Introduction
Colorectal cancer (CRC), the third leading cause of
cancer-related deaths in Japan, has an etiology linked to numerous
factors including genetic mutation, diet, life style, and
inflammatory process. The human gut is continually colonized by
complex microbial communities in which the combined number of cells
(1011–13 cells/g in the colon) is greater than the total
number of the host cells (1).
Therefore, the human body harbors 10 times more exogenous cells
than human cells. The human gut microbiota becomes relatively
stable around 1 week after birth, begins to resemble that of an
adult after weaning, and once established remains stable over
lifetime (2). It is generally
believed that each healthy individual has his or her own unique gut
microbiota (3,4).
Numerous researchers have catalogued the gut
microbiota of healthy humans and the gut microbiota associated with
inflammatory bowel disease (IBD) (5–7) or
obesity (8–10). In addition, a recent study suggests
that the gut microbiota is associated with CRC development. Several
plausible mechanisms in which the gut microbiota could interface
with CRC have been proposed; for example, inflammation,
DNA-damaging effects, and non-DNA-damaging effects could each be
mechanistically important (11).
There is a considerable amount of research
confirming that the gut microbiota is a primary driver of
inflammation in the colon and the inflammatory environment is
related to CRC development (12).
Microbial dysbiosis (i.e., disturbance of the normal microbial
community) can increase the proportion of facultative anaerobic
bacteria, which include potentially harmful inflammation-inducing
microorganisms. Inflammation driven by such bacteria (e.g.,
Bacteroides fragilis and Streptococcus bovis) is
thought to affect carcinogenesis because these bacteria can
activate immune cells to release promitogenic and proangiogenic
cytokines such as IL-6 and IL-17 (13–15).
In fact, there is epidemiological data that suggest up to 15% of
human cancer incidence is inflammation-associated (16,17).
DNA-damaging effects of microbiota in CRC are
thought to be induced by microbes that produce numerous genotoxic
substances and are thus linked to CRC development. For example,
microbial-derived nitric oxide has the capacity to damage DNA
(18,19). Reactive oxygen species (ROS) are
also powerful instigators of mutation and could contribute to
chromosomal instability and risk of CRC (20,21).
Carcinogenic effects of CRC-associated microbiota,
effects that are unrelated to DNA damage, may be attributable to a
number of bacterial metabolites. For example, hydrogen sulfide
(H2S) has been linked to CRC as a potential
tumor-promoting agent. H2S is produced by
sulfate-reducing commensal bacteria as part of their normal
metabolism (22). Although
H2S does not act as a direct DNA damaging agent, it
modulates proliferation, apoptosis, and differentiation of colonic
epithelial cells (23). Moreover,
the gut microbiota metabolizes different dietary components to
influence CRC development. For example, the gut microbiota
metabolizes proteins from red meat to nitrosamine and heterocycle
amines, and these metabolites are risk factors for CRC development
(24,25). In contrast, a high intake of dietary
fiber has been considered to be protective against CRC development
(26,27); nevertheless, the beneficial effect
of microbial fermentation of fiber and production of butyrate on
CRC development is still an area of substantial controversy.
The gut microbiota and its products are clearly
linked to CRC. However, most research has focused on the
association between the gut microbiota and advanced CRC, rather
than early-stage cancer. Thus, it is not clear whether the gut
micro-biota plays a role at an early stage of colorectal
carcinogenesis. In addition, most studies in this field have been
carried out in Western countries, and it is unknown whether certain
members of the gut microbiota particularly associated with CRC also
exist in the Japanese population, whose dietary habits and
lifestyles are different from those of Western populations. In this
study, we used next-generation sequencing (NGS) subsequent to
terminal restriction fragment length polymorphism (T-RFLP) analysis
to investigate the human gut microbiota in a Japanese population.
We specifically selected patients with carcinoma in adenoma for the
NGS analysis to evaluate possible associations of gut microbiota
with early-stage cancer. We identified several potential bacterial
genera and species uniquely associated with control specimens or
with carcinoma-in-adenoma specimens.
Materials and methods
Human subjects
Subjects who were under 65 years of age and had
undergone colonoscopy at the Mie Prefectural General Medical
Center, Yokkaichi, Japan, between 2012 and 2013 were enrolled in
the study. To evaluate differences in gut microflora via T-RFLP
analysis, the subjects were classified into three groups as
follows: i), control subjects, who had normal colonoscopy; ii),
adenoma patients, who were diagnosed with colon adenoma bases on
the colonoscopy; and iii), cancer patients, who had recently been
diagnosed with CRC. Exclusion criteria for all participants
included current use of antibiotics, history of or current chronic
bowel or liver disease, history of chemotherapy or radiation
therapy, and regular use of immunomodulators (steroids,
interferons, etc.) or probiotics. Assignment of the patients is
shown in Fig. 1. All patients
received an explanation of the procedures and possible risks
associated with the study, and they gave their written informed
consent to participate. This study was performed in accordance with
the Declaration of Helsinki and was approved by our Institutional
Ethics Committee (authorized no. 2011-5; Mie Prefectural General
Medical Center, Yokkaichi, Japan). Stool samples were collected
from each participant prior to polyethylene-glycol preparation of
the bowel for colonoscopy; each sample was stored at 4°C and
submitted to Technosuruga Laboratory (Shizuoka, Japan) for the
T-RFLP analysis, which is described below.
