Introduction
Hepatocellular carcinoma (HCC) is the second most
common cause of cancer-related deaths worldwide, with an estimated
782,500 new liver cancer cases and 745,500 related deaths occurring
worldwide in 2012 (1). Chemotherapy
is a reasonable treatment option for patients with advanced HCC.
However, traditional chemotherapy has shown modest efficacy with
severe side-effects. Therefore, it is necessary to identify new
agents or therapeutic strategies to improve the treatment of liver
cancer. One of the important methods is to understand the detailed
effect of traditional drugs for drug repositioning (2).
Chloroquine (CQ) is a classic drug for the treatment
of malarial (3). Recently, CQ has
been widely used as an enhancing agent in cancer therapies and has
a synergistic effect with ionizing radiation or chemotherapeutic
agents in a cancer-specific manner (4–9). In
addition, CQ was reported to inhibit cell growth and/or induce cell
death in several tumor models (10–13),
and showed lower toxicity to non-tumorigenic epithelial cells
(14). However, the effects of
single treatment of CQ on liver cancer have not been
investigated.
In the present study, we examined the effects of CQ
on the growth and viability of liver cancer cells in vitro
and in vivo, and revealed that in vitro treatment of
liver cancer cells with CQ inhibited cell proliferation and
viability, induced G0/G1 cell cycle arrest, DNA damage and
apoptosis with upregulation of pro-apoptotic protein Bim. Moreover,
administration of CQ to tumor-bearing mice inhibited the tumor
growth in an orthotopic xenograft model of liver cancer.
Materials and methods
Cell lines, culture and reagents
Human liver cancer cell lines HepG2 and Huh7 were
cultured in Dulbecco's modified Eagle's medium (DMEM) (HyClone),
and were supplemented with 10% fetal bovine serum (FBS) (Biochrom
AG) at 37°C with 5% CO2. CQ was purchased from
Sigma-Aldrich and was dissolved in phosphate-buffered saline
(PBS).
Cell proliferation and clonogenic
assays
HepG2 and Huh7 cells were seeded into 96-well plates
(2.5×103 cells/well) and were treated with different
concentrations of CQ as indicated for 24, 48 or 72 h. Cell
proliferation was determined using the ATPLite Luminescence assay
kit (Perkin-Elmer) according to the manufacturer's protocol. Cell
Counting Kit-8 (CCK-8) (Dojindo) was used to quantify drug-induced
cytotoxicity as follows. Cells were seeded in 96-well plates,
exposed to different concentrations of CQ for 72 h and were then
treated with CCK-8 reagent for assessment of cytotoxicity.
For the clonogenic assay, cells were seeded into
6-well plates with 500 cells/well in triplicate, treated with the
indicated concentrations of CQ for 24 h, and then washed with PBS
twice, followed by incubation for 9 days. The colonies formed were
fixed, stained and counted. Colonies with >50 cells were
counted.
Western blotting
HepG2 and Huh7 cell lysates treated with CQ were
prepared for western blot analysis, using antibodies against
cleaved caspase-3, cleaved poly(ADP-ribose) polymerase (PARP), the
Pro-Apoptosis Bcl-2 Family Antibody Sampler Kit, the Pro-Survival
Bcl-2 Family Antibody Sampler kit, IAP Family Antibody Sampler kit
and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Cell
Signaling, Boston, MA, USA).
Cell cycle analysis
Cells treated with CQ at the indicated
concentrations were harvested, fixed in 70% ethanol at −20°C, and
were then stained with propidium iodide (PI; 50 µg/ml)
containing RNase A (30 µg/ml) (both from Sigma) at 37°C for
30 min. The cells were then analyzed for cell cycle profile by flow
cytometry (FACScan; Becton-Dickinson). Data were analyzed with
ModFit LT software (verity).
Apoptosis assay
HepG2 and Huh7 cells were treated with CQ for 72 h.
Apoptosis was determined with the Annexin V-FITC/PI apoptosis kit
(Biovision, Inc. Milpitas, CA, USA) as per the manufacturer's
instructions. The early apoptotic (Annexin V-FITC-positive) and
necrotic/late apoptotic (Annexin V-FITC-positive, PI-positive)
cells were quantified as apoptotic cells. Caspase-3 activity was
assessed by the CaspGLOW Fluorescein Active Caspase-3 Staining kit
(Biovision, Inc.) according to the manufacturer's instructions.
Evaluation of mitochondrial membrane
depolarization
HepG2 and Huh7 cells were treated with CQ at the
indicated concentrations. Mitochondrial membrane depolarization was
detected with the mitochondrial membrane potential assay kit with
JC-1 according to the manufacturer's protocol (Yeasen Inc.,
Shanghai, China). The data were acquired and analyzed by flow
cytometry as previously described (15).
