrastre L, Li P, Lumeau S, Cassar
G, Dupont MA, Chevolleau S, Gasc N, Tulliez J and Tercé F:
Sulforaphane, a naturally occurring isothiocyanate, induces cell
cycle arrest and apoptosis in HT29 human colon cancer cells. Cancer
Res. 60:1426–1433. 2000.PubMed/NCBI
|
|
12
|
Snoek BC, de Wilt LH, Jansen G and Peters
GJ: Role of E3 ubiquitin ligases in lung cancer. World J Clin
Oncol. 4:58–69. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Myatt SS, Brosens JJ and Lam EW: Sense and
sensitivity: FOXO and ROS in cancer development and treatment.
Antioxid Redox Signal. 14:675–687. 2011. View Article : Google Scholar
|
|
14
|
Trachootham D, Alexandre J and Huang P:
Targeting cancer cells by ROS-mediated mechanisms: a radical
therapeutic approach? Nat Rev Drug Discov. 8:579–591. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kong D, Zheng T, Zhang M, Wang D, Du S, Li
X, Fang J and Cao X: Static mechanical stress induces apoptosis in
rat endplate chondrocytes through MAPK and mitochondria-dependent
caspase activation signaling pathways. PLoS One. 8:e694032013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Uchakina ON, Ban H and McKallip RJ:
Targeting hyaluronic acid production for the treatment of leukemia:
treatment with 4-methylumbelliferone leads to induction of
MAPK-mediated apoptosis in K562 leukemia. Leuk Res. 37:1294–1301.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Leber B, Geng F, Kale J and Andrews DW:
Drugs targeting Bcl-2 family members as an emerging strategy in
cancer. Expert Rev Mol Med. 12:e282010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yuan Z, Feng W, Hong J, Zheng Q, Shuai J
and Ge Y: p38MAPK and ERK promote nitric oxide production in
cultured human retinal pigmented epithelial cells induced by high
concentration glucose. Nitric Oxide. 20:9–15. 2009. View Article : Google Scholar
|
|
19
|
Pozarowski P and Darzynkiewicz Z: Analysis
of cell cycle by flow cytometry. Methods Mol Biol. 281:301–311.
2004.PubMed/NCBI
|
|
20
|
Rastogi RP, Singh SP, Häder DP and Sinha
RP: Detection of reactive oxygen species (ROS) by the
oxidant-sensing probe 2′, 7′-dichlorodihydrofluorescein diacetate
in the cyanobacterium Anabaena variabilis PCC 793. Biochem Biophys
Res Commun. 397:603–607. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Laemmli UK: Cleavage of structural
proteins during the assembly of the head of bacteriophage T4.
Nature. 227:680–685. 1970. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kyhse-Andersen J: Electroblotting of
multiple gels: a simple apparatus without buffer tank for rapid
transfer of proteins from polyacrylamide to nitrocellulose. J
Biochem Biophys Methods. 10:203–209. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lan T, Wang L, Xu Q, Liu W, Jin H, Mao W
and Wang X and Wang X: Growth inhibitory effect of Cucurbitacin E
on breast cancer cells. Int J Clin Exp Pathol. 6:1799–1805.
2013.PubMed/NCBI
|
|
24
|
Drexler HG: Review of alterations of the
cyclin-dependent kinase inhibitor INK4 family genes p15, p16, p18
and p19 in human leukemia-lymphoma cells. Leukemia. 12:845–859.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Padua MB and Hansen PJ: Changes in
expression of cell-cycle-related genes in PC-3 prostate cancer
cells caused by ovine uterine serpin. J Cell Biochem.
107:1182–1188. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wyllie AH, Kerr JF and Currie AR: Cell
death: the significance of apoptosis. Int Rev Cytol. 68:251–306.
1980. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cooper WA, Kohonen-Corish MR, Zhuang L,
McCaughan B, Kennedy C, Screaton G, Sutherland RL and Lee CS: Role
and prognostic significance of tumor necrosis factor-related
apoptosis-inducing ligand death receptor DR5 in nonsmall-cell lung
cancer and precursor lesions. Cancer. 113:135–142. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dewaele M, Maes H and Agostinis P:
ROS-mediated mechanisms of autophagy stimulation and their
relevance in cancer therapy. Autophagy. 6:838–854. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Madureira PA, Hill R, Miller VA,
Giacomantonio C, Lee PW and Waisman DM: Annexin A2 is a novel
cellular redox regulatory protein involved in tumorigenesis.
Oncotarget. 2:1075–1093. 2011.PubMed/NCBI
|
|
30
|
Kirshner JR, He S, Balasubramanyam V,
Kepros J, Yang CY, Zhang M, Du Z, Barsoum J and Bertin J:
Elesclomol induces cancer cell apoptosis through oxidative stress.
Mol Cancer Ther. 7:2319–2327. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
You BR and Park WH: Zebularine-induced
apoptosis in Calu-6 lung cancer cells is influenced by ROS and GSH
level changes. Tumour Biol. 34:1145–1153. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Strickertsson JA, Desler C,
Martin-Bertelsen T, Machado AM, Wadstrøm T, Winther O, Rasmussen LJ
and Friis-Hansen L: Enterococcus faecalis infection causes
inflammation, intra-cellular oxphos-independent ROS production, and
DNA damage in human gastric cancer cells. PLoS One. 8:e631472013.
View Article : Google Scholar
|
|
33
|
Lee SJ, Kim MS, Park JY, Woo JS and Kim
YK: 15-Deoxy-delta 12,14-prostaglandin J2 induces apoptosis via
JNK-mediated mitochondrial pathway in osteoblastic cells.
Toxicology. 248:121–129. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lane DP: Cancer. p53, guardian of the
genome. Nature. 358:15–16. 1992. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang Q, Su L, Liu N, Zhang L, Xu W and
Fang H: Cyclin dependent kinase 1 inhibitors: a review of recent
progress. Curr Med Chem. 18:2025–2043. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lee K, Hart MR, Briehl MM, Mazar AP and
Tome ME: The copper chelator ATN-224 induces caspase-independent
cell death in diffuse large B cell lymphoma. Int J Oncol.
45:439–447. 2014.PubMed/NCBI
|