DNA extraction
Fecal samples (~4 mg each) were suspended in a
solution containing 100 mM Tris-HCI, pH 9.0, 40 mM Tris-EDTA, pH
8.0, and 4 M guanidine thiocyanate. A 0.8-ml aliquot of each
suspension was homogenized with zirconia beads in a 2.0-ml screw
cap tube with a FastPrep 24 Instrument (MP Biomedicals, Santa Ana,
CA, USA) run at 5 m/sec for 2 min and placed on ice for 5 min. Each
sample was spun at 5,000 × g for 1 min; an automatic nucleic acid
extractor (Precision System Science, Chiba, Japan) was then used to
extract DNA from a 200-µl aliquot of each sample. MagDEA DNA
200 (GC; Precision System Science) was used as the reagent for
automated nucleic acid extraction.
T-RFLP
The 16S rDNA was amplified from human fecal DNA
using the fluorescent-labeled 516f primer (5′-TGCCAGCAGCCGCGGTA-3′;
Escherichia coli positions 516–532) and 1510r primer
(5′-GGTTACCTTGTTACGACTT-3′; E. coli positions 1,510–1,492).
HotStarTaq DNA polymerase by Gene Amp PCR system 9600 (Applied
Biosystems, Foster City, CA, USA) was used for each amplification
reaction. The amplification program was as follows: preheating at
95°C for 15 min and then 30 cycles of i), denaturation at 95°C for
30 sec, ii), annealing at 50°C for 30 sec, and iii), extension at
72°C for 1 min, and finally, a terminal extension at 72°C for 10
min. Amplified DNA was purified by a MultiScreen PCR96 Filter Plate
(Millipore, Billerica, MA, USA) and verified by electrophoresis.
The restriction enzymes were selected according to Nagashima et
al (28,29). In brief, 16S-rDNA PCR products were
purified and digested with 10 U of BslI (New England
BioLabs, Ipswich, MA, USA) at 55°C for 3 h. An ABI PRISM 3130×l
genetic analyzer was used to analyze the resultant DNA fragments,
namely fluorescent-labeled terminal restriction fragments (T-RFs),
and GeneMapper software (Applied Biosystems) was used to determine
T-RF length and peak area for each sample. T-RFs were divided into
29 operational taxonomic units (OTUs). The OTUs were quantified as
the percentage of individual OTU per total OTU area, which were
expressed as the percentage of the area under the curve (% AUC).
The reference database, Human Fecal Microbiota T-RFLP profiling
(http://www.tecsrg-lab.jp/), was used to
putatively identify the bacteria in each classification unit and
the corresponding OTU.
To evaluate differences in gut microbiota
composition at the species level, samples from six control subjects
and six patients with carcinoma in adenoma were selected for NGS;
IBM SPSS software ver. 22 was used to match control and patient
samples based on age, gender, and BMI. Subjects with carcinoma in
adenoma were selected and subjects with advanced cancer were
excluded to avoid the possibility of gut microbial environment
alterations due to cancer progression.
Illumina library generation
NGS analysis of microbial community structure in
each feces sample was performed with MiSeq (Illumina, San Diego,
CA, USA), as previously described by Takahashi et al
(30). Briefly, the V3–V4 region of
16S rDNA was amplified using 341F (5′-CCTACGGGAGGCAGCAG-3′)
(31) and 806R
(5′-GGACTACHVGGGTWTCTAAT-3′) (32)
primers. In addition to the V3–V4-specific priming regions, these
primers were complementary to standard Illumina forward and reverse
primers. The reverse primer also contained a 6-bp indexing sequence
(CAGATC, ACTTGA, GATCAG, TAGCTT, GGCTAC, CTTGTA, ATCACG, CGATGT,
TTAGGC, TGACCA, ACAGTG and GCCAAT) to allow for multiplexing. The
touchdown PCR method was used with a GeneAmp PCR system 9700
(Applied Biosystems) for thermal cycling. Each PCR reaction mixture
(25 µl) contained 20 ng genomic DNA, 2X MightyAmp Buffer
ver. 2 (Takara), 0.25 µM of each primer, and 1.25 units of
MightyAmp DNA Polymerase (Takara). Each PCR amplification and
preparation of amplicon pool were performed as described by
Takahashi et al (30).
Illumina sequencing and quality
filtering
As recommended by Illumina for the pooling of two
libraries and described by Takahashi et al (30), each multiplexed library pool was
spiked with 30% PhiX control to improve base calling during
sequencing. Sequencing was conducted using a paired-end, 2×251-bp
cycle run on an Illumina MiSeq sequencing system and MiSeq reagent
Nano kit version 2 (500 cycle) chemistry. Paired-end sequencing
with read lengths of ~251 bp was performed. After demultiplexing, a
clear overlap in the paired-end reads was observed. This overlap
allowed paired reads to be joined together with the fastqjoin
program (http://code.google.com/p/ea-utils/). Only reads that
had quality value (QV) scores of ≥20 for >99% of the sequence
were extracted for further analysis. All sequences with ambiguous
base calls were discarded (30).
Bioinformatics analysis
Metagenome@Kin software (World Fusion Co., Ltd.,
Tokyo, Japan) was used to conduct homology searches of the
TechnoSuruga Lab Microbial Identification Database DB-BA9.0
(TechnoSuruga Laboratory Co., Ltd., Shizuoka, Japan), which
contains only bacteria with standing in the taxonomic nomenclature,
with the 16S rDNA sequences.
Statistical analysis
Data were analyzed using the Kruskal-Wallis test or
the Mann-Whitney test (two-sided) for continuous variables and
Fisher's exact test for categorical variables using IBM SPSS
software ver. 22. P-values <0.05 were considered
significant.