Immunofluorescence
HepG2 and Huh7 cells were plated on chamber slides
and treated with CQ for 36 h. Cells were fixed with 4%
paraformaldehyde, permeabilized using 0.2% Triton X-100, and
incubated overnight with the γ-H2AX antibody (Cell Signaling). Goat
anti-rabbit Alexa 488 fluorescent secondary antibody was used to
visualize γ-H2AX foci, and DAPI was used to visualize the
nuclei.
Antitumor effect of CQ in vivo
An orthotopic xenograft model of liver cancer was
established by AntiCancer Biotech as previously described (15). Briefly, HepG2-GFP human liver cancer
tissues that originated from subcutaneous tumors of nude mice were
harvested and carefully inspected to remove necrotic tissue. The
harvested tumor tissues were then equally divided into small pieces
of 1 mm3 each. One 1-mm piece of the above tumor tissue
fragments was inserted into the incision of the liver of each
mouse. The tumor-bearing mice were randomized into 2 groups (7
mice/group) and treated with PBS or CQ (80 mg/kg, s.c.), twice a
day respectively, on a 3-day-on/2-day-off schedule for 25 days.
Tumor growth was observed, and the tumor area was recorded twice a
week with a Fluorvivo Model-300 imaging system as previously
described (16,17). Briefly, whole-body images of each
mouse were obtained with a Fluorvivo Model-300 imaging system
(INDEC BioSystems, Santa Clara, CA, USA) in live animals. High
resolution images were directly captured on a personal computer
(Axis 945GM) and were analyzed using Power Analysis Station (INDEC