Results
Differences in bacterial community
profiles between control, adenoma, and cancer subjects as
determined by T-RFLP analysis
Demographic and clinical characteristics of the
subject groups are shown in Table
I. A total of 49 control subjects, 50 adenoma subjects, and 9
CRC subjects (3 with invasive cancer and 6 with carcinoma in
adenoma) were enrolled in this study. Blood test results showed
that total cholesterol and high-density-lipoprotein cholesterol
levels were significantly lower in the cancer subjects. The average
age and body mass index of the cancer subjects were each higher
than those of the control and adenoma subjects. Differences in
bacterial flora between the three groups are summarized in Table II. There were no significant
differences in bacterial composition between each pair of
groups.
 | Table IDemographic and clinical
characteristics of the study groups. |
Table I
Demographic and clinical
characteristics of the study groups.
| Control (n=49) | Adenoma (n=50) | Cancer
(n=9)b | P-value |
|---|
| Age (years)a | 48.8±8.2 | 53.5±9.3 | 54.3±7.9 | 0.011 |
| Gender, male; n
(%) | 21 (42.9) | 28 (56) | 4 (44.4) | 0.399 |
| BMI
(kg/m2)a | 22.5±3.7 | 24.2±3.9 | 24.4±2.8 | 0.030 |
| Constipation; yes,
n (%) | 11 (22.4) | 8 (16.0) | 5 (55.6) | 0.042 |
| Alcohol intake;
yes, n (%) | 23 (48.9) | 26 (53.1) | 3 (33.3) | 0.646 |
| Smoking; yes, n
(%) | 9 (18.8) | 13 (26.5) | 3 (33.3) | 0.480 |
| Laboratory
dataa | | | | |
| HbA1c (JDS; %) | 5.4±0.8 | 5.4±0.6 | 5.4±0.7 | 0.219 |
| Total cholesterol
(mg/dl) | 206.2±34.7 | 195.2±32.1 | 174.6±39.5 | 0.021 |
| Triglyceride
(mg/dl) | 113.9±85.5 | 128.7±80.0 | 135.0±104.0 | 0.229 |
| HDL-cholesterol
(mg/dl) | 70.7±22.1 | 64.0±16.9 | 52.1±14.5 | 0.028 |
| Table IIDifferences in bacterial flora based
on T-RFLP analysis. |
Table II
Differences in bacterial flora based
on T-RFLP analysis.
| Control | Adenoma | Cancer | P-value |
|---|
|
Bifidobacterium | 7.8±7.6 | 8.1±7.4 | 5.6±5.5 | 0.838 |
|
Lactobacillales | 5.7±8.1 | 6.3±8.9 | 2.3±2.2 | 0.516 |
|
Bacteroides | 40.1±12.9 | 37.5±15.0 | 39.1±7.0 | 0.835 |
|
Prevotella | 2.5±6.8 | 2.7±7.2 | 0.6±1.1 | 0.637 |
|
Clostridium | 8.0±5.4 | 7.9±7.8 | 6.9±5.7 | 0.662 |
| cluster IV | | | | |
|
Clostridium | 21.5±7.9 | 21.6±7.5 | 22.4±10.2 | 0.979 |
| subcluster
XIVa | | | | |
|
Clostridium | 2.0±4.0 | 1.4±3.0 | 2.9±2.5 | 0.144 |
| cluster XI | | | | |
|
Clostridium | 1.7±2.4 | 2.0±1.8 | 1.8±2.8 | 0.215 |
| cluster XVIII | | | | |
Differences in bacterial communities
between control and carcinoma in adenoma subjects by 16S rRNA
sequencing
Our T-RFLP analysis showed no significant
differences in bacterial population between the control, adenoma,
and cancer groups. However, in order to determine the possible
presence of bacteria correlated with health and cancer, we selected
12 subjects (six control and six carcinoma in adenoma) from the
initial groups for NGS (Table
III). Using our primer set and MiSeq platform combination, an
average of 24,084 reads were obtained for each sequencing reaction.
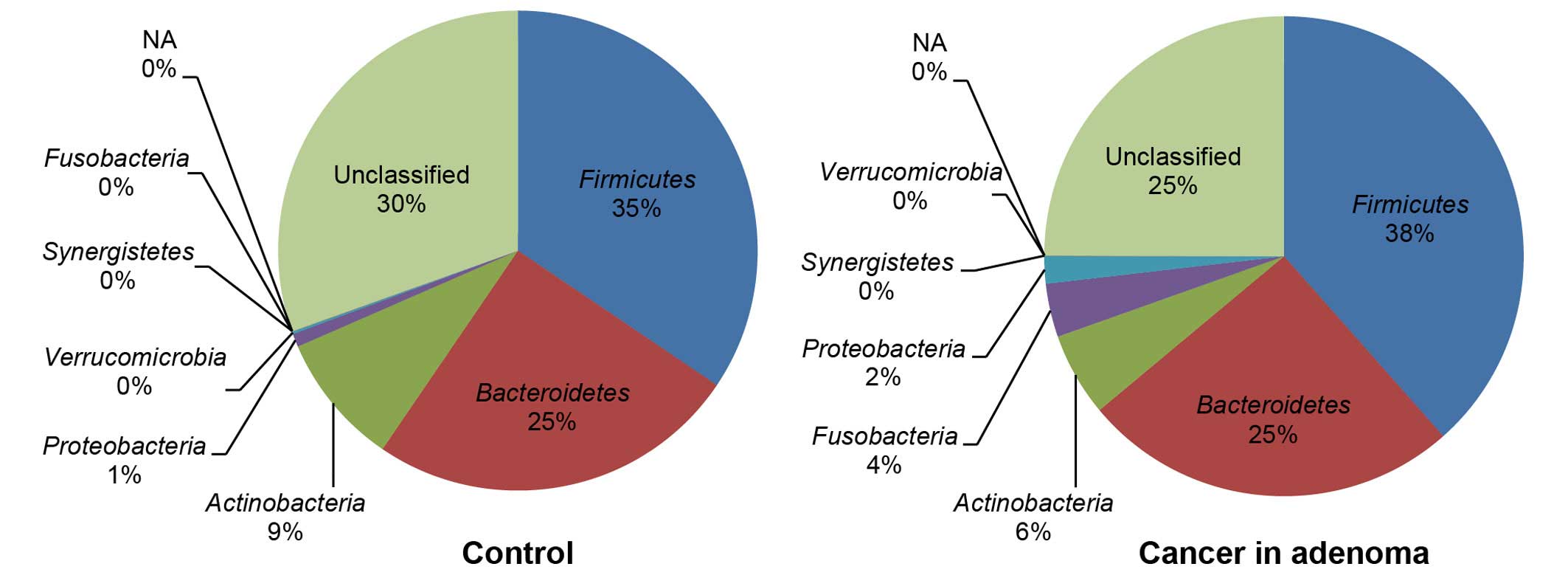
Fig. 2 shows the phylotype
distribution for individual subjects in this study. The composition
and relative abundance of the major bacterial phyla were similar,
with Bacteroidetes and Firmicutes being the dominant
phyla. However, after dividing the samples into two groups (control
vs. carcinoma in adenoma) and performing statistical analyses, a
significant increase in the proportion of Fusobacteria
(control 0% vs. carcinoma in adenoma 4%) was observed in the
carcinoma-in-adenoma group relative to the control group (Fig. 3). There were no between-group
differences with regard to other bacteria.
| Table IIICharacteristics of the study subjects
participating in next-generation sequencing analysis. |
Table III
Characteristics of the study subjects
participating in next-generation sequencing analysis.
| Participant ID | Health status
Gender | Age (years) | BMI | Tumor size
(mm) | Tumor location |
|---|
| N1 | Healthy M | 46 | 25.1 | | |
| N2 | Healthy M | 39 | 19.6 | | |
| N3 | Healthy F | 55 | 24.7 | | |
| N4 | Healthy F | 49 | 20.3 | | |
| N5 | Healthy M | 56 | 25.63 | | |
| N6 | Healthy M | 57 | 23.15 | | |
| C1 | Cancer M | 48 | 24.5 | 10 | Sigmoid |
| C2 | Cancer F | 64 | 26.9 | 20 | Sigmoid |
| C3 | Cancer M | 40 | 20.5 | 13 | Sigmoid |
| C4 | Cancer M | 62 | 24.9 | 20 | Sigmoid |
| C5 | Cancer M | 61 | 24.3 | 15 | Sigmoid |
| C6 | Cancer F | 49 | 19.46 | 10 | Descending |
Comparison of microbiomes at the genus
level
Genus-level analyses identified one bacterial genus
that was significantly associated with the control group
(Slackia), and four bacterial genera were significantly
associated with the carcinoma-in-adenoma group (Actinomyces,
Atopobium, Fusobacterium, and Heamophilus)
(Table IV).
| Table IVBacterial genera with significantly
different group-specific representation. |
Table IV
Bacterial genera with significantly
different group-specific representation.
| Ave. N (%) | Ave. C (%) | P-valuea |
|---|
|
Actinomyces | 0.022 | 0.116 | 0.037 |
|
Atopobium | ND | 0.005 | 0.022 |
|
Fusobacterium | 0.004 | 3.84 | 0.004 |
|
Heamophilus | 0.002 | 0.027 | 0.020 |
| Slackia | 0.162 | 0.009 | 0.049 |
Comparison of microbiomes at the species
level
Species-level analyses identified one bacterial
species (Eubacterium coprostanoligens) that was
significantly associated with the control group and eight bacterial
species (Actinomyces odontolyticus, Bacteroides
fragilis, Clostridium nexile, Fusobacterium
varium, Heamophilus parainfluenzae, Prevotella
stercorea, Streptococcus gordonii, and Veillonella
dispar) that were significantly associated with the
carcinoma-in-adenoma group (Table
V).
| Table VBacterial species with significantly
different group-specific representation. |
Table V
Bacterial species with significantly
different group-specific representation.
| Ave. N (%) | Ave. C (%) | P-valuea |
|---|
|
Actinomyces | ND | 0.036 | 0.007 |
|
odontolyticus | | | |
| Bacteroides
fragilis | 0.015 | 0.658 | 0.0046 |
| Clostridium
nexile | 0.067 | 0.661 | 0.036 |
|
Eubacterium | 0.650 | ND | 0.022 |
|
coprostanoligens | | | |
| Fusobacterium
varium | ND | 0.268 | 0.022 |
|
Heamophilus | 0.001 | 0.022 | 0.020 |
|
parainfluenzae | | | |
| Prevotella
stercorea | ND | 1.186 | 0.022 |
| Streptococcus
gordonii | 0.002 | 0.031 | 0.014 |
| Veillonella
dispar | 0.004 | 0.198 | 0.042 |
Most notably, the proportions of Actinomyces
odontolyticus, Bacteriodes fragilis, and Heamophilus
parainfluenzae were significantly higher in feces from
carcinoma-in-adenoma subjects than in those from control subjects;
in fact, these bacteria were barely detectable in feces from
control subjects (Fig. 4A–C). In
contrast, the proportions of Eubacterium coprostanoligens
were significantly higher in feces from control subjects than in
those from carcinoma-in-adenoma subjects; this bacteria was barely
detectable in feces of carcinoma-in-adenoma subjects (Fig. 4D).