BioSystems). At the time of sacrifice, tumor tissues of the mice
were collected, photographed and weighed.
Statistical analysis
The statistical significance of differences between
groups was assessed using GraphPad Prism 5 software. The unpaired
two-tailed t-test was used for the comparison of parameters between
groups. The level of significance was set at P<0.05.
Results
CQ inhibits the proliferation of liver
cancer cells
To assess the anticancer activity of CQ on liver
cancer, we first investigated the effect of CQ on the proliferation
of two liver cancer cell lines HepG2 and Huh7. Morphologically, we
found that both HepG2 and Huh7 cells shrunk and floated with
increasing concentrations of CQ (Fig.
1A). As a result, the viability of both cell lines decreased
with CQ treatment in a time- and dose-dependent manner using
ATPLite assay (Fig. 1B). At the
same time, CQ induced a dose-dependent inhibition of cell colony
formation (Fig. 1C) of liver cancer
cells. Cytotoxicity of CQ was assessed in both cell lines (Fig. 1D) using the CCK-8 assay, which was
in accordance with the results of the ATPLite assay (Fig. 1B).
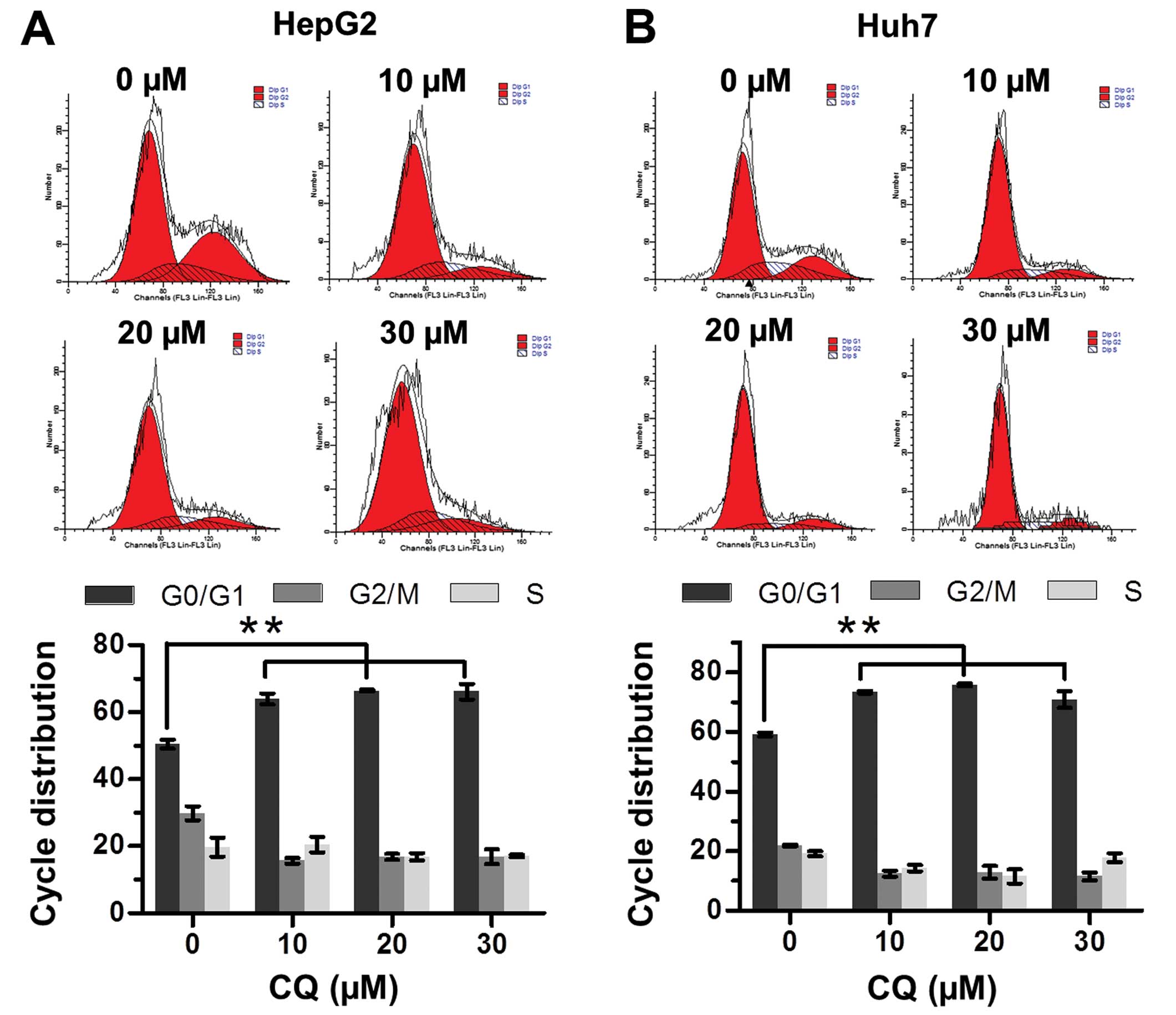
CQ induces G0/G1 cell cycle arrest in
liver cancer cells
The effect of CQ on cell cycle progression was
examined to elucidate the mechanism of its antiproliferative
activity. Flow cytometric analysis showed that CQ triggered G0/G1
cell cycle arrest in both the HepG2 and Huh7 cells in a
dose-dependent manner (Fig. 2A and
B).