Although the genus Slackia was significantly
associated with control subjects (Fig.
5A), there were no statistically significant between-group
differences in the relative proportion of each individual
Slackia species. Slackia species as a whole, however,
were more abundant in feces from control subjects compared with
that from carcinoma-in-adenoma subjects (Fig. 5B).
Discussion
Using NGS, we found that the gut microbiota differs
between control and carcinoma-in-adenoma subjects; however, our
initial T-RFLP analysis did not reveal any statistically
significant differences in relative proportions of bacterial flora
between control, adenoma, and carcinoma-in-adenoma subjects. We
identified several potential gut microbial members significantly
associated with the control and carcinoma-in-adenoma groups.
Phylum-level analyses revealed that the relative
proportion of Fusobacterium was significantly higher in
carcinoma-in-adenoma subjects than in control subjects.
Fusobacterium has been studied recently because of its
correlations with CRC (33,34). There are two studies (35,36)
that investigated the mechanisms by which Fusobacterium
nucleatum in the gut could be associated with CRC. The first
study was conducted by Kostic et al; it suggested that F.
nucleatum induced a nuclear factor-κB (NF-κB)-driven
proinflammatory response to promote CRC (35). The second study was by Rubinstein
et al; it provided mechanistic insights, most notably that
the actions of Fusobacterium spp. were presumably mediated
via binding of FadA, a virulence factor expressed on the bacterial
cell surface, to receptors on host epithelial cells; this
FadA-receptor binding seemed to modify barrier function, increase
inflammation through the modulation of the tumor microenvironment,
and activate pro-oncogenic signals to promote CRC (36).
Genus-level and species-level analyses showed that
the genus Slackia and the species Eubacterium
coprostanoligens were present in significantly higher
proportions in control subjects compared with carcinoma-in-adenoma
subjects. Slackia is one of the few characterized
equol-forming gut bacteria isolated from humans (37). Equol is produced from daidzein (a
soy isoflavone) by intestinal bacteria in some, but not all, adults
(38). An individual's capacity for
equol production depends on the representation of equol-forming
bacteria in the individual's intestine (39). Equol is produced in only 20–30% of
adults who consume soy diets containing isoflavones in Western
countries; in contrast, it is produced in no less than 50–60% of
adults in Asian countries, and these adults more commonly consume
soy diets (40–42). Soy isoflavones are often referred to
as phytoestrogens; they have an estrogen-like chemical structure
and can bind to estrogen receptors (43). Equol is the active form of soy
isoflavones in the human intestine, and equol shows a stronger
estrogen-like activity than daizein because it affects
hormone-dependent diseases (44).
Equol is anticipated to have a protective effect on prostate cancer
development and to reduce the risk of mammary tumors (45,46).
We expect that the equol-forming bacteria Slackia have a
preventive effect against CRC, as well.
Eubacterium, a beneficial genus of fecal
bacteria, includes many species that produce butyrate (47,48).
Butyrate is regarded as the most important nutrient for epithelial
cells of the colon, and it plays an essential role in the energy
metabolism and normal development of these cells (49). Several studies have shown that
butyrate is a beneficial inhibitor of colon carcinoma cell
proliferation because it induces apoptosis in human colon carcinoma
cells (50–52). Eubacterium coprostanoligens
is a cholesterol-reducing bacterium (53). Cholesterol-reducing bacteria convert
cholesterol to coprostanol, which is not absorbed by the human
gastrointestinal system, thereby leading to reduced cholesterol
levels. There is strong epidemiological evidence that links high
fat consumption to increased risk of CRC (54,55).
We thus expect Eubacterium coprostanoligens to be another
prospective inhibitor of CRC.
Genus-level analyses showed that four genera
(Actinomyces, Atopobium, Fusobacterium, and
Haemophilus) were present in significantly higher
proportions in carcinoma-in-adenoma subjects than in control
subjects. Species-level analyses showed that eight species
(Actinomyces odontolyticus, Bacteroides fragilis,
Clostridium nexile, Fusobacterium varium,
Haemophilus parainfluenzae, Prevotella stercorea,
Streptococcus gordonii, and Veillonella dispar) were
present in significantly higher proportions in carcinoma-in-adenoma
subjects than in control subjects. Here we focused on only three of
these species (Actinomyces odontolyticus, Bacteroides
fragilis, Haemophilus parainfluenzae sp); each was
highly represented in carcinoma-in-adenoma subjects, but barely
detected in control subjects.
Actinomyces odontolyticus are often present
in the oral cavity and gastrointestinal tract of healthy humans.
Some Actinomyces spp. are known to be opportunist pathogens
associated with several colon-related diseases such as CRC and
Crohn's disease. Bacteroides fragilis is a Gram-negative
obligate anaerobe persistently preset in the colon of nearly all
humans. It accounts for only 0.5% of the human gut micro-biota;
nevertheless, it has enterotoxigenicity and is considered to be
pathogen important to CRC. Chronic inflammation may lead to the
hypermethylation of DNA and drive cells to malignancy (56). Persistent enterotoxigenic
Bacteroides fragilis (ETBF) infection may increase the risk
of colon carcinogenesis (57).
Haemophilus parainfluenzae is a commensal species, which
belongs to the phylum Proteobacteria. It is an opportunistic
pathogen that may induce invasive infections such as pneumonia and
endocarditis.
The control subjects in our study harbored
beneficial bacterial species, whereas the carcinoma-in-adenoma
subjects harbored harmful bacteria species that could act as
opportunist pathogens and/or inflammation drivers. In the case of
progressive CRC, the colonic environment can be modified by
multiple factors such as epithelial cell apoptosis and cancer
cachexia; therefore, it is often complicated whether the current
microbial environment is a sequel of CRC or the gut microbiota is a
driver of CRC development by way of inflammatory responses.
However, the fact that the gut microbial profiles differed
significantly even between control subjects and
carcinoma-in-adenoma (i.e., relatively-early-stage cancer) subjects
on the genus and species levels suggested that the microbial
environment including the gut microbiota was an important etiologic
factor for CRC. In order to determine the exact triggers of CRC, we
should also carefully observe non-CRC patients harboring
inflammation-driving microbes in a future long-term study of CRC
progression.
We acknowledge this research was limited to the
characterization of microbiota and that stool metabolites were not
analyzed. We believe further related research that includes
analysis of stool metabolites will definitely improve our
understanding of the mechanisms that lead to CRC.
In conclusion, the results of the present study in a
Japanese population showed that gut microbiota differed between
control and carcinoma-in-adenoma subjects. In particular, the
results suggested that the gut microbiota served as a driver of
carcinogenesis because changes in the composition of the gut
microbiota were observed even in carcinoma in adenoma, which is an
early-stage cancer. However, further study will be necessary to
clarify the precise mechanisms by which the gut microbiota drives
carcinogenesis and to identify the cancer-associated microbial
members. An improved understanding of mechanisms that cause gut
microbiota metabolites to interface with carcinogenesis should lead
to improved diagnostic, preventative, and therapeutic approaches;
for example, probiotics may become useful as more natural and less
disruptive treatments for the prevention of CRC and/or other
GI-related disorders.
Acknowledgments
The authors appreciate TechnoSuruga Laboratory Co.,
Ltd. (Shizuoka, Japan) for technical assistance.
Abbreviations:
|
T-RFLP
|
terminal restriction fragment length
polymorphism
|
|
CRC
|
colorectal cancer
|
|
NGS
|
next-generation sequencing
|
References
|
1
|
Savage DC: Microbial ecology of the
gastrointestinal tract. Annu Rev Microbiol. 31:107–133. 1977.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mitsuoka T and Hayakawa K: The fecal flora
in man. I. Composition of the fecal flora of various age groups.
Zentralbl Bakteriol Orig A. 223:333–342. 1973.In German. PubMed/NCBI
|
|
3
|
Zoetendal EG, Akkermans AD and De Vos WM:
Temperature gradient gel electrophoresis analysis of 16S rRNA from
human fecal samples reveals stable and host-specific communities of
active bacteria. Appl Environ Microbiol. 64:3854–3859.
1998.PubMed/NCBI
|
|
4
|
Rajilić-Stojanović M, Heilig HG, Tims S,
Zoetendal EG and de Vos WM: Long-term monitoring of the human
intestinal microbiota composition. Environ Microbiol. 15:1146–1159.
2013. View Article : Google Scholar
|
|
5
|
Frank DN, St Amand AL, Feldman RA,
Boedeker EC, Harpaz N and Pace NR: Molecular-phylogenetic
characterization of microbial community imbalances in human
inflammatory bowel diseases. Proc Natl Acad Sci USA.
104:13780–13785. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Martinez C, Antolin M, Santos J, Torrejon
A, Casellas F, Borruel N, Guarner F and Malagelada JR: Unstable
composition of the fecal microbiota in ulcerative colitis during
clinical remission. Am J Gastroenterol. 103:643–648. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Morgan XC, Tickle TL, Sokol H, Gevers D,
Devaney KL, Ward DV, Reyes JA, Shah SA, LeLeiko N, Snapper SB, et
al: Dysfunction of the intestinal microbiome in inflammatory bowel
disease and treatment. Genome Biol. 13:R792012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini
V, Mardis ER and Gordon JI: An obesity-associated gut microbiome
with increased capacity for energy harvest. Nature. 444:1027–1031.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Duncan SH, Lobley GE, Holtrop G, Ince J,
Johnstone AM, Louis P and Flint HJ: Human colonic microbiota
associated with diet, obesity and weight loss. Int J Obes.
32:1720–1724. 2008. View Article : Google Scholar
|
|
10
|
Schwiertz A, Taras D, Schäfer K, Beijer S,
Bos NA, Donus C and Hardt PD: Microbiota and SCFA in lean and
overweight healthy subjects. Obesity (Silver Spring). 18:190–195.