CQ induces apoptosis in liver cancer
cells
We next examined whether apoptosis was also
responsible for the anticancer activity of CQ. The results showed
that CQ treatment led to the accumulation of cells in early-
(Annexin v+/PI−) and late-stage (Annexin
v+/PI+) apoptosis in a dose-dependent manner
(Fig. 3A). At the same time, CQ
treatment also led to the increased activity of caspase-3 (Fig. 3B), and proteolytic cleavage of PARP
and caspase-3 (Fig. 3C).
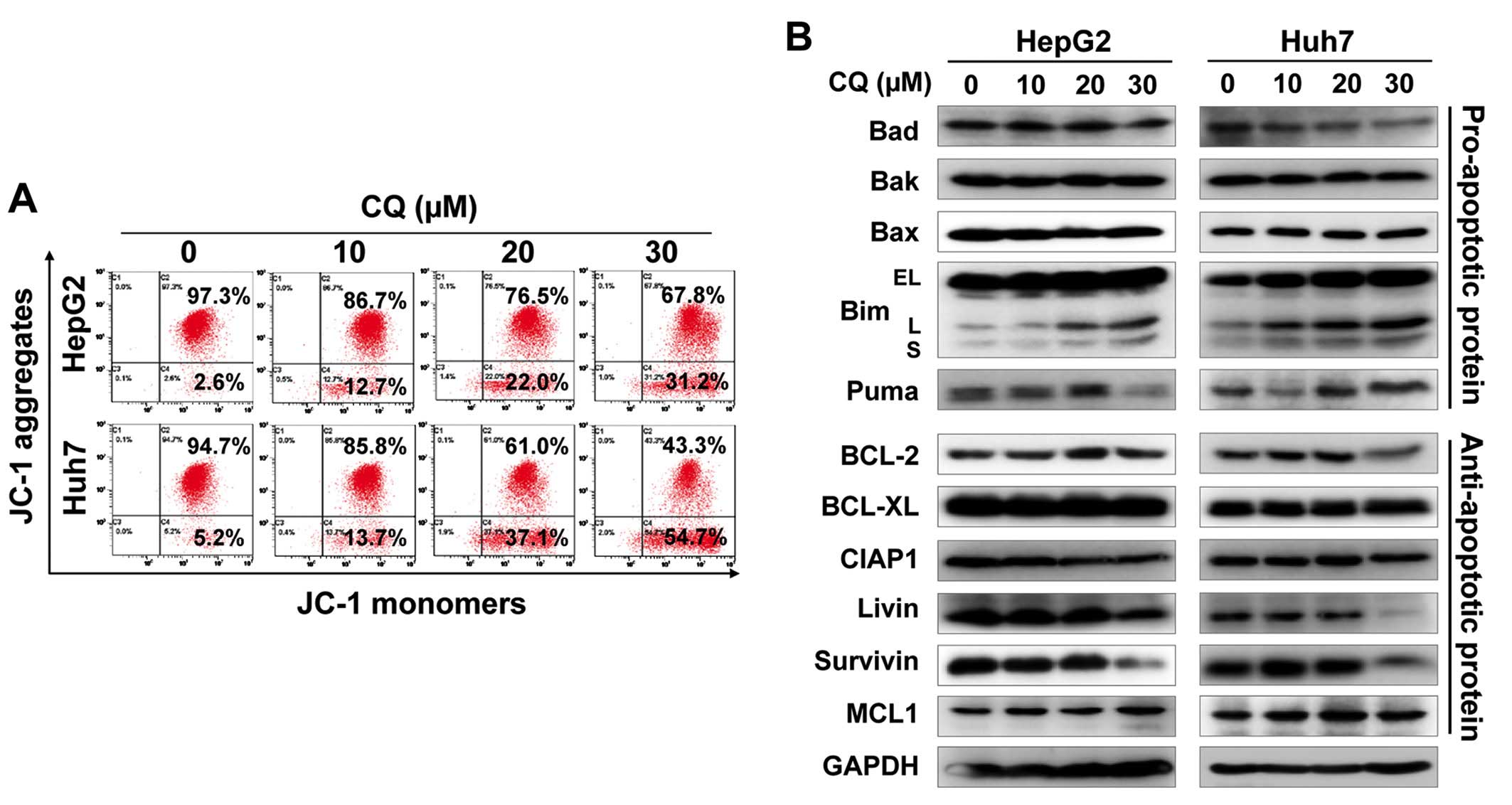
Furthermore, we found that CQ induced the loss of mitochondrial
membrane potential (ΔΨm), a classical marker of the activation of
intrinsic apoptosis (Fig. 4A),
which suggested that CQ triggered mitochondrial apoptosis.
To further explore the potential mechanism of
apoptosis, we systematically investigated the effect of CQ on the
expression of pro-apoptotic and anti-apoptotic proteins. Among
these proteins, pro-apoptotic protein Bim was substantially
upregulated in both cell lines in a dose-dependent manner (Fig. 4B), suggesting that Bim may be
critical for CQ-mediated apoptosis.
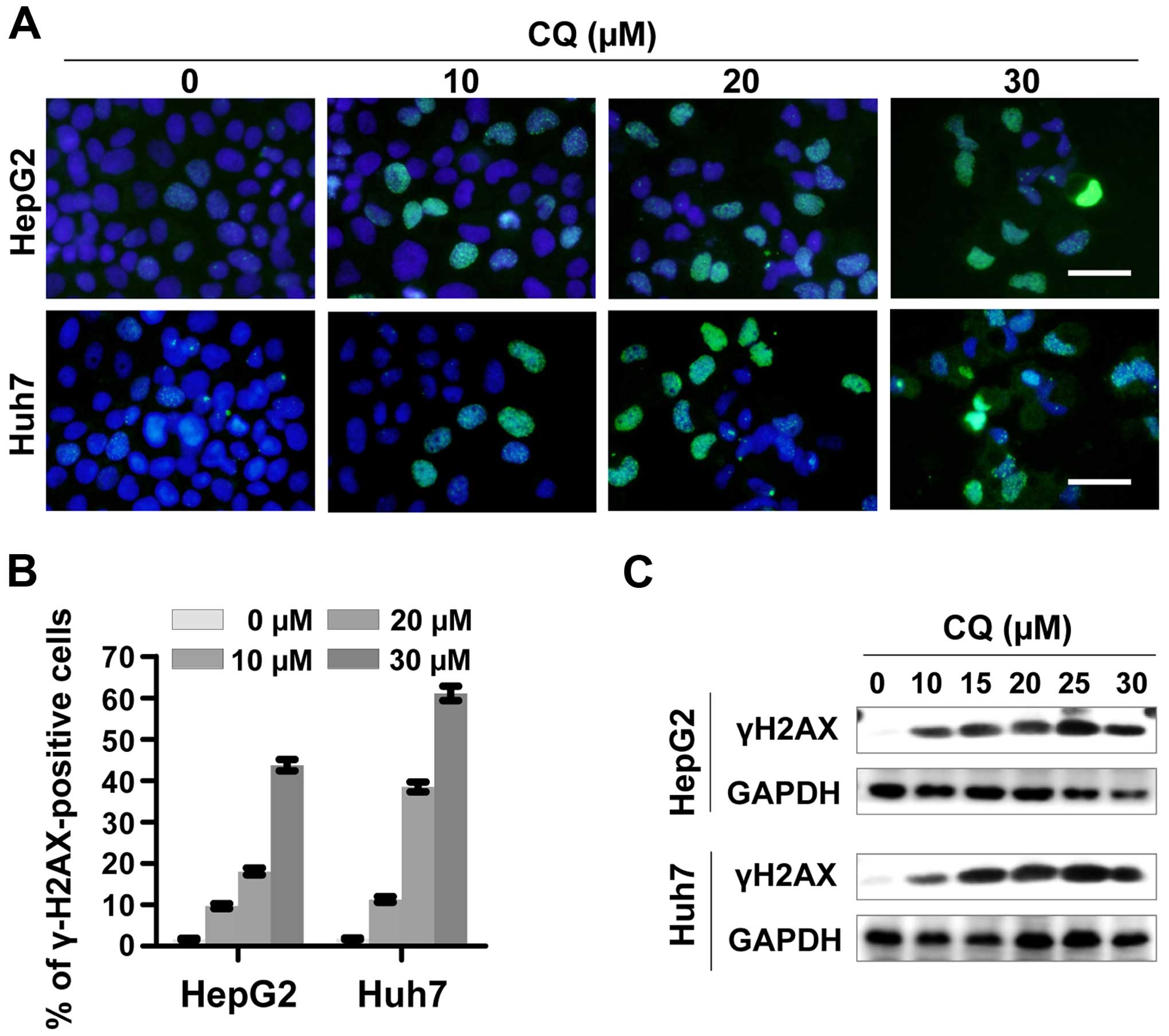
CQ induces DNA damage response
To determine whether CQ induces DNA damage response,
we examined the expression of phosphorylated histone H2AX at Ser139
(γH2AX), a surrogate marker of DNA double-strand breaks (DSBs) by
immunofluorescence and western blotting. CQ induced rapid and
sustained γH2AX foci in the HepG2 and Huh7 cells in a
dose-dependent manner (Fig. 5A and
B), which was further confirmed by western blot analysis
(Fig. 5C).
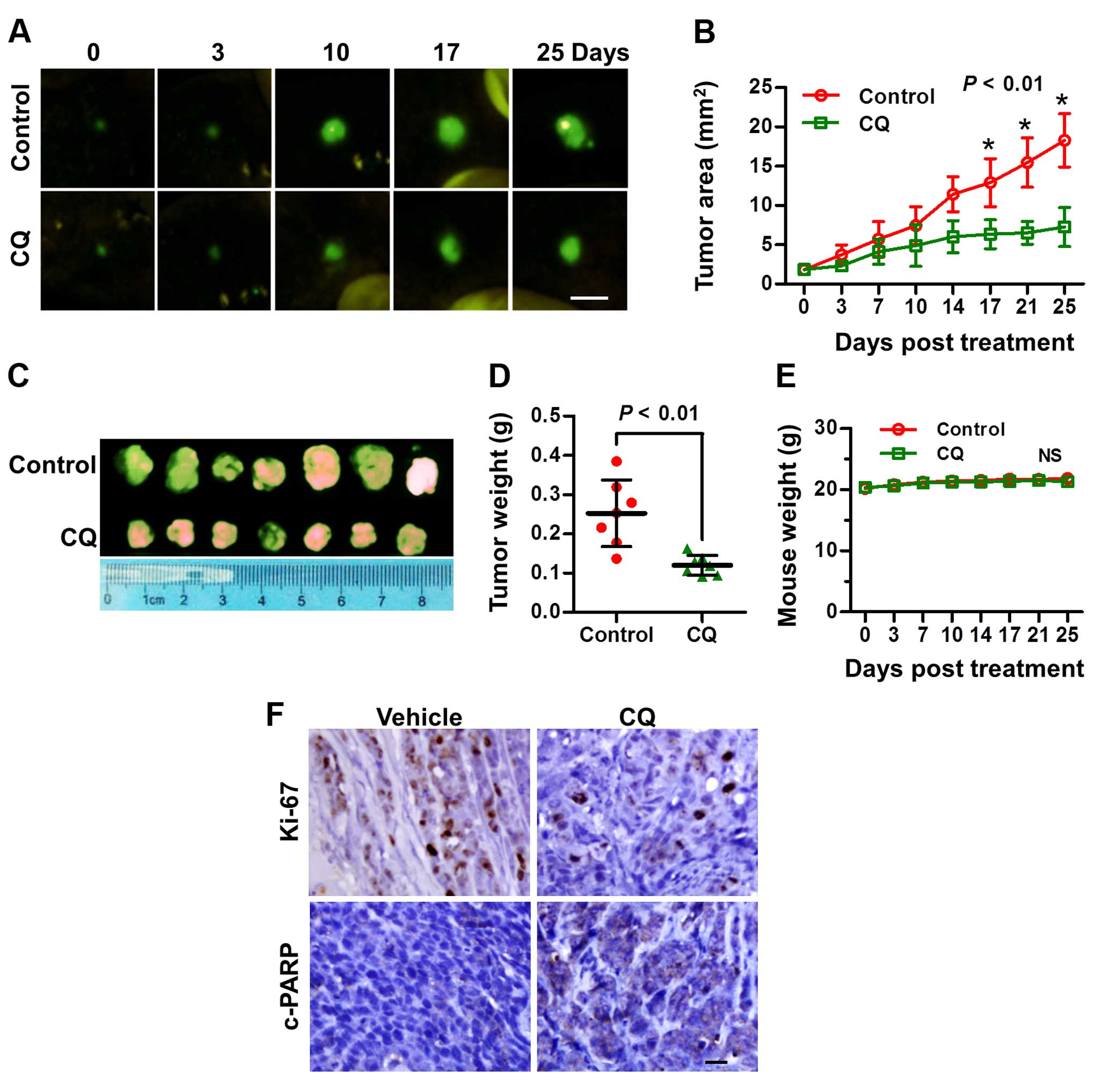
CQ inhibits the tumor growth of liver
cancer in vivo
Based on the above in vitro results, we next
explored the potential anticancer effect of CQ in vivo in an
orthotopic xenograft model of liver cancer. As expected, CQ led to
a substantial decrease in tumor growth and weight, compared with
the vehicle control (Fig. 6A–D),
while little effect on the body weight of mice was noted (Fig. 6E). Moreover, a significant reduction
in the proliferation marker Ki-67 and an increase in cleaved PARP
were observed in the mouse tumors following treatment with CQ
(Fig. 6F), suggesting that CQ
effectively inhibited tumor growth in vivo by inhibiting
liver cancer cell proliferation and inducing apoptosis.
Discussion
Recently, CQ has been widely used as a sensitizer of
radiotherapy and chemotherapy (5,6,8,9).
CQ was found to significantly promote the efficiency of traditional
chemotherapy drug [such as oxaliplatin (5,6)] or
tumor targeting drugs [such as sorafenib (18,19),
MLN4924 (15), proteasome
inhibitors (4)] in HCC xenografts.
CQ was also found to enhance the efficacy of transcatheter arterial
chemoembolization (TACE) in a rabbit VX2 liver tumor model
(7). However, the antitumor effects
and the related mechanisms of single CQ treatment for liver cancer
have not been defined. In the present study, we assessed and
validated the efficacy of a single treatment of CQ on liver cancer
cells in vitro and in an orthotopic xenograft of human liver