2010. View Article : Google Scholar
|
|
11
|
Irrazábal T, Belcheva A, Girardin SE,
Martin A and Philpott DJ: The multifaceted role of the intestinal
microbiota in colon cancer. Mol Cell. 54:309–320. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schwabe RF and Jobin C: The microbiome and
cancer. Nat Rev Cancer. 13:800–812. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ernst M, Najdovska M, Grail D,
Lundgren-May T, Buchert M, Tye H, Matthews VB, Armes J, Bhathal PS,
Hughes NR, et al: STAT3 and STAT1 mediate IL-11-dependent and
inflammation-associated gastric tumorigenesis in gp130 receptor
mutant mice. J Clin Invest. 118:1727–1738. 2008.PubMed/NCBI
|
|
14
|
Wu S, Rhee KJ, Albesiano E, Rabizadeh S,
Wu X, Yen HR, Huso DL, Brancati FL, Wick E, McAllister F, et al: A
human colonic commensal promotes colon tumorigenesis via activation
of T helper type 17 T cell responses. Nat Med. 15:1016–1022. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Winter SE, Lopez CA and Bäumler AJ: The
dynamics of gut-associated microbial communities during
inflammation. EMBO Rep. 14:319–327. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kuper H, Adami HO and Trichopoulos D:
Infections as a major preventable cause of human cancer. J Intern
Med. 248:171–183. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mantovani A, Garlanda C and Allavena P:
Molecular pathways and targets in cancer-related inflammation. Ann
Med. 42:161–170. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lundberg JO, Weitzberg E, Cole JA and
Benjamin N: Nitrate, bacteria and human health. Nat Rev Microbiol.
2:593–602. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Belcheva A, Green B, Weiss A, Streutker C
and Martin A: Elevated incidence of polyp formation in
APC(Min/+)Msh2−/− mice is independent of
nitric oxide-induced DNA mutations. PLoS One. 8:e652042013.
View Article : Google Scholar
|
|
20
|
Cooke MS, Evans MD, Dizdaroglu M and Lunec
J: Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB
J. 17:1195–1214. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Evans MD, Dizdaroglu M and Cooke MS:
Oxidative DNA damage and disease: Induction, repair and
significance. Mutat Res. 567:1–61. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Christl SU, Scheppach W and Kasper H:
Hydrogen metabolism in the large intestine - physiology and
clinical implications. Z Gastroenterol. 33:408–413. 1995.In German.
PubMed/NCBI
|
|
23
|
Deplancke B, Finster K, Graham WV, Collier
CT, Thurmond JE and Gaskins HR: Gastrointestinal and microbial
responses to sulfate-supplemented drinking water in mice. Exp Biol
Med (Maywood). 228:424–433. 2003.
|
|
24
|
Hughes R, Cross AJ, Pollock JR and Bingham
S: Dose-dependent effect of dietary meat on endogenous colonic
N-nitrosation. Carcinogenesis. 22:199–202. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Norat T and Riboli E: Meat consumption and
colorectal cancer: A review of epidemiologic evidence. Nutr Rev.
59:37–47. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hague A, Manning AM, Hanlon KA, Huschtscha
LI, Hart D and Paraskeva C: Sodium butyrate induces apoptosis in
human colonic tumour cell lines in a p53-independent pathway:
Implications for the possible role of dietary fibre in the
prevention of large-bowel cancer. Int J Cancer. 55:498–505. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Heerdt BG, Houston MA and Augenlicht LH:
Potentiation by specific short-chain fatty acids of differentiation
and apoptosis in human colonic carcinoma cell lines. Cancer Res.
54:3288–3293. 1994.PubMed/NCBI
|
|
28
|
Nagashima K, Mochizuki J, Hisada T, Suzuki
S and Shimomura K: Phylogenetic analysis of 16S ribosomal RNA gene
sequences from human fecal microbiota and improved utility of
terminal restriction fragment length polymorphism profiling. Biosci
Microflora. 25:99–107. 2006. View Article : Google Scholar
|
|
29
|
Nagashima K, Hisada T, Sato M and
Mochizuki J: Application of new primer-enzyme combinations to
terminal restriction fragment length polymorphism profiling of
bacterial populations in human feces. Appl Environ Microbiol.
69:1251–1262. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Takahashi S, Tomita J, Nishioka K, Hisada
T and Nishijima M: Development of a prokaryotic universal primer
for simultaneous analysis of Bacteria and Archaea using
next-generation sequencing. PLoS One. 9:e1055922014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Muyzer G, de Waal EC and Uitterlinden AG:
Profiling of complex microbial populations by denaturing gradient
gel electrophoresis analysis of polymerase chain reaction-amplified
genes coding for 16S rRNA. Appl Environ Microbiol. 59:695–700.
1993.PubMed/NCBI
|
|
32
|
Caporaso JG, Lauber CL, Walters WA,
Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N and Knight R:
Global patterns of 16S rRNA diversity at a depth of millions of
sequences per sample. Proc Natl Acad Sci USA. 108(Suppl 1):
4516–4522. 2011. View Article : Google Scholar :
|
|
33
|
Castellarin M, Warren RL, Freeman JD,
Dreolini L, Krzywinski M, Strauss J, Barnes R, Watson P,
Allen-Vercoe E, Moore RA, et al: Fusobacterium nucleatum infection
is prevalent in human colorectal carcinoma. Genome Res. 22:299–306.