cancer in vivo. CQ had a profound effect on liver cancer
cell viability, induced G0/G1 cell cycle arrest and promoted liver
cancer cell apoptosis in both HepG2 [wild-type-p53 (20,21)]
and Huh7 [mutant-p53 (20,21)] cells.
Previous studies have shown that single treatment of
CQ exerted an antitumor effect in several types of tumors in a cell
type-dependent manner (22–24). CQ could induce cell death in a
subset of tumor cell lines; but the underlying molecular target and
mechanism are still not fully understood. Recently, Lakhter et
al reported that CQ promoted the apoptosis of melanoma cells by
stabilizing PUMA in a lysosomal protease-independent manner
(10). In the present study, we
found that treatment with CQ induced DNA damage, which is in
accordance with previous studies that CQ induces a genotoxic effect
(25,26). Further investigation of the
mechanism showed that CQ treatment led to loss of mitochondrial
membrane potential, which suggests that CQ treatment induces
mitochondrial apoptosis in liver cancer cells. By analyzing the
balance between pro-apoptotic and anti-apoptotic proteins, we found
that CQ treatment led to significant upregulation of pro-apoptotic
protein Bim in a dose-dependent manner. As a member of the BH3-only
proteins, Bim upregulation triggered cytochrome c release
from mitochondria and consequently induced the activation of
pro-caspase-9 (27). Previous
studies have shown that targeting Bim may be an effective
therapeutic strategy (27).
Treatment of tumor cells, such as colorectal cancer and melanoma
cell lines, with an inhibitor of the BRAF-MEK-ERK signaling pathway
increases the expression of Bim and induces Bim-dependent cell
death (28–30). It was also reported that Bim plays
an important role in gefitinibinduced cell death (31). These studies suggest that Bim is a
critical mediator of drug-induced apoptosis, which perhaps plays an
important role in CQ-induced apoptosis in liver cancer cells.
Together, our studies showed that single treatment
of CQ effectively suppressed the growth of liver cancer cells in
vitro and in vivo by triggering G0/G1 cell cycle arrest,
inducing DNA damage and apoptosis in liver cancer cells. These
findings extend our understanding and propose the use of CQ for the
treatment of liver cancer in single treatment or in
combination.
Acknowledgments
The present study was supported by the National
Natural Science Foundation Grant of China (grant nos. 81001102 and
81101894), and the Research Foundation of Education Bureau of Henan
Province, China (grant no. 15A310024)
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015.
|
|
2
|
Chong CR and Sullivan DJ Jr: New uses for
old drugs. Nature. 448:645–646. 2007.
|
|
3
|
Al-Bari MA: Chloroquine analogues in drug
discovery: New directions of uses, mechanisms of actions and toxic
manifestations from malaria to multifarious diseases. J Antimicrob
Chemother. 70:1608–1621. 2015.
|
|
4
|
Hui B, Shi YH, Ding ZB, Zhou J, Gu CY,
Peng YF, Yang H, Liu WR, Shi GM and Fan J: Proteasome inhibitor
interacts synergistically with autophagy inhibitor to suppress
proliferation and induce apoptosis in hepatocellular carcinoma.
Cancer. 118:5560–5571. 2012.
|
|
5
|
Ding ZB, Hui B, Shi YH, Zhou J, Peng YF,
Gu CY, Yang H, Shi GM, Ke AW, Wang XY, et al: Autophagy activation
in hepatocellular carcinoma contributes to the tolerance of
oxaliplatin via reactive oxygen species modulation. Clin Cancer
Res. 17:6229–6238. 2011.
|
|
6
|
Du H, Yang W, Chen L, Shi M, Seewoo V,
Wang J, Lin A, Liu Z and Qiu W: Role of autophagy in resistance to
oxaliplatin in hepatocellular carcinoma cells. Oncol Rep.
27:143–150. 2012.
|
|
7
|
Gao L, Song JR, Zhang JW, Zhao X, Zhao QD,
Sun K, Deng WJ, Li R, Lv G, Cheng HY, et al: Chloroquine promotes
the anticancer effect of TACE in a rabbit VX2 liver tumor model.
Int J Biol Sci. 9:322–330. 2013.
|
|
8
|
Liang X, Tang J, Liang Y, Jin R and Cai X:
Suppression of autophagy by chloroquine sensitizes
5-fluorouracil-mediated cell death in gallbladder carcinoma cells.
Cell Biosci. 4:102014.
|
|
9
|
Ratikan JA, Sayre JW and Schaue D:
Chloroquine engages the immune system to eradicate irradiated
breast tumors in mice. Int J Radiat Oncol Biol Phys. 87:761–768.
2013.
|
|
10
|
Lakhter AJ, Sahu RP, Sun Y, Kaufmann WK,
Androphy EJ, Travers JB and Naidu SR: Chloroquine promotes
apoptosis in melanoma cells by inhibiting BH3 domain-mediated PuMA
degradation. J Invest Dermatol. 133:2247–2254. 2013.
|
|
11
|
Geng Y, Kohli L, Klocke BJ and Roth KA:
Chloroquine-induced autophagic vacuole accumulation and cell death
in glioma cells is p53 independent. Neuro Oncol. 12:473–481.
2010.
|
|
12
|
Fan C, Wang W, Zhao B, Zhang S and Miao J:
Chloroquine inhibits cell growth and induces cell death in A549
lung cancer cells. Bioorg Med Chem. 14:3218–3222. 2006.
|
|
13
|
Jiang PD, Zhao YL, Deng XQ, Mao YQ, Shi W,
Tang QQ, Li ZG, Zheng YZ, Yang SY and Wei YQ: Antitumor and
antimetastatic activities of chloroquine diphosphate in a murine
model of breast cancer. Biomed Pharmacother. 64:609–614. 2010.
|
|
14
|
Rahim R and Strobl JS: Hydroxychloroquine,
chloroquine, and all-trans retinoic acid regulate growth,
survival, and histone acetylation in breast cancer cells.
Anticancer Drugs. 20:736–745. 2009.
|
|
15
|
Chen P, Hu T, Liang Y, Jiang Y, Pan Y, Li
C, Zhang P, Wei D, Li P, Jeong LS, et al: Synergistic inhibition of
autophagy and neddylation pathways as a novel therapeutic approach
for targeting liver cancer. Oncotarget. 6:9002–9017. 2015.
|
|
16
|
Hoffman RM: The multiple uses of
fluorescent proteins to visualize cancer in vivo. Nat Rev Cancer.