2012. View Article : Google Scholar :
|
|
34
|
Kostic AD, Gevers D, Pedamallu CS, Michaud
M, Duke F, Earl AM, Ojesina AI, Jung J, Bass AJ, Tabernero J, et
al: Genomic analysis identifies association of Fusobacterium with
colorectal carcinoma. Genome Res. 22:292–298. 2012. View Article : Google Scholar :
|
|
35
|
Kostic AD, Chun E, Robertson L, Glickman
JN, Gallini CA, Michaud M, Clancy TE, Chung DC, Lochhead P, Hold
GL, et al: Fusobacterium nucleatum potentiates intestinal
tumorigenesis and modulates the tumor-immune microenvironment. Cell
Host Microbe. 14:207–215. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rubinstein MR, Wang X, Liu W, Hao Y, Cai G
and Han YW: Fusobacterium nucleatum promotes colorectal
carcinogenesis by modulating E-cadherin/β-catenin signaling via its
FadA adhesin. Cell Host Microbe. 14:195–206. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Matthies A, Blaut M and Braune A:
Isolation of a human intestinal bacterium capable of daidzein and
genistein conversion. Appl Environ Microbiol. 75:1740–1744. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Setchell KD and Clerici C: Equol: History,
chemistry, and formation. J Nutr. 140:1355S–1362S. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Watanabe S, Yamaguchi M, Sobue T,
Takahashi T, Miura T, Arai Y, Mazur W, Wähälä K and Adlercreutz H:
Pharmacokinetics of soybean isoflavones in plasma, urine and feces
of men after ingestion of 60 g baked soybean powder (kinako). J
Nutr. 128:1710–1715. 1998.PubMed/NCBI
|
|
40
|
Setchell KD and Cole SJ: Method of
defining equol-producer status and its frequency among vegetarians.
J Nutr. 136:2188–2193. 2006.PubMed/NCBI
|
|
41
|
Setchell KD, Zhao X, Shoaf SE and Ragland
K: The pharmacokinetics of S-(−)equol administered as SE5-OH
tablets to healthy postmenopausal women. J Nutr. 139:2037–2043.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Song KB, Atkinson C, Frankenfeld CL,
Jokela T, Wähälä K, Thomas WK and Lampe JW: Prevalence of
daidzein-metabolizing phenotypes differs between Caucasian and
Korean American women and girls. J Nutr. 136:1347–1351.
2006.PubMed/NCBI
|
|
43
|
Setchell KD: Phytoestrogens: The
biochemistry, physiology, and implications for human health of soy
isoflavones. Am J Clin Nutr. 68(Suppl 6): 1333S–1346S.
1998.PubMed/NCBI
|
|
44
|
Cai Y, Guo K, Chen C, Wang P, Zhang B,
Zhou Q, Mei F and Su Y: Soya isoflavone consumption in relation to
carotid intimamedia thickness in Chinese equol excretors aged 40–65
years. Br J Nutr. 108:1698–1704. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lund TD, Munson DJ, Haldy ME, Setchell KD,
Lephart ED and Handa RJ: Equol is a novel anti-androgen that
inhibits prostate growth and hormone feedback. Biol Reprod.
70:1188–1195. 2004. View Article : Google Scholar
|
|
46
|
Brown NM, Belles CA, Lindley SL,
Zimmer-Nechemias L, Witte DP, Kim MO and Setchell KD: Mammary gland
differentiation by early life exposure to enantiomers of the soy
isoflavone metabolite equol. Food Chem Toxicol. 48:3042–3050. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Uematsu H, Sato N, Hossain MZ, Ikeda T and
Hoshino E: Degradation of arginine and other amino acids by
buty-rate-producing asaccharolytic anaerobic Gram-positive rods in
periodontal pockets. Arch Oral Biol. 48:423–429. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Barcenilla A, Pryde SE, Martin JC, Duncan
SH, Stewart CS, Henderson C and Flint HJ: Phylogenetic
relationships of butyrate-producing bacteria from the human gut.
Appl Environ Microbiol. 66:1654–1661. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wong JM, de Souza R, Kendall CW, Emam A
and Jenkins DJ: Colonic health: Fermentation and short chain fatty
acids. J Clin Gastroenterol. 40:235–243. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Dronamraju SS, Coxhead JM, Kelly SB and
Mathers JC: Differential antineoplastic effects of butyrate in
cells with and without a functioning DNA mismatch repair. Nutr
Cancer. 62:105–115. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ooi CC, Good NM, Williams DB, Lewanowitsch
T, Cosgrove LJ, Lockett TJ and Head RJ: Efficacy of butyrate
analogues in HT-29 cancer cells. Clin Exp Pharmacol Physiol.
37:482–489. 2010. View Article : Google Scholar
|
|
52
|
Roy MJ, Dionne S, Marx G, Qureshi I, Sarma
D, Levy E and Seidman EG: In vitro studies on the inhibition of
colon cancer by butyrate and carnitine. Nutrition. 25:1193–1201.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Freier TA, Beitz DC, Li L and Hartman PA:
Characterization of Eubacterium coprostanoligenes sp nov, a
cholesterol-reducing anaerobe. Int J Syst Bacteriol. 44:137–142.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Stadler J, Stern HS, Yeung KS, McGuire V,
Furrer R, Marcon N and Bruce WR: Effect of high fat consumption on
cell proliferation activity of colorectal mucosa and on soluble
faecal bile acids. Gut. 29:1326–1331. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ou J, Carbonero F, Zoetendal EG, DeLany
JP, Wang M, Newton K, Gaskins HR and O'Keefe SJ: Diet, microbiota,
and microbial metabolites in colon cancer risk in rural Africans
and African Americans. Am J Clin Nutr. 98:111–120. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Mantovani A, Allavena P, Sica A and
Balkwill F: Cancer-related inflammation. Nature. 454:436–444. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Toprak NU, Yagci A, Gulluoglu BM, Akin ML,
Demirkalem P, Celenk T and Soyletir G: A possible role of
Bacteroides fragilis enterotoxin in the aetiology of colorectal
cancer. Clin Microbiol Infect. 12:782–786. 2006. View Article : Google Scholar : PubMed/NCBI
|