5:796–806. 2005.
|
|
17
|
Hoffman RM and Yang M: Whole-body imaging
with fluorescent proteins. Nat Protoc. 1:1429–1438. 2006.
|
|
18
|
Fischer TD, Wang JH, Vlada A, Kim JS and
Behrns KE: Role of autophagy in differential sensitivity of
hepatocarcinoma cells to sorafenib. World J Hepatol. 6:752–758.
2014.
|
|
19
|
Shi YH, Ding ZB, Zhou J, Hui B, Shi GM, Ke
AW, Wang XY, Dai Z, Peng YF, Gu CY, et al: Targeting autophagy
enhances sorafenib lethality for hepatocellular carcinoma via ER
stress-related apoptosis. Autophagy. 7:1159–1172. 2011.
|
|
20
|
Brito AF, Abrantes AM, Pinto-Costa C,
Gomes AR, Mamede AC, Casalta-Lopes J, Gonçalves AC,
Sarmento-Ribeiro AB, Tralhão JG and Botelho MF: Hepatocellular
carcinoma and chemotherapy: The role of p53. Chemotherapy.
58:381–386. 2012.
|
|
21
|
Müller M, Strand S, Hug H, Heinemann EM,
Walczak H, Hofmann WJ, Stremmel W, Krammer PH and Galle PR:
Drug-induced apoptosis in hepatoma cells is mediated by the CD95
(APo-1/Fas) receptor/ligand system and involves activation of
wild-type p53. J Clin Invest. 99:403–413. 1997.
|
|
22
|
Balic A, Sørensen MD, Trabulo SM, Sainz B
Jr, Cioffi M, Vieira CR, Miranda-Lorenzo I, Hidalgo M, Kleeff J,
Erkan M, et al: Chloroquine targets pancreatic cancer stem cells
via inhibition of CXCR4 and hedgehog signaling. Mol Cancer Ther.
13:1758–1771. 2014.
|
|
23
|
Zheng Y, Zhao YL, Deng X, Yang S, Mao Y,
Li Z, Jiang P, Zhao X and Wei Y: Chloroquine inhibits colon cancer
cell growth in vitro and tumor growth in vivo via induction of
apoptosis. Cancer Invest. 27:286–292. 2009.
|
|
24
|
Kim EL, Wüstenberg R, Rübsam A,
Schmitz-Salue C, Warnecke G, Bücker EM, Pettkus N, Speidel D, Rohde
V, Schulz-Schaeffer W, et al: Chloroquine activates the p53 pathway
and induces apoptosis in human glioma cells. Neuro oncol.
12:389–400. 2010.
|
|
25
|
Farombi EO: Genotoxicity of chloroquine in
rat liver cells: Protective role of free radical scavengers. Cell
Biol Toxicol. 22:159–167. 2006.
|
|
26
|
Krajewski WA: Alterations in the
internucleosomal DNA helical twist in chromatin of human
erythroleukemia cells in vivo influences the chromatin higher-order
folding. FEBS Lett. 361:149–152. 1995.
|
|
27
|
Akiyama T, Dass CR and Choong PF:
Bim-targeted cancer therapy: A link between drug action and
underlying molecular changes. Mol Cancer Ther. 8:3173–3180.
2009.
|
|
28
|
Gillings AS, Balmanno K, Wiggins CM,
Johnson M and Cook SJ: Apoptosis and autophagy: BIM as a mediator
of tumour cell death in response to oncogene-targeted therapeutics.
FEBS J. 276:6050–6062. 2009.
|
|
29
|
Wickenden JA, Jin H, Johnson M, Gillings
AS, Newson C, Austin M, Chell SD, Balmanno K, Pritchard CA and Cook
SJ: Colorectal cancer cells with the BRAFv600E mutation
are addicted to the ERK1/2 pathway for growth factor-independent
survival and repression of BIM. Oncogene. 27:7150–7161. 2008.
|
|
30
|
Cartlidge RA, Thomas GR, Cagnol S, Jong
KA, Molton SA, Finch AJ and McMahon M: Oncogenic
BRAFv600E inhibits BIM expression to promote melanoma
cell survival. Pigment Cell Melanoma Res. 21:534–544. 2008.
|
|
31
|
Cragg MS, Kuroda J, Puthalakath H, Huang
DC and Strasser A: Gefitinib-induced killing of NSCLC cell lines
expressing mutant EGFR requires BIM and can be enhanced by
BH3 mimetics. PLoS Med. 4:1681–1690. 2007.
|